
Theoretical Insights into the Chemical Bonding, Electronic Structure, and Spectroscopic Properties of the Lanarkite Pb2SO5 Structure

 , , and
, , and
Abstract
1. Introduction
2. Computational Method and Model System
3. Results and Discussion
3.1. Structural Properties
3.2. Bond Analysis
3.3. Electronic Properties
3.4. Mechanical Properties and Dynamical Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- La Porta, F.A.; Taft, C.A. Functional Properties of Advanced Engineering Materials and Biomolecules; Springer International Publishing: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- La Porta, F.A.; Taft, C.A. Emerging Research in Science and Engineering Based on Advanced Experimental and Computational Strategies; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Longo, E.; La Porta, F.A. Recent Advances in Complex Functional Materials; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- de Jesus, J.P.A.; Santos, A.C.L.; Pinto, F.M.; Taft, C.A.; La Porta, F.A. Review: Theoretical and experimental investigation of the intrinsic properties of Zn2GeO4 nanocrystals. J. Mater. Sci. 2021, 56, 4552–4568. [Google Scholar] [CrossRef]
- de Conti, M.C.M.D.; Dey, S.; Pottker, W.E.; La Porta, F.A. An overview into advantages and applications of conventional and unconventional hydro(solvo)thermal approaches for novel advanced materials design. Mater. Today Sustain. 2023, 23, 100458. [Google Scholar] [CrossRef]
- de Pablo, J.J.; Jackson, N.E.; Webb, M.A.; Chen, L.Q.; Moore, J.E.; Morgan, D.; Jacobs, R.; Pollock, T.; Schlom, D.G.; Toberer, E.S.; et al. New frontiers for the materials genome initiative. npj Comput. Mater. 2019, 5, 41. [Google Scholar] [CrossRef]
