Abstract
Liver fibrosis is a complex, dynamic process associated with a broad spectrum of chronic liver diseases and acute liver failure, characterised by the dysregulated intrahepatic production of extracellular matrix proteins replacing functional liver cells with scar tissue. Fibrosis progresses due to an interrelated cycle of hepatocellular injury, triggering a persistent wound-healing response. The accumulation of scar tissue and chronic inflammation can eventually lead to cirrhosis and hepatocellular carcinoma. Currently, no therapies exist to directly treat or reverse liver fibrosis; hence, it remains a substantial global disease burden. A better understanding of the intricate inflammatory network that drives the initiation and maintenance of liver fibrosis to enable the rationale design of new intervention strategies is required. This review clarifies the most current understanding of the hepatic fibrosis cellular network with a focus on the role of regulatory T cells, and a possible trajectory for T cell immunotherapy in fibrosis treatment. Despite good progress in elucidating the role of the immune system in liver fibrosis, future work to better define the function of different immune cells and their mediators at different fibrotic stages is needed, which will enhance the development of new therapies.
Keywords:
liver; immunity; fibrosis; immune modulation; T cell immunotherapy; regulatory T cells; biomaterials 1. Introduction
Liver fibrosis is characterised by the progressive deposition and intrahepatic accretion of extracellular matrix (ECM) proteins, partly in response to the fibrogenic factors produced by hepatic stellate cells (HSCs) responding to liver injury or inflammatory mediators. Ineffective regulation of fibroplasia leads to excessive accumulation of scar tissue, stemming from repeated inflammatory activation as the liver attempts to repair and replace damaged hepatic cells. While fibrogenesis does not directly cause symptoms, severe scarring can result in liver cirrhosis if the source of injury persists. Cirrhosis, often referred to as end-stage liver disease, can prevent normal liver functionality, resulting in organ failure with the need for a liver transplant. This disease advancement is estimated to affect approximately 2% of the world population, causing over 1 million deaths per year [1,2]. In some cases, cirrhosis even induces a microenvironment supportive of hepatocellular carcinoma (HCC) development, the most common primary liver malignancy, of which 80–90% of cases occur within cirrhotic livers [3,4].
Most cases of hepatic fibrogenesis are derived from a causal liver disease; the epidemiological data identify a striking disparity in liver disease burden depending on geographical location and socioeconomic echelons, alongside individual distinctions such as ethnicity and gender. Plausibly, the incidence of hepatitis B virus (HBV), hepatitis C virus (HCV), alcoholic liver disease (ALD), and non-alcoholic fatty liver disease (NAFLD) are attributed as the leading causes of global liver diseases [5]. Systemic analyses of global cirrhosis burden suggest that chronic HBV and HCV infections instigate the highest affliction [6,7]. However, rising occurrence of NAFLD and progression to non-alcoholic steatohepatitis (NASH), the severest form, encompassing extensive liver inflammation and cirrhosis, may become most prominent in the near future [7], conceivably due to the escalating obesity and diabetes pandemics [8,9]. The impact of other liver diseases are also major public health concerns, including autoimmune conditions such as autoimmune hepatitis [2]. Cholestatic-induced injury to the biliary network, which includes the liver, additionally causes hepatic fibrosis. Impaired bile flow, or cholestasis, can occur within several pathologies, such as biliary atresia and biliary cholangitis [10]. Thus, a better understanding and early treatment of liver fibrogenesis has extensive clinical implications for global healthcare.
Determining the degree of liver fibrosis is fundamental for diagnosis and assessing disease severity. Highly invasive liver biopsies and histological scoring systems are the gold standard for estimating liver inflammation and scarring. Two of the most used systems are the Metavir score and Ishak score, defining a different number of progressive steps. The former demarcates liver fibrosis into four major stages: F1–F4, with F0 indicating a normal, non-fibrotic liver and F4 signifying cirrhosis [11]. As earlier stages of liver fibrosis are typically asymptomatic, they could go undetected unless picked up; they are picked up during routine blood tests that may show subtle changes in liver function tests, which could be followed up with non-invasive imaging strategies such as magnetic resonance imaging (MRI) and sonography, enabling detailed imaging of the liver’s appearance and stiffness, indicating the level of fibrosis [12]. Serum biomarker measurement can also provide insight into progressing hepatic fibrogenesis [13], falling into two major categories: direct and indirect markers. The former constitutes the turnover of ECM proteins produced by various hepatic cells. These include fibrillar type I and III collagen, α-smooth muscle actin (α-SMA), and tissue inhibitors of metalloproteins (TIMPs), which become elevated with fibrotic progression. A limitation of these biomarkers is that they are not organ-specific and alone cannot diagnose hepatic fibrosis severity. Hence, the detection of indirect biomarkers, namely products of liver function such as bilirubin and aminotransferases, are required in conjunction. While diagnostic imaging and serum biomarkers have a more limited predictive ability of intermediate fibrotic stages, their less-invasive nature allows for fewer complications caused by biopsy, such as pain and hypertension, as well as higher reproducibility and availability. Therefore, these procedures can give a greater insight into disease progression and offer a more accurate prognosis [14].
