
Yeast Surface Display of Protein Addresses Confers Robust Storage and Access of DNA-Based Data

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
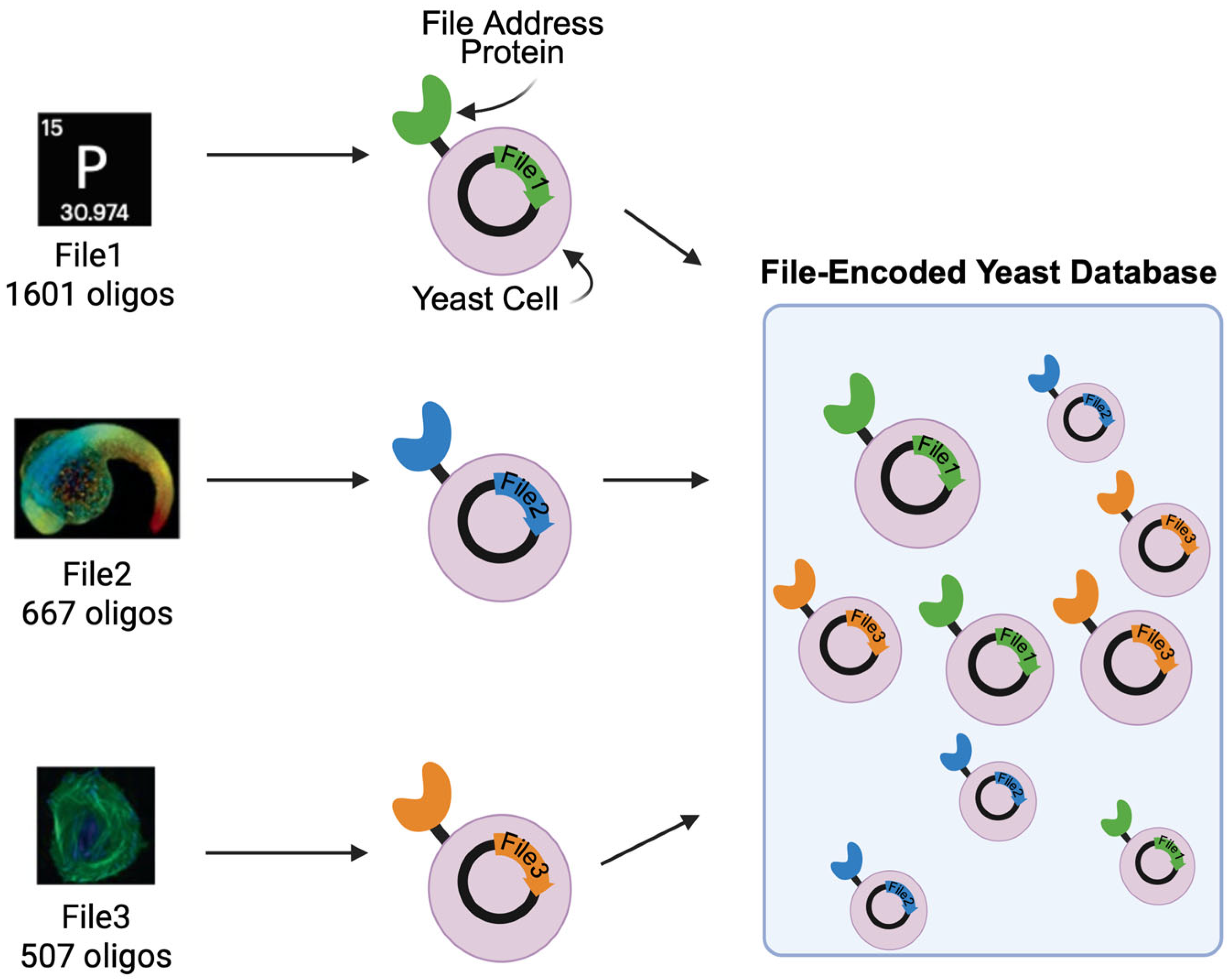
1. Introduction
2. Materials and Methods
2.1. Yeast Strains
2.2. Labeling Epitope Tag Yeast Before and After File Transformation with Flow Cytometry
2.3. File DNA Synthesis
2.4. Inserting Files into Selected Yeast
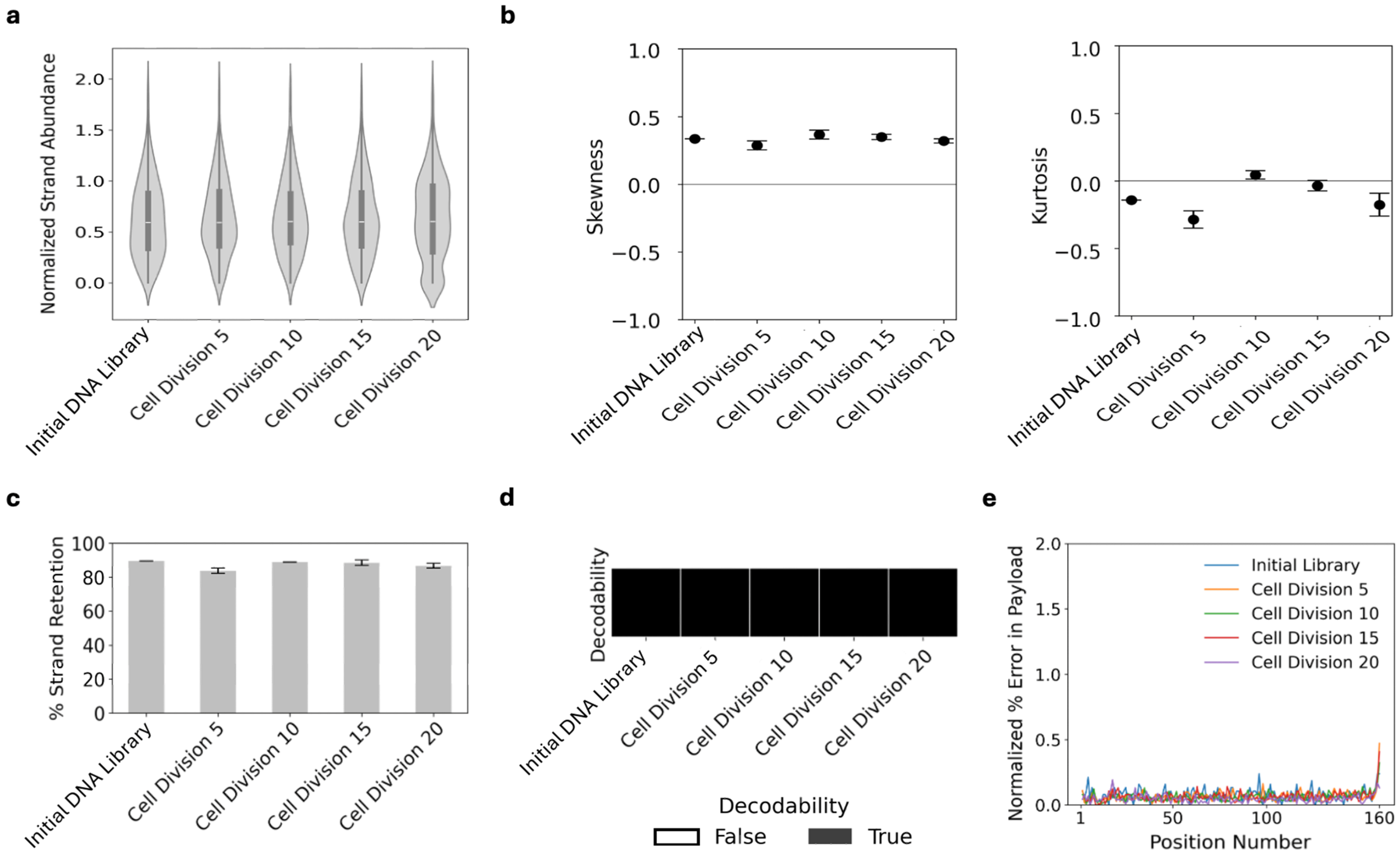
2.5. Analyzing the File3 Yeast Through Multiple Cell Divisions
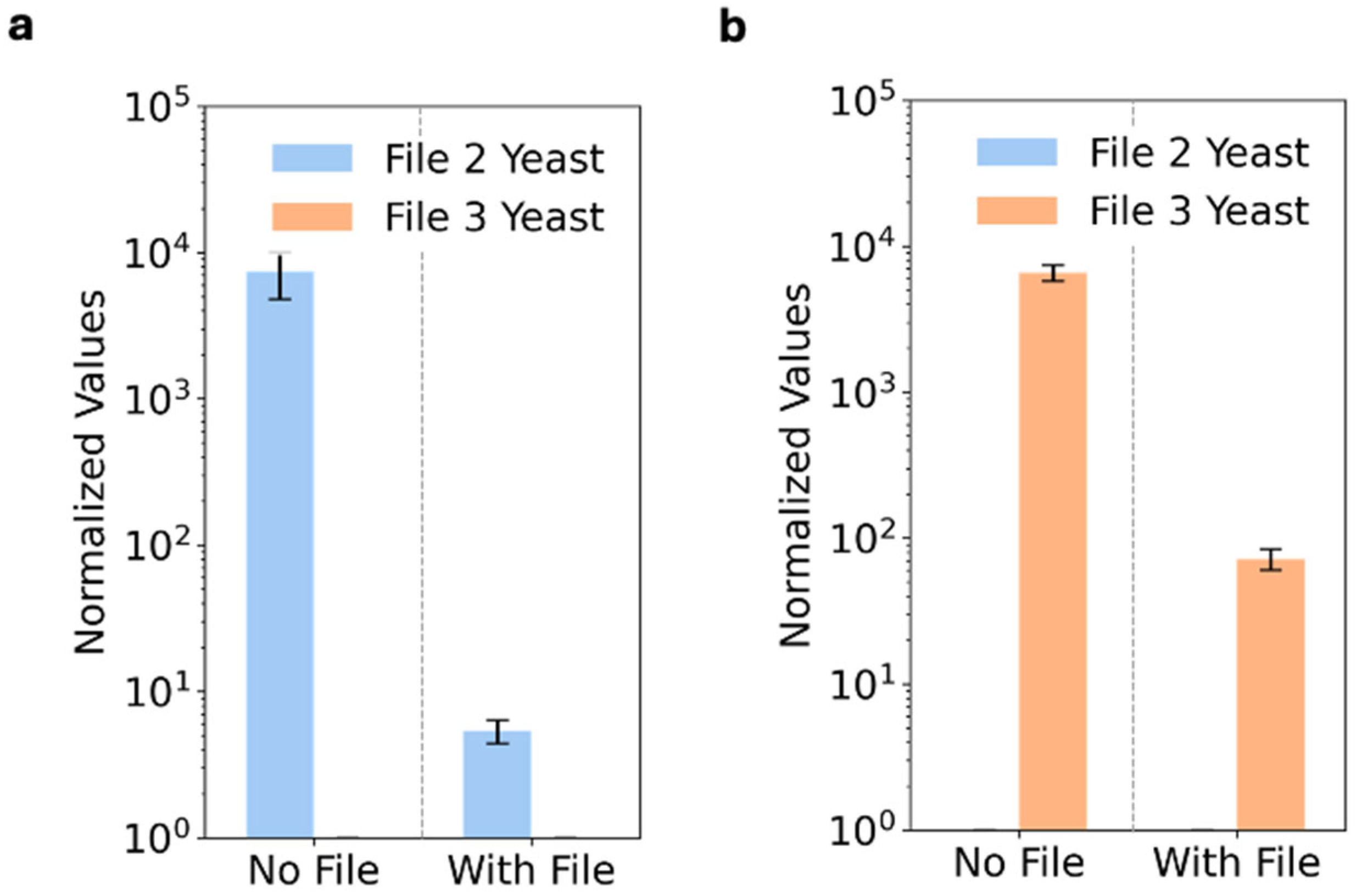
2.6. Yeast Sorting and File Access via Epitope Tag
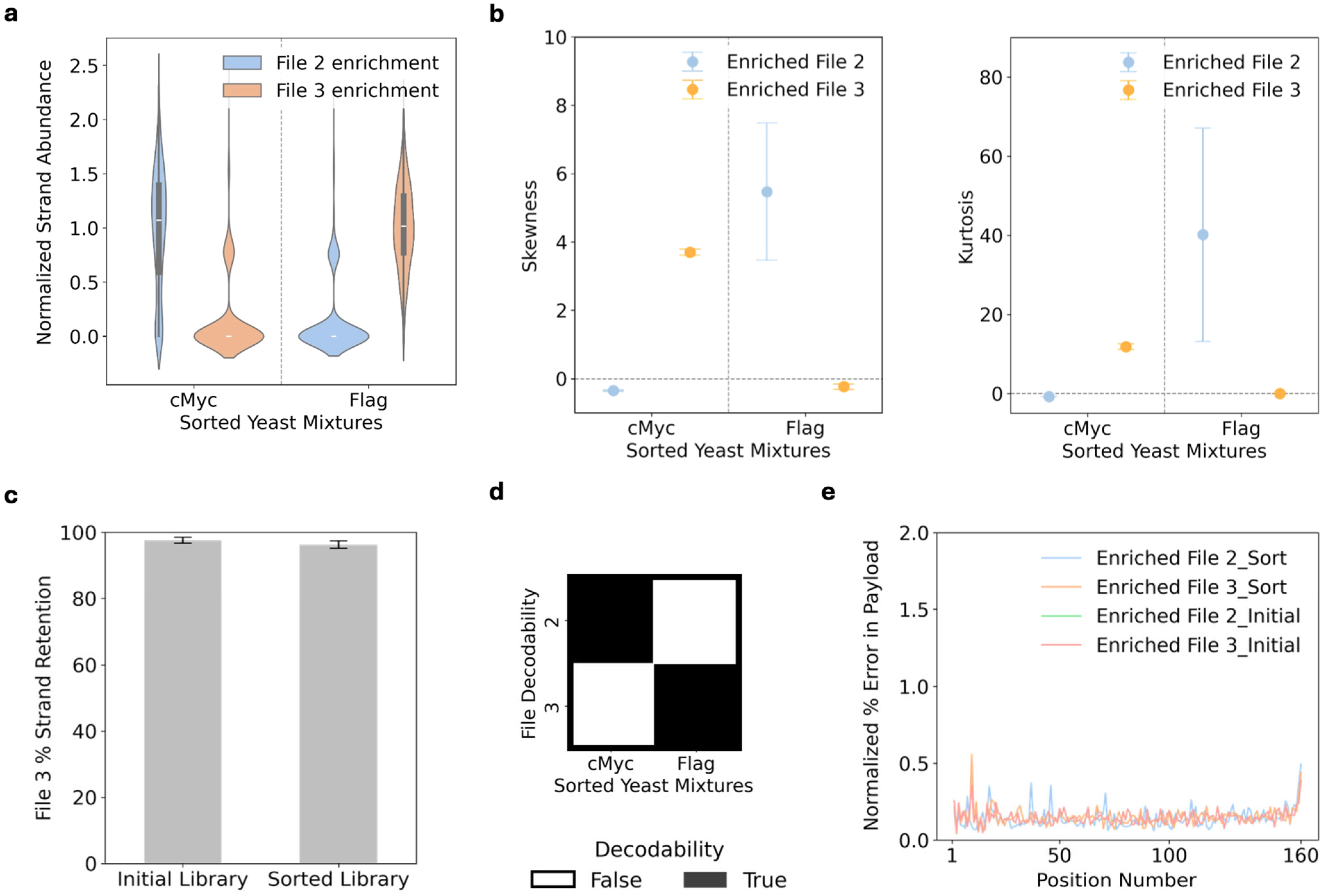
2.7. Labeling the Histone Binder Yeast After File Transformation with Flow Cytometry
3. Results
3.1. Data Transformed into Yeast Populations Maintain Their Fidelity Through Cell Divisions
3.2. Transformation of Yeast with File DNA Partially Impacts Display Effiicency but Not Labeling Specificity
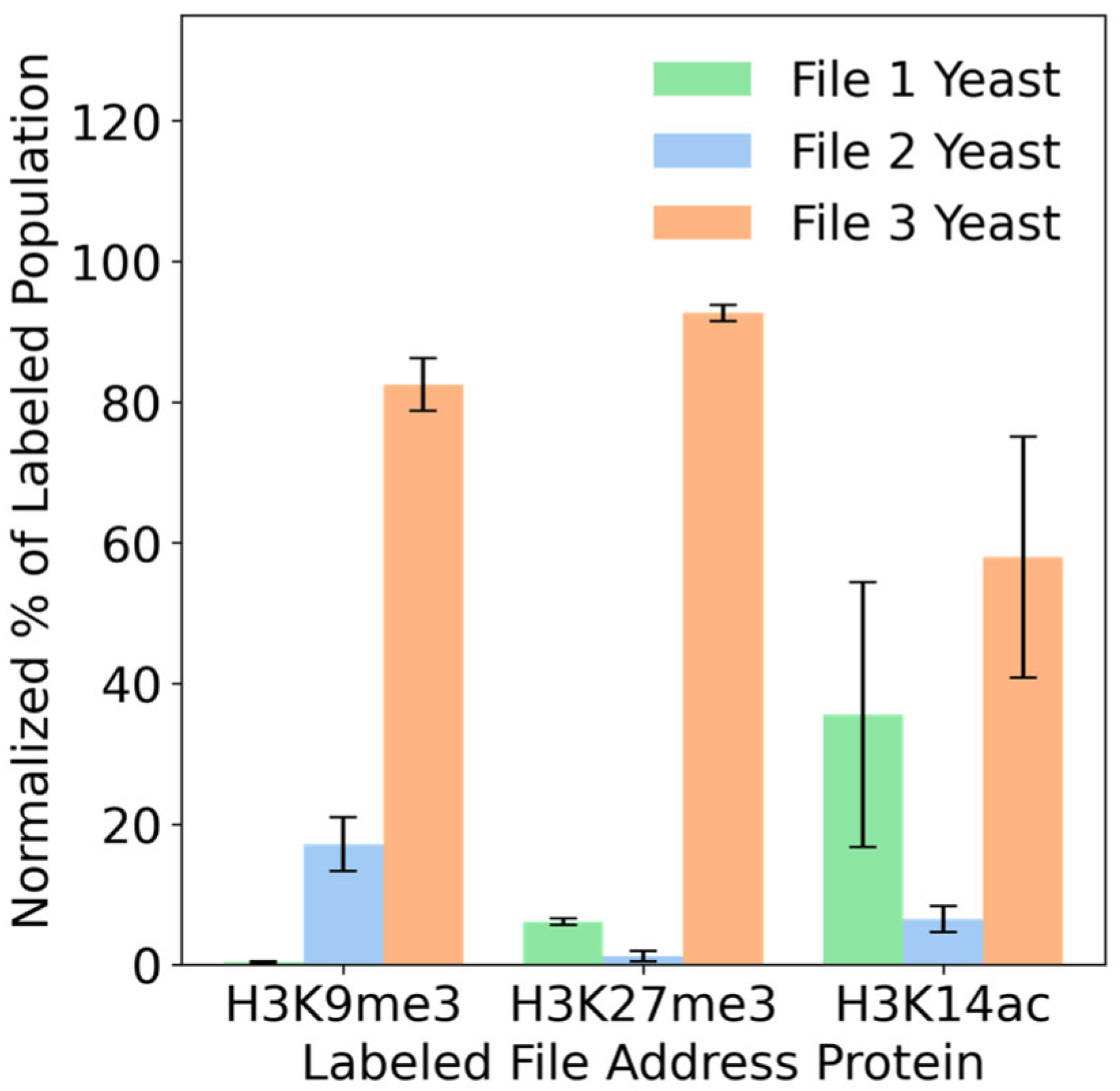
3.3. Files Accessed with Specificity via Protein Addresses
3.4. Combinatorial Peptide Binding Enables Multiplexed File Access
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Organick, L.; Chen, Y.-J.; Ang, S.D.; Lopez, R.; Liu, X.; Strauss, K.; Ceze, L. Probing the Physical Limits of Reliable DNA Data Retrieval. Nat. Commun. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Tomek, K.J.; Volkel, K.; Simpson, A.; Hauss, A.G.; Indermaur, E.W.; Tuck, J.M.; Keung, A.J. Driving the Scalability of DNA-Based Information Storage Systems. ACS Synth. Biol. 2019, 8, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Tabatabaei, S.K.; Hossein Tabatabaei Yazdi, S.M.; Hernandez, A.G.; Schroeder, C.M.; Milenkovic, O. Rewritable Two-Dimensional DNA-Based Data Storage with Machine Learning Reconstruction. Nat. Commun. 2022, 13, 2984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, F.; Chao, J.; Xie, M.; Liu, H.; Pan, M.; Kopperger, E.; Liu, X.; Li, Q.; Shi, J.; et al. DNA Origami Cryptography for Secure Communication. Nat. Commun. 2019, 10, 5469. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, R.; Sun, F.; Lin, Y.; Liang, Y.; Teng, J.; Liu, N.; Ouyang, Q.; Qian, L.; Yan, H. Parallel Molecular Data Storage by Printing Epigenetic Bits on DNA. Nature 2024, 634, 824–832. [Google Scholar] [CrossRef]
- Banal, J.L.; Shepherd, T.R.; Berleant, J.; Huang, H.; Reyes, M.; Ackerman, C.M.; Blainey, P.C.; Bathe, M. Random Access DNA Memory Using Boolean Search in an Archival File Storage System. Nat. Mater. 2021, 20, 1272–1280. [Google Scholar] [CrossRef]
