
Challenges in the Identification of Environmental Bacterial Isolates from a Pharmaceutical Industry Facility by 16S rRNA Gene Sequences

 , ,
, ,
Abstract
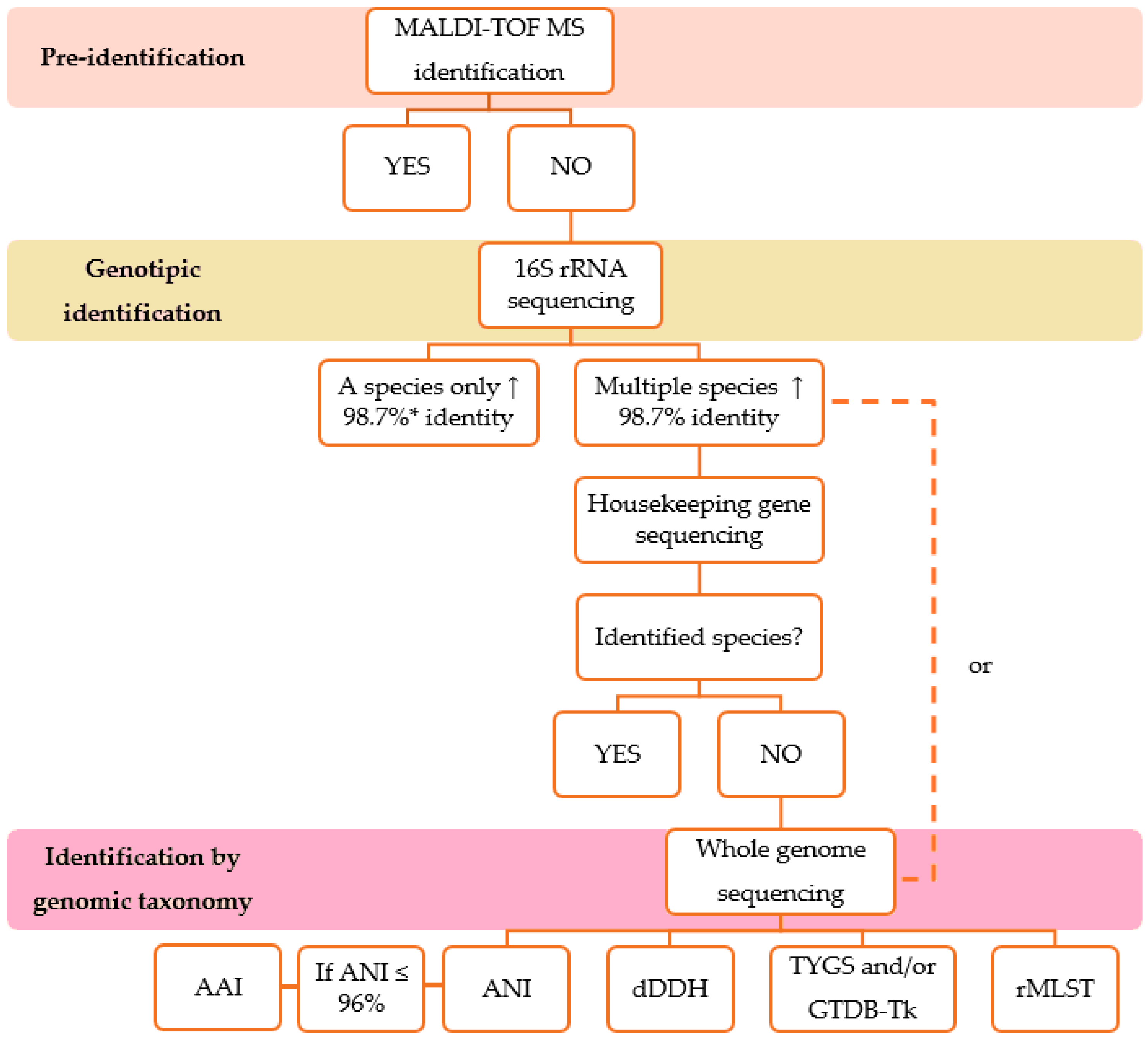
1. Background
2. Phenotypic Identification
3. MALDI-TOF MS
4. Analysis of the 16S Ribosomal Gene Sequences
5. Analysis of the Housekeeping Gene Sequences
6. Genomic Taxonomy Tools
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAI | Average Amino Acid Identity |
| ANI | Average Nucleotide Identity |
| ATCC | American Type Culture Collection |
| BacDive | Bacterial Diversity Metadatabase |
| BLAST | Basic Local Alignment Search Tool |
| bp | Base pairs |
| CFU | Colony-Forming Unit |
| DDBJ | DNA Data Bank of Japan |
| dDDH | Digital DNA-DNA Hybridization |
| DDH | Hibridization DNA-DNA |
| DSMZ | Deutsche Sammlung von Mikroorganismen und Zellkulturen |
| EMA | European Medicines Agency |
| EMBL-EBI | European Molecular Biology Laboratory—European Bioinformatics Institute |
| FDA | Food and Drug Administration |
| G+C | Guanine and cytosine |
| GBDP | Genome BLAST Distance Phylogeny |
| GGDC | Genome-to-Genome Distance Calculator |
| GMPs | Good Manufacturing Practices |
| GTDB | Genome Taxonomy Database |
| GTDB-Tk | Genome Taxonomy Database Toolkit |
| JGI | Joint Genome Institute |
| LPSN | List of Prokaryotic Names with Standing in Nomenclature |
| MALDI-TOF MS | Matrix-Assisted Laser Desorption Ionization–Time of Flight/Mass Spectrometry |
| MBP | Multigene-Based Phylogenies |
| MLSA | Multilocus Sequence Analysis |
| MLST | Multilocus Sequence Typing |
| NCBI | National Center for Biotechnology Information |
| ORFs | Open Reading Frames |
| PCR | Polymerase Chain Reaction |
| RED | Relative Evolutionary Divergence |
| rMLST | Ribosomal Multilocus Sequence Typing |
| rRNA | Ribosomal RNA |
| TYGS | Type Strain Genome Server |
| WGS | Whole-Genome Sequencing |
References