- Zhou, T.; Song, Z.; Sundmacher, K. Big Data Creates New Opportunities for Materials Research: A Review on Methods and Applications of Machine Learning for Materials Design. Engineering 2019, 5, 1017–1026. [Google Scholar] [CrossRef]
- Nosengo, N. Can artificial intelligence create the next wonder material? Nature 2016, 533, 22–25. [Google Scholar] [CrossRef]
- Alberi, K.; Nardelli, M.B.; Zakutayev, A.; Mitas, L.; Curtarolo, S.; Jain, A.; Fornari, M.; Marzari, N.; Takeuchi, I.; Green, M.L.; et al. The 2019 materials by design roadmap. J. Phys. D Appl. Phys. 2019, 52, 013001. [Google Scholar] [CrossRef]
- Choudhary, K.; DeCost, B.; Chen, C.; Jain, A.; Tavazza, F.; Cohn, R.; Park, C.W.; Choudhary, A.; Agrawal, A.; Billinge, S.J.; et al. Recent advances and applications of deep learning methods in materials science. npj Comput. Mater. 2022, 8, 59. [Google Scholar] [CrossRef]
- Resasco, J.; Abild-Pedersen, F.; Hahn, C.; Bao, Z.; Koper, M.T.; Jaramillo, T.F. Enhancing the connection between computation and experiments in electrocatalysis. Nat. Catal. 2022, 5, 374–381. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.B.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef]
- Chen, X.; Hou, T.; Persson, K.A.; Zhang, Q. Combining theory and experiment in lithium–sulfur batteries: Current progress and future perspectives. Mater. Today 2019, 22, 142–158. [Google Scholar] [CrossRef]
- Yang, R.X.; McCandler, C.A.; Andriuc, O.; Siron, M.; Woods-Robinson, R.; Horton, M.K.; Persson, K.A. Big Data in a Nano World: A Review on Computational, Data-Driven Design of Nanomaterials Structures, Properties, and Synthesis. ACS Nano 2022, 16, 19873–19891. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Jain, A.; Hautier, G.; Moore, C.J.; Ong, S.P.; Fischer, C.C.; Mueller, T.; Persson, K.A.; Ceder, G. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 2011, 50, 2295–2310. [Google Scholar] [CrossRef]