- Bögels, B.W.A.; Nguyen, B.H.; Ward, D.; Gascoigne, L.; Schrijver, D.P.; Makri Pistikou, A.-M.; Joesaar, A.; Yang, S.; Voets, I.K.; Mulder, W.J.M.; et al. DNA Storage in Thermoresponsive Microcapsules for Repeated Random Multiplexed Data Access. Nat. Nanotechnol. 2023, 18, 912–921. [Google Scholar] [CrossRef]
- Lin, K.N.; Volkel, K.; Cao, C.; Hook, P.W.; Polak, R.E.; Clark, A.S.; San Miguel, A.; Timp, W.; Tuck, J.M.; Velev, O.D.; et al. A Primordial DNA Store and Compute Engine. Nat. Nanotechnol. 2024, 19, 1654–1664. [Google Scholar] [CrossRef]
- Zhong, W.; Wang, S.; Geng, C.; Zheng, Y.; Bai, S.; Cao, X.; Liu, K.; Yang, Y.; Lu, C.; Jiang, X. Multiplexed Random Access Approach to DNA Microspheres for High-Capacity Data Storage. Adv. Funct. Mater. 2024, 34, 2408852. [Google Scholar] [CrossRef]
- Ball, N.; Kagawa, H.; Hindupur, A.; Hogan, J.A. Development of Storage Methods for Saccharomyces Strains to Be Utilized for In Situ Nutrient Production in Long-Duration Space Missions. In Proceedings of the 47th International Conference on Environmental Systems, Charleston, SC, USA, 16–20 July 2017. [Google Scholar]
- Shusta, E.V.; Pepper, L.R.; Cho, Y.K.; Boder, E.T. A Decade of Yeast Surface Display Technology: Where Are We Now? Comb. Chem. High Throughput Screen. 2008, 11, 127. [Google Scholar] [CrossRef]