- da Costa, L.V.; da Silva Lage de Miranda, R.V.; da Fonseca, E.L.; Gonçalves, N.P.; dos Reis, C.M.F.; Frazão, A.M.; Cruz, F.V.; Brandão, M.L.L.; Ramos, J.N.; Vieira, V.V. Assessment of VITEK® 2, MALDI-TOF MS and Full Gene 16S RRNA Sequencing for Aerobic Endospore-Forming Bacteria Isolated from a Pharmaceutical Facility. J. Microbiol. Methods 2022, 194, 106419. [Google Scholar] [CrossRef] [PubMed]
- United States Pharmacopeial Convention. The United States Pharmacopeia, 43rd ed.; United States Pharmacopeial Convention: Rockville, MD, USA, 2021. [Google Scholar]
- Santos, A.M.C.; Doria, M.S.; Meirinhos-Soares, L.; Almeida, A.J.; Menezes, J.C. A QRM Discussion of Microbial Contamination of Non-Sterile Drug Products, Using FDA and EMA Warning Letters Recorded between 2008 and 2016. PDA J. Pharm. Sci. Technol. 2018, 72, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L. Microbial Diversity in Pharmaceutical Product Recalls and Environments. PDA J. Pharm. Sci. Technol. 2007, 61, 383–399. [Google Scholar] [PubMed]
- Sandle, T. Review of FDA Warning Letters for Microbial Bioburden Issues (2001–2011). Pharma Times 2012, 44, 29–30. [Google Scholar]
- Jain, S.K.; Jain, R.K. Review of FDA Warning Letters to Pharmaceuticals: Cause and Effect Analysis. Res. J. Pharm. Technol. 2018, 11, 3219. [Google Scholar] [CrossRef]
- da Costa, L.V.; da Silva Lage de Miranda, R.V.; dos Reis, C.M.F.; de Andrade, J.M.; Cruz, F.V.; Frazão, A.M.; da Fonseca, E.L.; Ramos, J.N.; Brandão, M.L.L.; Vieira, V.V. MALDI-TOF MS Database Expansion for Identification of Bacillus and Related Genera Isolated from a Pharmaceutical Facility. J. Microbiol. Methods 2022, 203, 106625. [Google Scholar] [CrossRef]
- Caldeira, N.G.S.; de Souza, M.L.S.; da Silva Lage de Miranda, R.V.; da Costa, L.V.; Forsythe, S.J.; Zahner, V.; Brandão, M.L.L. Characterization by MALDI-TOF MS and 16S RRNA Gene Sequencing of Aerobic Endospore-Forming Bacteria Isolated from Pharmaceutical Facility in Rio de Janeiro, Brazil. Microorganisms 2024, 12, 724. [Google Scholar] [CrossRef]
- Food and Drug Administration—FDA. Guidance for Industry Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice; FDA: Silver Spring, MD, USA, 2004.