- Walsh, A.; Zunger, A. Instilling defect tolerance in new compounds. Nat. Mater. 2017, 16, 964–967. [Google Scholar] [CrossRef]
- Laranjeira, J.A.S.; Martins, N.F.; Azevedo, S.A.; Fabris, G.S.L.; Sambrano, J.R. Novel octa-graphene-like structures based on GaP and GaAs. J. Mol. Model. 2023, 29, 202. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, V.Y.; Amorin, L.H.; Fabris, G.S.; Dey, S.; Sambrano, J.R.; Cohen, H.; Oron, D.; La Porta, F.A. Enhanced Photocatalytic and Photoluminescence Properties Resulting from Type-I Band Alignment in the Zn2GeO4/g-C3N4 Nanocomposites. Catalysts 2022, 12, 692. [Google Scholar] [CrossRef]
- Zhu, H.; Hautier, G.; Aydemir, U.; Gibbs, Z.M.; Li, G.; Bajaj, S.; Pöhls, J.H.; Broberg, D.; Chen, W.; Jain, A.; et al. Computational and experimental investigation of TmAgTe 2 and XYZ 2 compounds, a new group of thermoelectric materials identified by first-principles high-throughput screening. J. Mater. Chem. C 2015, 3, 10554–10565. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.; Im, S.; An, S.; Kwon, Y.W.; Auh, K.H. Consideration for the development of room-temperature ambient-pressure superconductor (LK-99). J. Korean Cryst. Growth Cryst. Technol. 2023, 33, 61–70. [Google Scholar]
- Lee, S.; Kim, J.; Kim, H.T.; Im, S.; An, S.; Auh, K.H. Superconductor Pb10−xCux(PO4)6O showing levitation at room temperature and atmospheric pressure and mechanism. arXiv 2023, arXiv:2307.12037. [Google Scholar]
- Griffin, S.M. Origin of correlated isolated flat bands in copper-substituted lead phosphate apatite. arXiv 2023, arXiv:2307.16892. [Google Scholar]