- Chen, W.; Han, M.; Zhou, J.; Ge, Q.; Wang, P.; Zhang, X.; Zhu, S.; Song, L.; Yuan, Y. An Artificial Chromosome for Data Storage. Natl. Sci. Rev. 2021, 8, 2021. [Google Scholar] [CrossRef]
- Annaluru, N.; Muller, H.; Mitchell, L.A.; Ramalingam, S.; Stracquadanio, G.; Richardson, S.M.; Dymond, J.S.; Kuang, Z.; Schifele, L.Z.; Cooper, E.M.; et al. Total Synthesis of a Functional Designer Eukaryotic Chromosome. Science 2014, 344, 55–58. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Ma, Y.; Tian, F.; Zhao, G.; Wu, Y.; Yuan, Y. YLC-Assembly: Large DNA Assembly via Yeast Life Cycle. Nucleic Acids Res. 2023, 51, 8283–8292. [Google Scholar] [CrossRef]
- Lin, Q.; Jia, B.; Luo, J.; Yang, K.; Zeller, K.I.; Zhang, W.; Xu, Z.; Stracquadanio, G.; Bader, J.S.; Boeke, J.; et al. RADOM, an Efficient in Vivo Method for Assembling Designed DNA Fragments up to 10 Kb Long in Saccharomyces Cerevisiae. ACS Synth. Biol. 2015, 4, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Qiao, H.; Gao, Y.; Wang, Z.; Qiao, X.; Chen, X.; Qi, H. A Mixed Culture of Bacterial Cells Enables an Economic DNA Storage on a Large Scale. Nat. Commun. Biol. 2020, 3, 416. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, Y.; Li, J.; Wang, F.; Wang, F.; Ma, C.; Chen, D.; Jiang, X.; Fan, C.; Zhang, H.; et al. In Vivo Processing of Digital Information Molecularly with Targeted Specificity and Robust Reliability. Sci. Adv. 2022, 8, 7415. [Google Scholar] [CrossRef]