- European Medicines Agency. The Rules Governing Medicinal Products in the European Union. In European Union Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use; Annex 1: Manufacture of Sterile Medicinal Products; European Medicines Agency: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Maruthamuthu, M.K.; Raffiee, A.H.; De Oliveira, D.M.; Ardekani, A.M.; Verma, M.S. Raman Spectra-Based Deep Learning: A Tool to Identify Microbial Contamination. Microbiologyopen 2020, 9, e1122. [Google Scholar] [CrossRef]
- Masucci, E.M.; Hauschild, J.E.; Gisler, H.M.; Lester, E.M.; Balss, K.M. Raman Spectroscopy as an Alternative Rapid Microbial Bioburden Test Method for Continuous, Automated Detection of Contamination in Biopharmaceutical Drug Substance Manufacturing. J. Appl. Microbiol. 2024, 135, lxae188. [Google Scholar] [CrossRef]
- Guest, M.; Pickard, B.; Smith, B.; Drinkwater, S. The Use of Amplified ATP Bioluminescence for Rapid Sterility Testing of Drug Product Formulations. PDA J. Pharm. Sci. Technol. 2023, 77, 402–411. [Google Scholar] [CrossRef]
- Brazilian Health Regulatory Agency. Brazilian Pharmacopeia, 7th ed.; Brazilian Health Regulatory Agency: Brasilia, Brazil, 2024; Volume I.
- European Directorate for the Quality of Medicines & HealthCare (EDQM). European Pharmacopoeia, 11th ed.; Council of Europe: Strasbourg, France, 2023. [Google Scholar]
- United States Pharmacopeial Convention. The United States Pharmacopeia, 1st ed.; United States Pharmacopeial Convention Inc: Rockville, MD, USA, 2024. [Google Scholar]
- European Medicines Agency. Guideline on the Sterilisation of the Medicinal Product, Active Substance, Excipient and Primary Container; EMA/CHMP/CVMP/QWP/850374/2015; European Medicines Agency: Amsterdam, The Netherlands, 2019; pp. 1–25. [Google Scholar]
- Mattoso, J.M.V.; Costa, L.V.; Vale, B.A.; Reis, C.M.F.; Andrade, J.M.; Braga, L.M.P.S.; Conceição, G.M.S.; Costa, P.B.M.; Silva, I.B.; Rodrigues, L.A.P.; et al. Quantitative and Qualitative Evaluation of Microorganism Profile Identified in Bioburden Analysis in a Biopharmaceutical Facility in Brazil: Criteria for Classification and Management of Results. PDA J. Pharm. Sci. Technol. 2024, 79, 125–156. [Google Scholar] [CrossRef] [PubMed]
- Deutschmann, S.; Paul, M.; Claassen-Willemse, M.; van den Berg, J.; IJzerman-Boon, P.; Grunert da Fonseca, V.; Brunbech, E.; Johnson, L.; Knutsen, C.; Plourde, L.; et al. Rapid Sterility Test Systems in the Pharmaceutical Industry: Applying a Structured Approach to Their Evaluation, Validation and Global Implementation. PDA J. Pharm. Sci. Technol. 2023, 77, 211–235. [Google Scholar] [CrossRef] [PubMed]
- Stamatoski, B.; Ilievska, M.; Babunovska, H.; Sekulovski, N.; Panov, S. Optimized Genotyping Method for Identification of Bacterial Contaminants in Pharmaceutical Industry. Acta Pharm. 2016, 66, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.C.; Vidal, L.; Salazar, V.; Swings, J.; Thompson, F.L. Microbial Genomic Taxonomy. In Trends in the Systematics of Bacteria and Fungi; CABI: Wallingford, UK, 2021; pp. 168–178. [Google Scholar] [CrossRef]