- Kumar, K.; Karn, N.K.; Kumar, Y.; Awana, V.P.S. Absence of superconductivity in LK-99 at ambient conditions. ACS Omega 2023, 8, 41737–41743. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Karn, N.K.; Awana, V.P.S. Synthesis of possible room temperature superconductor LK-99: Pb9Cu(PO4)6O. Supercond. Sci. Technol. 2023, 36, 10LT02. [Google Scholar] [CrossRef]
- Liu, L.; Meng, Z.; Wang, X.; Chen, H.; Duan, Z.; Zhou, X.; Yan, H.; Qin, P.; Liu, Z. Semiconducting Transport in Pb10−XCux(PO4)6O Sintered from Pb2SO5 and Cu3P. Adv. Funct. Mater. 2023, 33, 2308938. [Google Scholar] [CrossRef]
- Lai, J.; Li, J.; Liu, P.; Sun, Y.; Chen, X.-Q. First-principles study on the electronic structure of Pb10−xCux(PO4)6O (x = 0, 1). J. Mater. Sci. Technol. 2024, 171, 66–70. [Google Scholar] [CrossRef]
- Richmond, W.E.; Wolfe, C.W. Crystallography of lanarkite. Am. Mineral. J. Earth Planet. Mater. 1938, 23, 799–804. [Google Scholar]
- Mentzen, B.F.; Latrach, A. Crystal data for dilead(II) pentaoxosulfate(VI), Pb2SO5. J. Appl. Crystallogr. 1983, 16, 430. [Google Scholar] [CrossRef]
- Mentzen, B.F.; Latrach, A.; Bouix, J.; Hewat, A.W. The crystal structures of PbO.PbXO4 (X = S, Cr, Mo) at 5K by neutron powder profile refinement. Mater. Res. Bull. 1984, 19, 549–554. [Google Scholar] [CrossRef]
- Abdul-Samad, F.A.; Thomas, J.H.; Williams, P.A.; Symes, R.F. Chemistry of formation of lanarkite, Pb2OSO4. Mineral Mag. 1982, 46, 499–501. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Demichelis, R.; Civalleri, B.; Ferrabone, M.; Dovesi, R. On the performance of eleven DFT functionals in the description of the vibrational properties of aluminosilicates. Int. J. Quantum Chem. 2010, 110, 406–415. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Gomes, E.O.; Fabris, G.S.; Ferrer, M.M.; Motta, F.V.; Bomio, M.R.; Andres, J.; Longo, E.; Sambrano, J.R. Computational procedure to an accurate DFT simulation to solid state systems. Comput. Mater. Sci. 2019, 170, 109176. [Google Scholar] [CrossRef]