- Bhattarai-Kline, S.; Lear, S.K.; Shipman, S.L. One-Step Data Storage in Cellular DNA. Nat. Chem. Biol. 2021, 17, 232–233. [Google Scholar] [CrossRef]
- Liu, F.; Li, J.; Zhang, T.; Chen, J.; Ho, C.L. Engineered Spore-Forming Bacillus as a Microbial Vessel for Long-Term DNA Data Storage. ACS Synth. Biol. 2022, 11, 3583–3591. [Google Scholar] [CrossRef]
- Hou, Z.; Qiang, W.; Wang, X.; Chen, X.; Hu, X.; Han, X.; Shen, W.; Zhang, B.; Xing, P.; Shi, W.; et al. “Cell Disk” DNA Storage System Capable of Random Reading and Rewriting. Adv. Sci. 2024, 11, 2305921. [Google Scholar] [CrossRef]
- Luo, H.; Huang, W.; He, Z.; Fang, Y.; Tian, Y.; Xiong, Z. Engineered Living Memory Microspheroid-Based Archival File System for Random Accessible In Vivo DNA Storage. Adv. Mater. 2025, 37, 2415358. [Google Scholar] [CrossRef]
- Meanor, J.N.; Keung, A.J.; Rao, B.M. Modified Histone Peptides Linked to Magnetic Beads Reduce Binding Specificity. Int. J. Mol. Sci. 2022, 23, 1691. [Google Scholar] [CrossRef]
- Waldman, A.C.; Rao, B.M.; Keung, A.J. Mapping the Residue Specificities of Epigenome Enzymes by Yeast Surface Display. Cell Chem. Biol. 2021, 28, 1772–1779.e4. [Google Scholar] [CrossRef] [PubMed]