- Moreira, F.M.; de Araujo Pereira, P.; da Silva Lage de Miranda, R.V.; dos Reis, C.M.F.; da Silva Braga, L.M.P.; de Andrade, J.M.; do Nascimento, L.G.; Mattoso, J.M.V.; Forsythe, S.J.; da Costa, L.V.; et al. Evaluation of MALDI-TOF MS, Sequencing of D2 LSU RRNA and Internal Transcribed Spacer Regions (ITS) for the Identification of Filamentous Fungi Isolated from a Pharmaceutical Facility. J. Pharm. Biomed. Anal. 2023, 234, 115531. [Google Scholar] [CrossRef]
- da Costa, L.V.; Ramos, J.N.; de Sousa Albuquerque, L.; da Silva Lage de Miranda, R.V.; Valadão, T.B.; Veras, J.F.C.; Vieira, E.M.D.; Forsythe, S.; Brandão, M.L.L.; Vieira, V.V. Bacillus lumedeiriae sp. nov., a Gram-Positive, Spore-Forming Rod Isolated from a Pharmaceutical Facility Production Environment and Added to the MALDI Biotyper® Database. Microorganisms 2024, 12, 2507. [Google Scholar] [CrossRef]
- Obasi, A.; Nwachukwu, S.; Ugoji, E.; Kohler, C.; Göhler, A.; Balau, V.; Pfeifer, Y.; Steinmetz, I. Extended-Spectrum β-Lactamase-Producing Klebsiella Pneumoniae from Pharmaceutical Wastewaters in South-Western Nigeria. Microb. Drug Resist. 2017, 23, 1013–1018. [Google Scholar] [CrossRef]
- da Silva Lage de Miranda, R.V.; da Costa, L.V.; de Sousa Albuquerque, L.; dos Reis, C.M.F.; da Silva Braga, L.M.P.; de Andrade, J.M.; Ramos, J.N.; Mattoso, J.M.V.; Forsythe, S.J.; Brandão, M.L.L. Identification of Sutcliffiella Horikoshii Strains in an Immunobiological Pharmaceutical Industry Facility. Lett. Appl. Microbiol. 2023, 76, ovad056. [Google Scholar] [CrossRef]
- Sala-Comorera, L.; Vilaró, C.; Galofré, B.; Blanch, A.R.; García-Aljaro, C. Use of Matrix-Assisted Laser Desorption/Ionization–Time of Flight (MALDI–TOF) Mass Spectrometry for Bacterial Monitoring in Routine Analysis at a Drinking Water Treatment Plant. Int. J. Hyg. Environ. Health 2016, 219, 577–584. [Google Scholar] [CrossRef]
- Vithanage, N.R.; Yeager, T.R.; Jadhav, S.R.; Palombo, E.A.; Datta, N. Comparison of Identification Systems for Psychrotrophic Bacteria Isolated from Raw Bovine Milk. Int. J. Food Microbiol. 2014, 189, 26–38. [Google Scholar] [CrossRef]
- Biomérieux. VITEK® 2. Fully Integrated Identification and Antimicrobial Susceptibility Testing; Biomérieux: Marcy-l’Étoile, France, 2025. [Google Scholar]
- Bosshard, P.P.; Zbinden, R.; Abels, S.; Böddinghaus, B.; Altwegg, M.; Böttger, E.C. 16S RRNA Gene Sequencing versus the API 20 NE System and the VITEK 2 ID-GNB Card for Identification of Nonfermenting Gram-Negative Bacteria in the Clinical Laboratory. J. Clin. Microbiol. 2006, 44, 1359–1366. [Google Scholar] [CrossRef]
- Ligozzi, M.; Bernini, C.; Bonora, M.G.; De Fatima, M.; Zuliani, J.; Fontana, R. Evaluation of the VITEK 2 System for Identification and Antimicrobial Susceptibility Testing of Medically Relevant Gram-Positive Cocci. J. Clin. Microbiol. 2002, 40, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Vidal, L. Caracterização de Cocos Gram Positivos Provenientes de Análises Microbiológicas de Produtos Farmacêuticos Estéreis Realizadas No INCQS/FIOCRUZ; Oswaldo Cruz Foundation: Rio de Janeiro, Brazil, 2013. [Google Scholar]
- Ramos, J.N. Caracterização de Estirpes Sugestivas de Corinebactérias Isolados de Sítios Intravenosos; Oswaldo Cruz Foundation: Rio de Janeiro, Brazil, 2014. [Google Scholar]
- Stackebrandt, E.; Ebers, J. Taxonomic Parameters Revisited: Tarnished Gold Standards. Microb. Today 2006, 33, 152. Available online: https://www.scienceopen.com/document?vid=0cf4b084-5683-4ef4-a80c-c0df44a135dc (accessed on 17 March 2025).