- Fabris, G.S.L.; Marana, N.L.; Longo, E.; Sambrano, J.R. Theoretical study of porous surfaces derived from graphene and boron nitride. J. Solid State Chem. 2018, 258, 247–255. [Google Scholar] [CrossRef]
- Vilela Oliveira, D.; Laun, J.; Peintinger, M.F.; Bredow, T. BSSE-correction scheme for consistent gaussian basis sets of double- and triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2019, 40, 2364–2376. [Google Scholar] [CrossRef]
- Lima, E.F.; Bredow, T. Extended benchmark set for lattice parameters of inorganic solids. J. Comput. Chem. 2024, 45, 2702–2709. [Google Scholar] [CrossRef]
- Ferrero, M.; Rérat, M.; Kirtman, B.; Dovesi, R. Calculation of first and second static hyperpolarizabilities of one- to three-dimensional periodic compounds. Implementation in the CRYSTAL code. J. Chem. Phys. 2008, 129, 244110. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Rérat, M.; Orlando, R.; Dovesi, R. The calculation of static polarizabilities of 1-3D periodic compounds. the implementation in the crystal code. J. Comput. Chem. 2008, 29, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Rérat, M.; Orlando, R.; Dovesi, R. Coupled perturbed Hartree-Fock for periodic systems: The role of symmetry and related computational aspects. J. Chem. Phys. 2008, 128, 014110. [Google Scholar] [CrossRef]
- Casassa, S.; Erba, A.; Baima, J.; Orlando, R. Electron density analysis of large (molecular and periodic) systems: A parallel implementation. J. Comput. Chem. 2015, 36, 1940–1946. [Google Scholar] [CrossRef]
- Gatti, C.; Casassa, S. TOPOND User’s Manual; CNR-CSRSRC: Milano, Italy, 2013. [Google Scholar]
- Binnie, W.P. The crystal structure of lanarkite, PbO.PbSO4. Acta Crystallogr. 1951, 4, 471–472. [Google Scholar] [CrossRef]