- Bacon, K.; Bowen, J.; Reese, H.; Rao, B.M.; Menegatti, S. Use of Target-Displaying Magnetized Yeast in Screening MRNA-Display Peptide Libraries to Identify Ligands. ACS Comb. Sci. 2020, 22, 738–744. [Google Scholar] [CrossRef]
- Matange, K.; Tuck, J.M.; Keung, A.J. DNA Stability: A Central Design Consideration for DNA Data Storage Systems. Nat. Commun. 2021, 12, 1358. [Google Scholar] [CrossRef] [PubMed]
- Volkel, K.D.; Lin, K.N.; Hook, P.W.; Timp, W.; Keung, A.J.; Tuck, J.M. FrameD: Framework for DNA-Based Data Storage Design, Verification, and Validation. Bioinformatics 2023, 39, 572. [Google Scholar] [CrossRef]
- Cruz-Teran, C.A.; Bacon, K.; McArthur, N.; Rao, B.M. An Engineered Sso7d Variant Enables Efficient Magnetization of Yeast Cells. ACS Comb. Sci. 2018, 20, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.D.; Murphy, K.P. Protein Structure and the Energetics of Protein Stability. Chem. Rev. 1997, 97, 1251–1267. [Google Scholar] [CrossRef]
- Richardson, S.M.; Mitchell, L.A.; Stracquadanio, G.; Yang, K.; Dymond, J.S.; Dicarlo, J.E.; Lee, D.; Huang, C.L.V.; Chandrasegaran, S.; Cai, Y.; et al. Design of a Synthetic Yeast Genome. Science 2017, 355, 1040–1044. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 Genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef]