- Topić Popović, N.; Kazazić, S.P.; Bojanić, K.; Strunjak-Perović, I.; Čož-Rakovac, R. Sample Preparation and Culture Condition Effects on MALDI-TOF MS Identification of Bacteria: A Review. Mass Spectrom. Rev. 2023, 42, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF Mass Spectrometry: An Emerging Technology for Microbial Identification and Diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef]
- Seuylemezian, A.; Aronson, H.S.; Tan, J.; Lin, M.; Schubert, W.; Vaishampayan, P. Development of a Custom MALDI-TOF MS Database for Species-Level Identification of Bacterial Isolates Collected From Spacecraft and Associated Surfaces. Front. Microbiol. 2018, 9, 780. [Google Scholar] [CrossRef]
- Zasada, A.A.; Mosiej, E. Contemporary Microbiology and Identification of Corynebacteria spp. Causing Infections in Human. Lett. Appl. Microbiol. 2018, 66, 472–483. [Google Scholar] [CrossRef]
- Shah, H.N.; Shah, A.J.; Belgacem, O.; Ward, M.; Dekio, I.; Selami, L.; Duncan, L.; Bruce, K.; Xu, Z.; Mkrtchyan, H.V.; et al. MALDI-TOF MS and Currently Related Proteomic Technologies in Reconciling Bacterial Systematics. In Trends in the Systematics of Bacteria and Fungi; CABI: Wallingford, UK, 2021; pp. 93–118. [Google Scholar] [CrossRef]
- Ashfaq, M.Y.; Da’na, D.A.; Al-Ghouti, M.A. Application of MALDI-TOF MS for Identification of Environmental Bacteria: A Review. J. Environ. Manag. 2022, 305, 114359. [Google Scholar] [CrossRef]
- Rychert, J. Benefits and Limitations of MALDI-TOF Mass Spectrometry for the Identification of Microorganisms. J. Infect. Epidemiol. 2019, 2, 1–5. [Google Scholar] [CrossRef]
- Lasch, P.; Stämmler, M.; Schneider, A. A MALDI-TOF Mass Spectrometry Database for Identification and Classification of Highly Pathogenic Microorganisms from the Robert Koch-Institute (RKI); Zenodo: Geneva, Switzerland, 2016. [Google Scholar] [CrossRef]
- Caamaño-Antelo, S.; Fernández-No, I.C.; Böhme, K.; Ezzat-Alnakip, M.; Quintela-Baluja, M.; Barros-Velázquez, J.; Calo-Mata, P. Genetic Discrimination of Foodborne Pathogenic and Spoilage Bacillus spp. Based on Three Housekeeping Genes. Food Microbiol. 2015, 46, 288–298. [Google Scholar] [CrossRef]
- Rajendhran, J.; Gunasekaran, P. Microbial Phylogeny and Diversity: Small Subunit Ribosomal RNA Sequence Analysis and Beyond. Microbiol. Res. 2011, 166, 99–110. [Google Scholar] [CrossRef]
- Church, D.L.; Cerutti, L.; Gürtler, A.; Griener, T.; Zelazny, A.; Emler, S. Performance and Application of 16S RRNA Gene Cycle Sequencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin. Microbiol. Rev. 2020, 33, e00053-19. [Google Scholar] [CrossRef] [PubMed]
- Madigan, M.; Martinko, J.; Bender, K.; Buckley, D.; Stahl, D. Brock Biology of Microorganisms, 14th ed.; Benjamin Cummings: San Francisco, CA, USA, 2015. [Google Scholar]
- Mahato, N.K.; Gupta, V.; Singh, P.; Kumari, R.; Verma, H.; Tripathi, C.; Rani, P.; Sharma, A.; Singhvi, N.; Sood, U.; et al. Microbial Taxonomy in the Era of OMICS: Application of DNA Sequences, Computational Tools and Techniques. Antonie Leeuwenhoek 2017, 110, 1357–1371. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A Comprehensive Benchmarking Study of Protocols and Sequencing Platforms for 16S RRNA Community Profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.M.B.; Bronzato, G.F.; Santiago, G.S.; Botelho, L.A.B.; Moreira, B.M.; da Silva Coelho, I.; de Souza, M.M.S.; de Mattos de Oliveira Coelho, S. The Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry (MALDI-TOF MS) Identification versus Biochemical Tests: A Study with Enterobacteria from a Dairy Cattle Environment. Braz. J. Microbiol. 2016, 48, 132–138. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Mondotte, J.A.; Fazio, L.L.; Jetten, M. Authors Need to Be Prudent When Assigning Names to Microbial Isolates. Curr. Microbiol. 2021, 78, 4005–4008. [Google Scholar] [CrossRef]
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.J.; Ludwig, W.; Kämpfer, P. Notes on the Characterization of Prokaryote Strains for Taxonomic Purposes. Int. J. Syst. Evol. Microbiol. 2010, 60 Pt 1, 249–266. [Google Scholar] [CrossRef]