- Singh, H.; Gautam, A.; Singh, M.; Saha, P.; Kumar, P.; Das, P.; Lamba, M.; Yadav, K.; Mishra, P.K.; Patnaik, S.; et al. On the experimental evidence for possible superconductivity in LK99. arXiv 2023, arXiv:2308.06589. [Google Scholar]
- Izumi, F.; Momma, K. Three-Dimensional Visualization in Powder Diffraction. Solid State Phenom. 2007, 130, 15–20. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Silva, J.F.; Fabris, G.S.L.; Sambrano, J.R.; Albuquerque, A.R.; Maia, A.S. TopIso3D Viewer: Enhancing Topological Analysis through 3D Isosurfaces. J. Chem. Inf. Model. 2023, 63, 1999–2013. [Google Scholar] [CrossRef]
- Xiao, H.; Tahir-Kheli, J.; Goddard, W.A. Accurate Band Gaps for Semiconductors from Density Functional Theory. J. Phys. Chem. Lett. 2011, 2, 212–217. [Google Scholar] [CrossRef]
- Arı, H.; Büyükmumcu, Z. Comparison of DFT functionals for prediction of band gap of conjugated polymers and effect of HF exchange term percentage and basis set on the performance. Comput. Mater. Sci. 2017, 138, 70–76. [Google Scholar] [CrossRef]
- Ody, K.S.; Jesus, J.P.D.; Cava, C.E.; Albuquerque, A.R.; Maia, A.S.; Sambrano, J.R.; Porta, F.A.L. Avaliação da Estrutura Eletrônica da Fase Monoclinica do Óxido de Nióbio Com Base No Uso de Diferentes Funcionais de Densidade. Química Nova 2021, 44, 1124–1131. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Xu, Z.; Loo, J.S.C. Hybrid catalysts for photoelectrochemical reduction of carbon dioxide: A prospective review on semiconductor/metal complex co-catalyst systems. J. Mater. Chem. A 2014, 2, 15228. [Google Scholar] [CrossRef]
- Underwood, T.M.; Robinson, R.S. Adducing Knowledge Capabilities of Instrumental Techniques Through the Exploration of Heterostructures’ Modification Methods. ChemPhysChem 2022, 23, e202200241. [Google Scholar] [CrossRef]