1.1. Complications of Liver Regeneration Therapeutics
To date, the best treatment for liver fibrosis centres around the elimination of the causal liver disease, such as antiviral therapy for viral hepatitis, sobriety for ALD, and also immunosuppressants for autoimmune hepatitis, with the prospect that such interventions may hinder fibrosis progression [15]. Once cirrhosis is established, however, organ transplantation becomes the sole treatment to improve the condition of a patient. However, the systemic shortage of viable organ donation, alongside intrinsic immunological consequences of tissue incompatibility and infection, increases the risk of morbidity and mortality after transplantation. Prospectively, there is increasing evidence that fibrosis is a dynamic and reversible process; however, at which point fibrosis becomes irreversible remains unclear and could broadly depend on the disease ethology [16]. Hence, the development of regenerative medicine and anti-fibrotic therapies which can be administered upon diagnosis are exceedingly sought after. Due to the central role of the immune response within hepatic fibrogenesis, immunotherapeutics which can hinder specific cellular targets are of great research interest. Currently, broad-spectrum anti-inflammatory drugs, such as corticosteroids and non-steroidal anti-inflammatory drugs, have widespread use and can improve the outcome of many liver diseases. Yet, long-term treatment with these therapies shows aggregated hepatoxicity, augmenting fibrogenesis and thus increasing the prospect of cirrhosis [17,18]. As such, identifying therapeutic foci for targeted treatments to inhibit fibrogenesis and promote resolution is the leading clinical investigation.
Preliminary research into cell and tissue engineering approaches display promising results in the treatment of liver fibrosis. Conventional immunomodulatory methods have yielded promising early research, including the use of cytokine-based therapies and monoclonal antibodies. Following this success, there has been increasing momentum behind biomaterials for immunoengineering. This manipulation of the microenvironment is implemented via artificial cells and scaffolds which become integrated within the ECM. These scaffolds, constructed from natural materials and biopolymers such as peptides and sugars, are essential for maintaining proper cell functionality, and hence must be physically compatible with the natural liver framework and biological activity. Due to the intricacy of the liver microenvironment and its role in tissue homeostasis, these microtechnological applications are complex and still harbour many problems, such as the potential for tumorigenesis and inconsistent feasibility [19]. Hence, effective therapy delivery mechanisms remain a hindrance in administering fibrosis immunotherapeutics. Additionally, while several key targets of liver fibrosis have been identified, including the cytokines transforming growth factor (TGF)-β and interferon (IFN)-γ, the liver contains a complex network of intracellular signalling between the immune and mesenchymal systems. How the constituents of both are implicated within liver fibrosis, and diverge between fibrotic stages, will influence the result of such therapeutics. Indeed, even considering the remarkable regenerative capacity of the liver, chronic damage accompanied by fibrogenesis does not enable the full restoration of normal tissue’s microarchitecture and functionality, which must be considered in the development of cellular treatments. In particular, the progression of wound healing against liver fibrosis immunopathology is a delicate balance, involving analogous processes with differing outcomes. Hence, elucidation of the cellular network underpinning liver fibrogenesis can shed light on new avenues for treatment and effective delivery mechanisms; exogenous signalling methods may be able to mould a regenerative, antifibrotic microenvironment, causing fibrosis resolution.