- Sentausa, E.; Fournier, P.E. Advantages and Limitations of Genomics in Prokaryotic Taxonomy. Clin. Microbiol. Infect. 2013, 19, 790–795. [Google Scholar] [CrossRef]
- Vasconcellos, L.; Silva, S.V.; da Costa, L.V.; da Silva Lage de Miranda, R.V.; dos Reis, C.M.F.; da Silva Braga, L.M.P.; Silva, C.; Conceição, G.; Mattoso, J.; Silva, I.B.; et al. Phenotypical and Molecular Characterization of Acinetobacter spp. Isolated from a Pharmaceutical Facility. Lett. Appl. Microbiol. 2023, 76, ovad101. [Google Scholar] [CrossRef]
- de Almeida do Vale, B.; Costa de Lima, J.; de Souza, P.A.; da Silva Laje de Miranda, R.V.; Brandao, M.L.L.; da Costa, L.V.; Toma, H.K. Characterization of Staphylococcus Hominis Strains Isolated in an Immunobiological Pharmaceutical Unit. In Congresso Brasileiro de Microbiologia; Sociedade Brasileira de Microbiologia: Foz do Iguaçu, Brazil, 2023. [Google Scholar]
- Loreiro, J.M.P.; Guimarães, R.C.C.; Valadao, T.B.; Miranda, R.V.S.L.; Andrade, J.M.; Costa, L.V.; Brandao, M.L.L. Application of Fourier-Transform Infrared Spectroscopy (FT-IR) for Staphylococcus Epidermidis Typing as a Tool for Contamination Control Strategy in a Pharmaceutical Industry Facility. PDA J. Pharm. Sci. Technol. 2024, 78, 761–762. [Google Scholar] [CrossRef]
- Bazani, V.B.; da Silva, A.C.F.; de Pádua Silva, K.; Müller, K.C. Contaminação de Produtos Farmacêuticos Pelo Complexo Burkholderia Cepacia e Seus Possíveis Impactos Na Saúde e Na Indústria: Uma Revisão Bibliográfica. Res. Soc. Dev. 2024, 13, e10313245032. [Google Scholar] [CrossRef]
- Santana, G.; Aguiar, A.; Sales, F.; Miranda, R.; Valadão, T.; Costa, L.; Brandão, M. Polyphasic Characterization of Burkholderia cepacia Complex Strains Isolated from a Pharmaceutical Industry Facility. In Proceedings of the 8th International Symposium on Immunobiologicals, Rio de Janeiro, Brazil, 8–10 May 2024. [Google Scholar]
- Vlach, J.; Javůrková, B.; Karamonová, L.; Blažková, M.; Fukal, L. Novel PCR-RFLP System Based on RpoB Gene for Differentiation of Cronobacter Species. Food Microbiol. 2017, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.W.; Vandamme, P.; Morgan, S.H.; LiPuma, J.J.; Coenye, T.; Weightman, A.J.; Jones, T.H.; Mahenthiralingam, E. Development of a RecA Gene-Based Identification Approach for the Entire Burkholderia Genus. Appl. Environ. Microbiol. 2005, 71, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, S.; Kristl, J.; Zupančič, Š.; Berlec, A. Influence of Excipient Composition on Survival of Vaginal Lactobacilli in Electrospun Nanofibers. Pharmaceutics 2022, 14, 1155. [Google Scholar] [CrossRef]
- Chun, J.; Rainey, F.A. Integrating Genomics into the Taxonomy and Systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 2014, 64 Pt 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, S.P.; Kämpfer, P. Multilocus Sequence Analysis (MLSA) in Prokaryotic Taxonomy. Syst. Appl. Microbiol. 2015, 38, 237–245. [Google Scholar] [CrossRef]
- Maiden, M.C.J.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus Sequence Typing: A Portable Approach to the Identification of Clones within Populations of Pathogenic Microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Frederiksen, W.; Garrity, G.M.; Grimont, P.A.D.; Kämpfer, P.; Maiden, M.C.J.; Nesme, X.; Rosselló-Mora, R.; Swings, J.; Trüper, H.G.; et al. Report of the Ad Hoc Committee for the Re-Evaluation of the Species Definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 2002, 52, 1043–1047. [Google Scholar] [CrossRef]