- Sze, S.M.; Ng, K.K. Physics of Semiconductor Devices; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Cengage Learning: Boston, MA, USA, 1976. [Google Scholar]
- Yim, K.; Youn, Y.; Lee, M.; Yoo, D.; Lee, J.; Cho, S.H.; Han, S. Computational discovery of p-type transparent oxide semiconductors using hydrogen descriptor. NPJ Comput. Mater. 2018, 4, 17. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafe, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitrică, T.; Dominguez, A.; et al. DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Togo, A. First-principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 2023, 35, 353001. [Google Scholar] [CrossRef] [PubMed]
- Martins, N.F.; Laranjeira, J.A.; de Azevedo, S.A.; Fabris, G.S.L.; Sambrano, J.R. Tuning the electronic properties of the SiC graphenylene by transition metal (Fe, Mn and Co) doping. Phys. B Condens. Matter 2024, 691, 416369. [Google Scholar] [CrossRef]
- Martins, N.F.; Laranjeira, J.A.S.; Azevedo, S.A.; Fabris, G.S.L.; Sambrano, J.R. Structural, electronic and mechanical properties of a novel graphenylene-like structure based on GeC. J. Phys. Chem. Solids 2023, 181, 111518. [Google Scholar] [CrossRef]
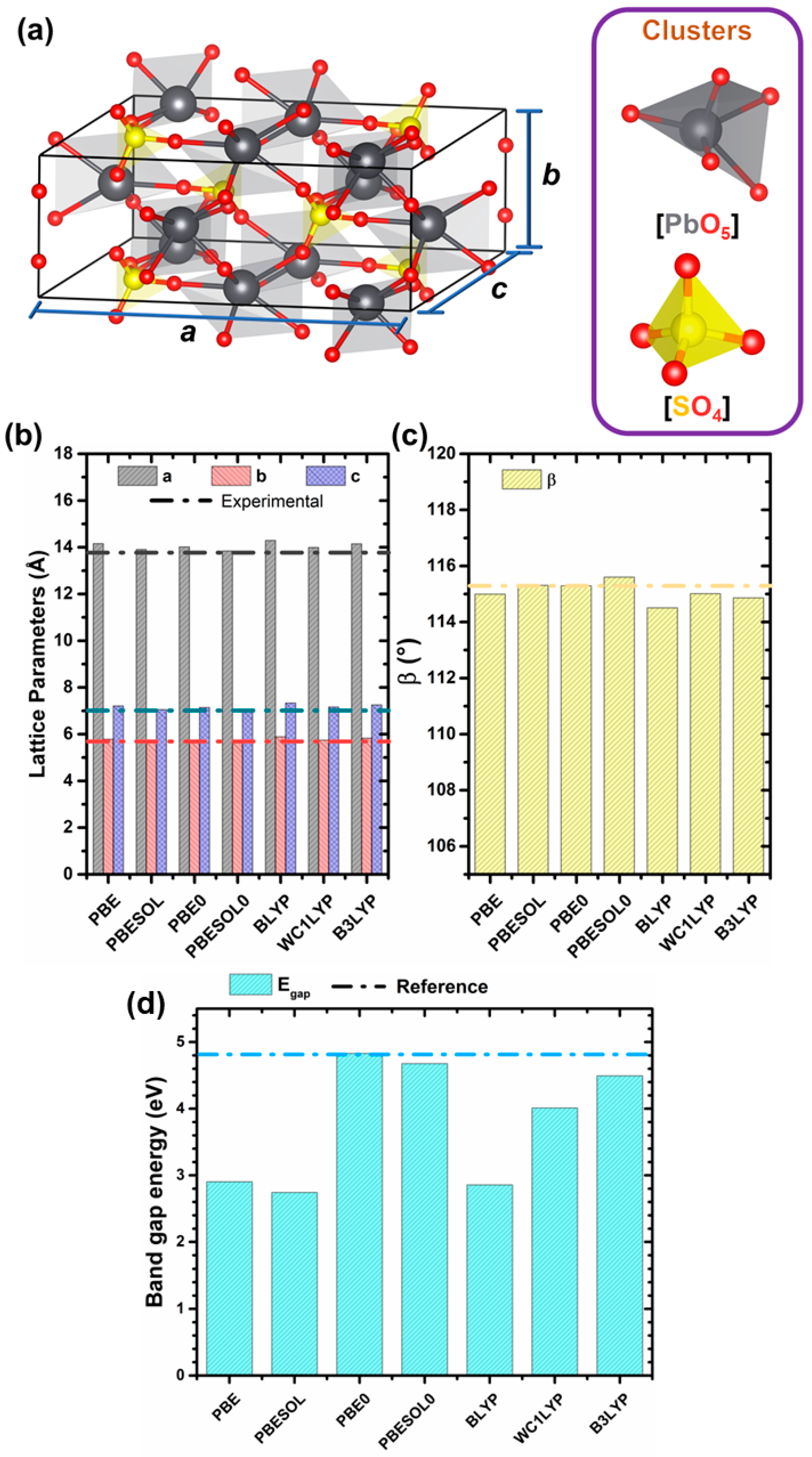
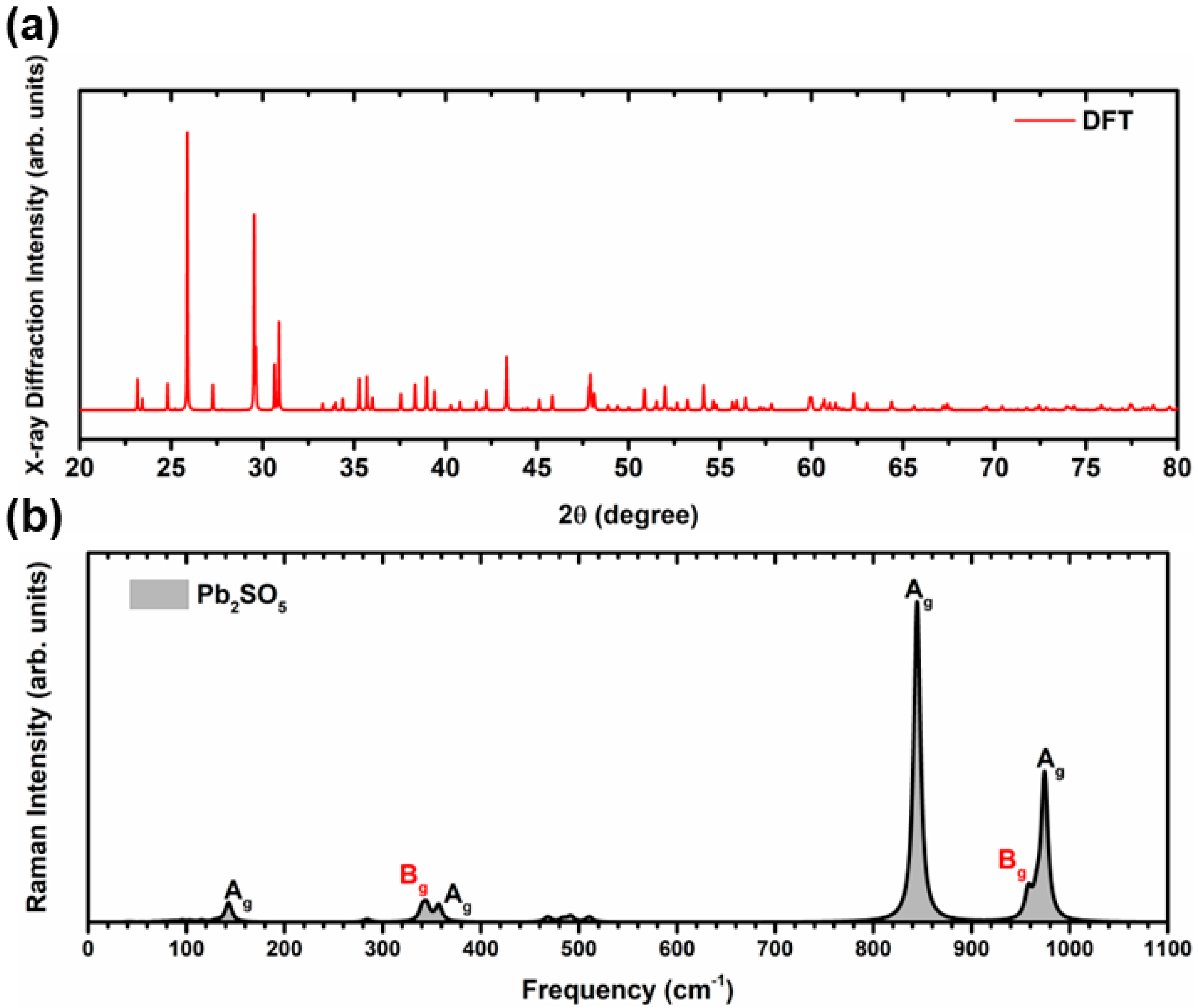
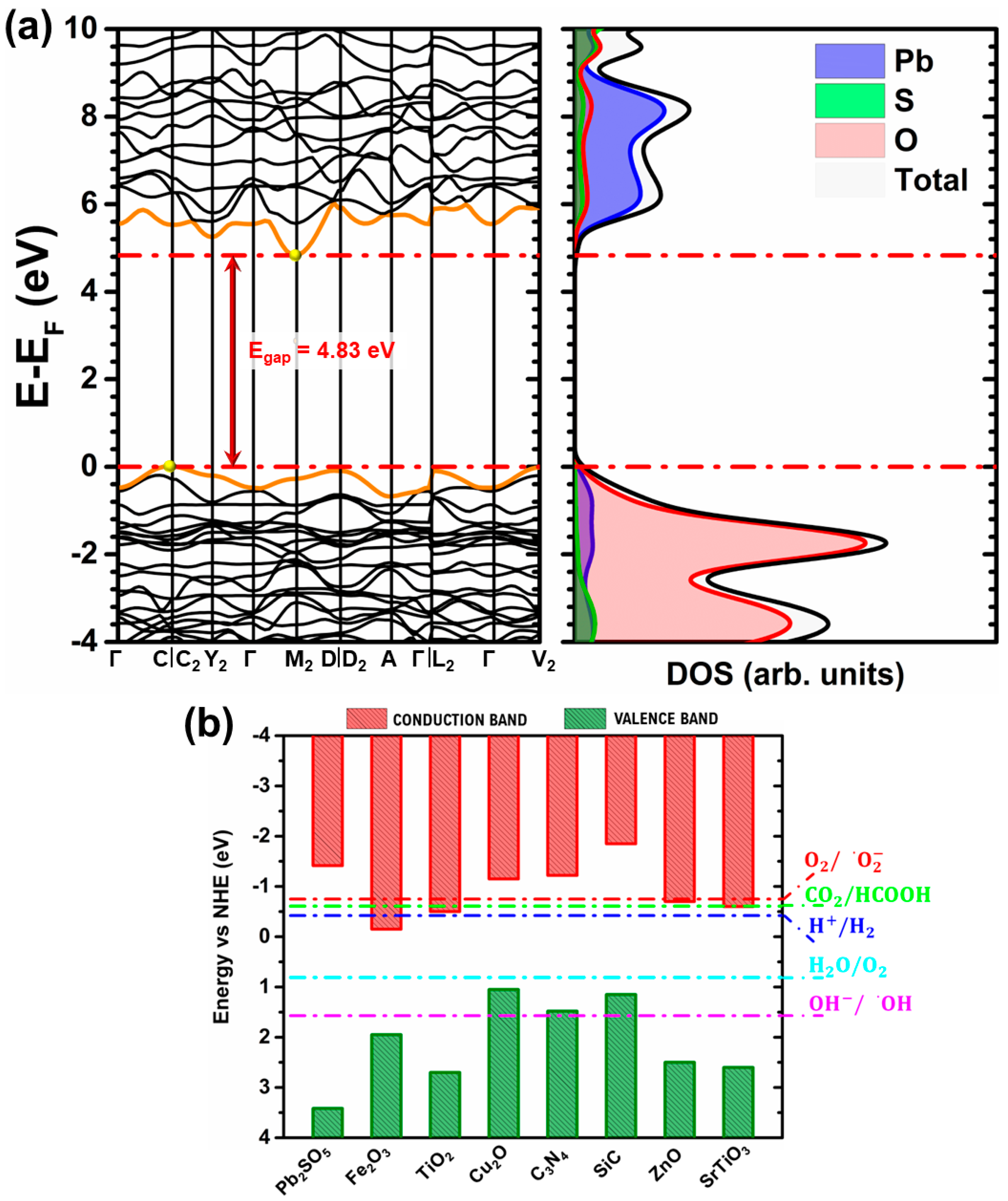
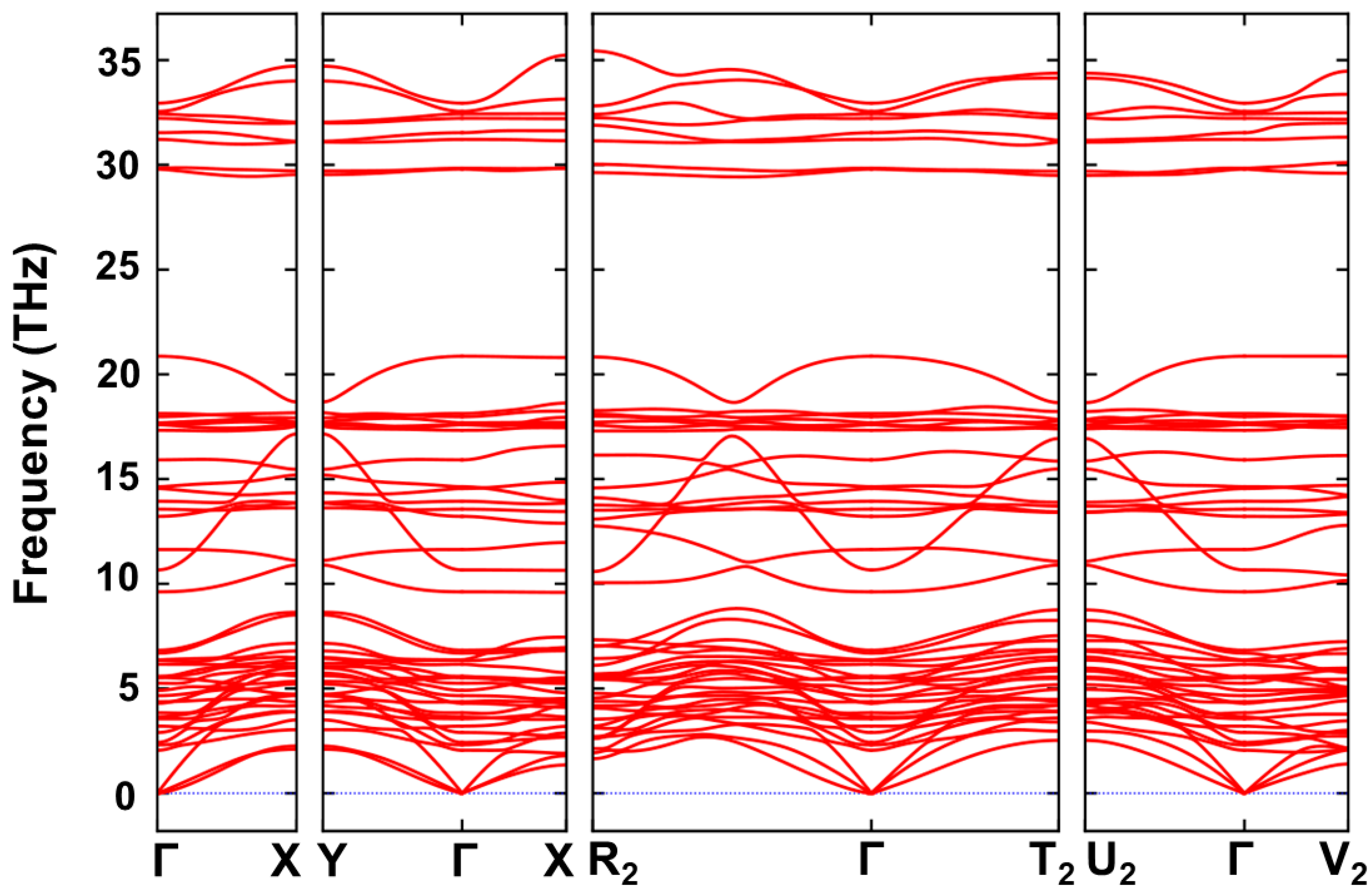

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Critical Points | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| A-B | d | ρ | ∇2ρ | G | V | |V|/G | H/ρ(r) | ε | Bond |
| Pb-O1 | 2.29 | 0.065 | 0.209 | 0.063 | −0.073 | 1.168 | −0.163 | 0.011 | T cov |
| Pb-O2 | 2.35 | 0.031 | 0.112 | 0.028 | −0.028 | 0.999 | 0.001 | 0.010 | T |
| Pb-O3 | 2.57 | 0.020 | 0.065 | 0.015 | −0.014 | 0.927 | 0.055 | 0.075 | ionic |