- Engel, S.R.; Dietrich, F.S.; Fisk, D.G.; Binkley, G.; Balakrishnan, R.; Costanzo, M.C.; Dwight, S.S.; Hitz, B.C.; Karra, K.; Nash, R.S.; et al. The Reference Genome Sequence of Saccharomyces Cerevisiae: Then and Now. G3 Genes Genomes Genet. 2014, 4, 389–398. [Google Scholar] [CrossRef]
- Caspeta, L.; Navarrete, P.C.S. Reduction of the Saccharomyces Cerevisiae Genome: Challenges and Perspectives. Minimal Cells: Des. Constr. Biotechnol. Appl. 2020, 117–139. [Google Scholar] [CrossRef]
- Briney, B.; Inderbitzin, A.; Joyce, C.; Burton, D.R. Commonality despite Exceptional Diversity in the Baseline Human Antibody Repertoire. Nature 2019, 566, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Boldridge, W.C.; Ljubetic, A.; Kim, H.; Lubock, N.; Szilagyi, D.; Lee, J.; Jerala, R.; Kosuri, S. A Multiplexed Bacterial Two-Hybrid for Rapid Characterization of Protein–Protein Interactions and Iterative Protein Design. Nat. Commun. 2023, 14, 1–11. [Google Scholar] [CrossRef]
- Chen, Z.; Boyken, S.E.; Jia, M.; Busch, F.; Flores-Solis, D.; Bick, M.J.; Lu, P.; VanAernum, Z.L.; Sahasrabuddhe, A.; Langan, R.A.; et al. Programmable Design of Orthogonal Protein Heterodimers. Nature 2019, 565, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Luo, Y.; Gao, H.; Li, F.; Li, J.; Chen, Y.; You, R.; Lv, H.; Hua, K.; Jiang, R.; et al. Toward a Unified Information Framework for Cell Atlas Assembly. Natl. Sci. Rev. 2022, 9, 179. [Google Scholar] [CrossRef]