- Hayashi Sant’Anna, F.; Bach, E.; Porto, R.Z.; Guella, F.; Hayashi Sant’Anna, E.; Passaglia, L.M.P. Genomic Metrics Made Easy: What to Do and Where to Go in the New Era of Bacterial Taxonomy. Crit. Rev. Microbiol. 2019, 45, 182–200. [Google Scholar] [CrossRef]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F.L. Microbial Genomic Taxonomy. BMC Genom. 2013, 14, 913. [Google Scholar] [CrossRef]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 Years of Bacterial Genome Sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.D.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.C.; Murray, R.G.E.; Stackebrandt, E.; et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Evol. Microbiol. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed Minimal Standards for the Use of Genome Data for the Taxonomy of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, S.; Fullmer, M.S.; Feng, Y.; Gogarten, J.P. Improving Phylogenies Based on Average Nucleotide Identity, Incorporating Saturation Correction and Nonparametric Bootstrap Support. Syst. Biol. 2022, 71, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ran, Q.; Du, X.; Wu, S.; Wang, J.; Sheng, D.; Chen, Q.; Du, Z.; Li, Y.-Z. Two New Polyangium Species, P. aurulentum sp. nov. and P. jinanense sp. nov., Isolated from a Soil Sample. Syst. Appl. Microbiol. 2021, 44, 126274. [Google Scholar] [CrossRef]
- Cuny, H.; Offret, C.; Boukerb, A.M.; Parizadeh, L.; Lesouhaitier, O.; Le Chevalier, P.; Jégou, C.; Bazire, A.; Brillet, B.; Fleury, Y. Pseudoalteromonas ostreae sp. nov., a New Bacterial Species Harboured by the Flat Oyster Ostrea Edulis. Int. J. Syst. Evol. Microbiol. 2021, 71, 005070. [Google Scholar] [CrossRef]
- Colston, S.M.; Fullmer, M.S.; Beka, L.; Lamy, B.; Peter Gogarten, J.; Graf, J. Bioinformatic Genome Comparisons for Taxonomic and Phylogenetic Assignments Using Aeromonas as a Test Case. mBio 2014, 5, e02136. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA Hybridization Values and Their Relationship to Whole-Genome Sequence Similarities. Int. J. Syst. Evol. Microbiol. 2007, 57 Pt 1, 81–91. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic Insights That Advance the Species Definition for Prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.-L.; Xie, B.-B.; Zhang, X.-Y.; Chen, X.-L.; Zhou, B.-C.; Zhou, J.; Oren, A.; Zhang, Y.-Z. A Proposed Genus Boundary for the Prokaryotes Based on Genomic Insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef]
- Kim, D.; Park, S.; Chun, J. Introducing EzAAI: A Pipeline for High Throughput Calculations of Prokaryotic Average Amino Acid Identity. J. Microbiol. 2021, 59, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Bypassing Cultivation To Identify Bacterial Species: Culture-Independent Genomic Approaches Identify Credibly Distinct Clusters, Avoid Cultivation Bias, and Provide True Insights into Microbial Species. Microbe Mag. 2014, 9, 111–118. [Google Scholar] [CrossRef]
- Nicholson, A.C.; Gulvik, C.A.; Whitney, A.M.; Humrighouse, B.W.; Bell, M.E.; Holmes, B.; Steigerwalt, A.G.; Villarma, A.; Sheth, M.; Batra, D.; et al. Division of the Genus Chryseobacterium: Observation of Discontinuities in Amino Acid Identity Values, a Possible Consequence of Major Extinction Events, Guides Transfer of Nine Species to the Genus Epilithonimonas, Eleven Species to the Genus Kaistella, and Three Species to the Genus Halpernia Gen. Nov., with Description of Kaistella daneshvariae sp. nov. and Epilithonimonas vandammei sp. nov. Derived from Clinical Specimens. Int. J. Syst. Evol. Microbiol. 2020, 70, 4432–4450. [Google Scholar] [CrossRef]
- Walter, J.M.; Coutinho, F.H.; Dutilh, B.E.; Swings, J.; Thompson, F.L.; Thompson, C.C. Ecogenomics and Taxonomy of Cyanobacteria Phylum. Front. Microbiol. 2017, 8, 2132. [Google Scholar] [CrossRef]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.A.P.; Harris, H.M.B.; Mattarelli, P.; O’toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A Taxonomic Note on the Genus Lactobacillus: Description of 23 Novel Genera, Emended Description of the Genus Lactobacillus Beijerinck 1901, and Union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Riesco, R.; Trujillo, M.E. Update on the Proposed Minimal Standards for the Use of Genome Data for the Taxonomy of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2024, 74, 006300. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An Ongoing Census of Bacterial and Archaeal Diversity through a Phylogenetically Consistent, Rank Normalized and Complete Genome-Based Taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bliss, C.M.; Bennett, J.S.; Bratcher, H.B.; Brehony, C.; Colles, F.M.; Wimalarathna, H.; Harrison, O.B.; Sheppard, S.K.; Cody, A.J.; et al. Ribosomal Multilocus Sequence Typing: Universal Characterization of Bacteria from Domain to Strain. Microbiology 2012, 158 Pt 4, 1005–1015. [Google Scholar] [CrossRef]