| Pb-O4 | 2.60 | 0.012 | 0.038 | 0.008 | −0.007 | 0.852 | 0.100 | 0.100 | ionic |
| S-O1 | 1.54 | 0.224 | 0.278 | 0.289 | −0.509 | 1.760 | −0.983 | 0.001 | T cov |
| S-O2 | 1.54 | 0.224 | 0.251 | 0.284 | −0.506 | 1.779 | −0.988 | 0.006 | T cov |
| C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 | C15 | C25 | C35 | C46 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 88.18 | 80.09 | 83.60 | 2.48 | 17.97 | 16.89 | 32.27 | 16.48 | 17.20 | 1.46 | 0.86 | −8.54 | 2.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabris, G.S.L.; Ferrer, M.M.; Almeida, C.R.R.; Paskocimas, C.A.; Sambrano, J.R.; La Porta, F.A. Theoretical Insights into the Chemical Bonding, Electronic Structure, and Spectroscopic Properties of the Lanarkite Pb2SO5 Structure. Physchem 2025, 5, 22. https://doi.org/10.3390/physchem5020022
Fabris GSL, Ferrer MM, Almeida CRR, Paskocimas CA, Sambrano JR, La Porta FA. Theoretical Insights into the Chemical Bonding, Electronic Structure, and Spectroscopic Properties of the Lanarkite Pb2SO5 Structure. Physchem. 2025; 5(2):22. https://doi.org/10.3390/physchem5020022
Chicago/Turabian StyleFabris, Guilherme S. L., Mateus M. Ferrer, Claudio R. R. Almeida, Carlos A. Paskocimas, Julio R. Sambrano, and Felipe A. La Porta. 2025. "Theoretical Insights into the Chemical Bonding, Electronic Structure, and Spectroscopic Properties of the Lanarkite Pb2SO5 Structure" Physchem 5, no. 2: 22. https://doi.org/10.3390/physchem5020022
APA StyleFabris, G. S. L., Ferrer, M. M., Almeida, C. R. R., Paskocimas, C. A., Sambrano, J. R., & La Porta, F. A. (2025). Theoretical Insights into the Chemical Bonding, Electronic Structure, and Spectroscopic Properties of the Lanarkite Pb2SO5 Structure. Physchem, 5(2), 22. https://doi.org/10.3390/physchem5020022