1.2. Initiation of Liver Fibrosis
Regardless of the aetiology, liver fibrosis results from a chronic wound-healing response, driven by a continuous and interrelated cycle of damage. While disease progression may differ contextual to the liver injury, the underlying pathology involves a similar, yet complex, interplay between dysregulated immune responses and distressed mesenchymal function [20]. Under healthy physiologic conditions, the liver maintains equilibrium between immune tolerance and inflammation, despite perpetual exposure to exogenous antigens derived from the diet, chemicals, and gut microbiota. This is achieved via the intricate, heterogenic network of resident immune cell populations and non-haematopoietic cells, which can produce and influence inflammatory mediators for local and systemic homeostasis. When a specific insult causes hepatocellular damage, compromising the liver microenvironment, danger-associated molecular patterns (DAMPs), or pathogen-associated molecular patterns (PAMPs) in the case of infection, are released. These DAMPs and PAMPs signal to pattern recognition receptors (PRRs) on the surface of resident immune cells and non-parenchymal cells, including the endothelium and HSCs. Notably, signalling by toll-like receptors (TLRs), an extensive family of PRRs, has been corroborated as driving liver fibrosis by propagating HSC activation in several disease settings [21]. Activating these cells via PRR and DAMP/PAMP interaction incites a cytokine and chemokine cascade, augmenting an inflammatory state and the recruitment of circulating innate and adaptive leukocytes into the hepatic microenvironment. In response, a healing reaction is initiated by the liver, causing ECM remodelling (Figure 1).
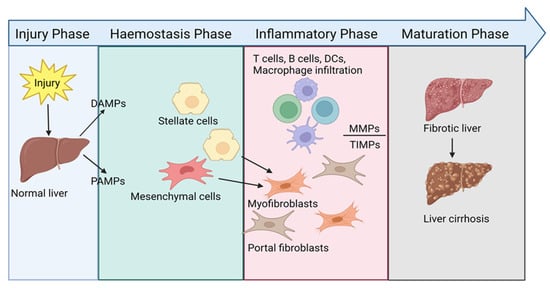
Figure 1.
Schematic showing the wound-healing cascade in liver disease.
During the natural course of wound healing, four distinct phases (injury, haemostasis, inflammatory, and maturation phase) after injury lead to the regeneration of damaged tissues. In the context of liver fibrosis, when the wound-healing response becomes pathogenic, the generation of fibrotic tissue replaces the structural foundations of the liver. Ultimately, scar formation distorts the liver tissue and impairs organ function [22].
Quiescent HSC activation is central to liver fibrosis initiation; as resident mesenchymal cells, HSCs differentiate into myofibroblasts, an intermediate cell lineage with a phenotype between smooth muscle cells and fibroblasts. These hyperproliferative cells are not present within homeostatic livers and develop a contractile ability for the secretion of ECM constituents. The current evidence proposes that perpetual HSC activation during liver fibrogenesis is the principal source of the hepatic myofibroblast pool [23], which comprises several other cell types, including resident fibroblasts and portal myofibroblasts [24]. Consequently, they are mainly responsible for signature fibrotic scar formation; thus, myofibroblast control and elimination is a desirable therapeutic target [25,26,27]. Besides increasing ECM mass, the topography of ECM constituents is also altered, particularly augmented expression of fibrotic collagen (i.e., type III, IV, and V), fibronectins, and hyaluronan. These pro-fibrotic components have been implicated as endogenous DAMPs which are recognised by PRRs, such as well have TLR2 and TLR4 recognition of fibronectin accumulation [28]. As such, upregulation of matrix metalloproteinases (MMPs) and the antagonistic action of TIMPs, the inhibitory counterparts, also ensues. MMPs are expressed by an array of immune and non-immune cells and degrade ECM components, including collagen and fibronectin; hence, they are fundamental for tissue remodelling [29]. The MMP/TIMP balance within the liver plays a vital role in the induction of liver fibrosis; however, the exact function and implication of individual types is yet to be fully understood due to conflicting evidence [30,31].
While unregulated mesenchymal activity is a critical mechanism initiating fibrosis, the advancement of inflammation is the driving force behind fibrogenesis and its evolution. Experimental mice evidence even demonstrates that self-limiting fibrosis and regeneration during an acute inflammatory response fosters liver protection; for example, upregulation of type I collagen in fibrotic scars safeguards hepatic cells from toxic insults [32]. However, with continuous aggressive liver damage, the recruitment of inflammatory cells into the hepatic environment becomes chronic and contributes to the pathogenesis of the original injury [33,34]. Subsequently, liver fibrosis advances until cirrhosis occurs and a potential transplant is required. Understanding this chronic inflammation is vital for improving liver disease therapeutics and reducing the global need for liver transplants.