{kind=link}
| Identification Method | Advantages | Limitations |
|---|---|---|
| Phenotypic (API®, VITEK® 2 | -Fast and easy to use -Applicable to clinical and environmental bacteria -Determines the biochemical profile of isolate analyzed -Automation or semi-automatic available | -Database originally clinical (limitations for environmental bacteria) -Low resolution for closely related species -Results may vary -Limited to existing databases -Difficulty with non-fermenting and environmental bacteria -May not identify new species without updating the database |
| MALDI-TOF MS | -Very fast -Low cost per analysis -High accuracy for many species -Allows database expansion | -Database originally clinical (limitations for environmental bacteria) -May not identify new species without updating the database -High initial cost of equipment -Requires comparison with well-characterized spectra |
| 16S rRNA gene sequencing | -Widely used -High conservation allows identification at genus level -Gold standard for general classification | -Low resolution between closely related species -There may be multiple copies in genome (intragenomic variability) -Analysis can be expensive and slow in routine use |
| Housekeeping gene sequencing and multilocus sequencing analysis (MLSA) | -Higher resolving power than 16S gene -Differentiates closely related species -Supports construction of robust phylogenetic trees | -Laborious process -Depends on appropriate choice of genes -Sequences are not always available in databases |
| Identification method | Advantages | Limitations |
| Comparative genomics: dDDH ANI AAI TYGS GTDB-Tk rMLST | -Greater taxonomy accuracy -Based on complete genome -Defines new species and genera -Good correlation with traditional DDH -Free tool (e.g., GGDC) -Easy interpretation -Strong correlation with DDH -Useful for delimiting species (≥96%) -Good tool for delimiting genera (≥60-65%) -Correlation with evolutionary relationships -Automated and updated platform -Compares with recognized type strains -Classifies based on global phylogenetic tree -Constant updates -Universal applicability -High resolution -Robustness against genetic recombination -Public database | -Requires complete genomic sequencing data -High initial cost -Complexity of analysis and need for bioinformatics -Requires high-quality genomic data -Low resolution for categories above species -Requires direct genomic comparison -Requires annotated genomes -Dependence on strain databases -Dependence on strain databases -Dependence of genomic data -Variability between loci -Need for continuous curation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes Ramos, J.; Veloso da Costa, L.; Viana Vieira, V.; Lima Brandão, M.L. Challenges in the Identification of Environmental Bacterial Isolates from a Pharmaceutical Industry Facility by 16S rRNA Gene Sequences. DNA 2025, 5, 33. https://doi.org/10.3390/dna5030033
Nunes Ramos J, Veloso da Costa L, Viana Vieira V, Lima Brandão ML. Challenges in the Identification of Environmental Bacterial Isolates from a Pharmaceutical Industry Facility by 16S rRNA Gene Sequences. DNA. 2025; 5(3):33. https://doi.org/10.3390/dna5030033
Chicago/Turabian StyleNunes Ramos, Juliana, Luciana Veloso da Costa, Verônica Viana Vieira, and Marcelo Luiz Lima Brandão. 2025. "Challenges in the Identification of Environmental Bacterial Isolates from a Pharmaceutical Industry Facility by 16S rRNA Gene Sequences" DNA 5, no. 3: 33. https://doi.org/10.3390/dna5030033
APA StyleNunes Ramos, J., Veloso da Costa, L., Viana Vieira, V., & Lima Brandão, M. L. (2025). Challenges in the Identification of Environmental Bacterial Isolates from a Pharmaceutical Industry Facility by 16S rRNA Gene Sequences. DNA, 5(3), 33. https://doi.org/10.3390/dna5030033