- Estridge, R.C.; Yagci, Z.B.; Sen, D.; Johnson, T.J.; Kelkar, G.R.; Ptacek, T.S.; Simon, J.M.; Keung, A.J. Loss of UBE3A Impacts Both Neuronal and Non-Neuronal Cells in Human Cerebral Organoids. Commun. Biol. 2025, 8, 838. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.N.; Brihadiswaran, G.; Rao, B.M.; Tuck, J.M.; Keung, A.J. Yeast Surface Display of Protein Addresses Confers Robust Storage and Access of DNA-Based Data. DNA 2025, 5, 34. https://doi.org/10.3390/dna5030034
Lee MN, Brihadiswaran G, Rao BM, Tuck JM, Keung AJ. Yeast Surface Display of Protein Addresses Confers Robust Storage and Access of DNA-Based Data. DNA. 2025; 5(3):34. https://doi.org/10.3390/dna5030034
Chicago/Turabian StyleLee, Magdelene N., Gunavaran Brihadiswaran, Balaji M. Rao, James M. Tuck, and Albert J. Keung. 2025. "Yeast Surface Display of Protein Addresses Confers Robust Storage and Access of DNA-Based Data" DNA 5, no. 3: 34. https://doi.org/10.3390/dna5030034
APA StyleLee, M. N., Brihadiswaran, G., Rao, B. M., Tuck, J. M., & Keung, A. J. (2025). Yeast Surface Display of Protein Addresses Confers Robust Storage and Access of DNA-Based Data. DNA, 5(3), 34. https://doi.org/10.3390/dna5030034