2. Immunopathology of Liver Fibrosis
In a non-fibrotic liver, myeloid and lymphoid cells of both innate and adaptive lineages reside in the sinusoidal system to maintain tissue homeostasis and protect the organ from blood-borne pathogens [35]. In response to hepatocellular injury, circulating immune cells migrate into the hepatic microenvironment to augment the inflammatory state via continual interaction and signalling between cell populations [33,34]. The interplay between these cells (Figure 2) therefore makes it challenging to definitively establish the individual contributions of each cell type to fibrosis; however, observing how cell populations behave under different disease settings can provide insight into the complex mechanisms of liver fibrosis.
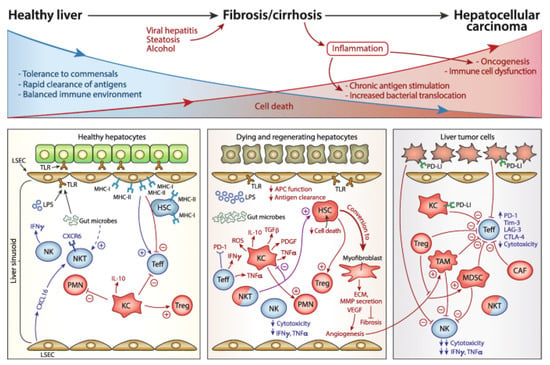
Figure 2.
Evolution of the immune response during liver fibrogenesis from healthy to late inflammation stage.
As hepatocellular injury persists, an aggravated immune response synergistically causes harm to the liver structure. In a healthy liver, a tolerogenic environment is maintained by the regular functioning of innate and adaptive immune constituents. At the onset of injury, cytotoxicity is directed towards the parenchymal structure and initiates a fibrotic cycle to regenerate the dying hepatocyte population. Continued secretion of pro-inflammatory and pro-fibrotic cytokines, alongside extracellular matrix deposition, enables progression to cirrhosis. In some cases, advancing cirrhosis can trigger oncogenesis of resident liver cells, eliciting the development of hepatocellular carcinoma [3].
2.1. Innate Immune Cells
It is well established that the innate immune system is the vanguard of rapid and initial immune responses. Neutrophils typically act as the first responders to inflammatory signals, performing clearance of apoptotic hepatic cells in response to damage. While the understanding of their recruitment to the liver and involvement in hepatic fibrosis is still within its infancy [36], the release of neutrophilic granules, including myeloperoxidase, and powerful chemoattractants, such as interleukin (IL)-8 (also known as CXCL8) and CXCL2, may exacerbate fibrosis progression [37,38]. More notably, the liver harbours the largest population of tissue-resident macrophages in the body; Kupffer cells (KCs) represent almost 15% of the total hepatic cell population. As sentinel antigen-presenting cells (APCs), KCs, alongside infiltrating monocytes and macrophages, perform critical phagocytic and scavenger functions, removing cellular debris from the sinusoidal blood flow via PRR recognition. Due to their highly plastic nature, KCs are the principal orchestrators of fibrogenesis and the resolution of inflammation, contingent on a phenotypic switch [20,39]. Macrophages present a spectrum of functional phenotypes in response to environmental cues [40]. At either ends of this spectrum, they can be broadly classified into pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes with distinct cytokine and transcriptional profiles, as well as surface markers.
During early fibrosis, KCs localise near activated myofibroblasts [41]. The release of type 1 pro-inflammatory cytokines, such as tumour necrosis factor (TNF)-α and IFN-γ [42], from other immune cells in the microenvironment enables M1 polarisation. In turn, M1 KCs produce high levels of signature type 1 cytokines, including TNF-α and IL-6, to facilitate the pro-inflammatory response. This activation enables myofibroblast proliferation by inducing injury in several liver diseases, including ALD, NAFLD, and HCC, thus instigating fibrogenesis [43]. Conversely, M2 macrophages are denoted as the healing-associated phenotype, which can be induced by mainly IL-4 and IL-13. As stated earlier, a spectrum of activation states exist amid these two main subsets [44]. While it is unclear whether these macrophage variations are subtle distinctions of M1/M2 phenotypes or independent populations, pro-fibrotic (M2a) and tissue repair (M2c) M2 phenotypes are well implicated within fibrogenesis immunopathology [22,45]. As regulated ECM deposition into tissues is a fundamental stage of the natural wound-healing response, macrophages are also vital during normal tissue repair [43]. Tissue repair and remodelling is prompted by the secretion of potent anti-inflammatory and regulatory cytokines, such as IL-10 and TGF-β, to suppress pro-inflammatory pathways. Interestingly, some studies have reported that the use of ECM-derived scaffolds can polarise macrophages towards a pro-regenerative state in both in vitro and in situ wound sites. This polarisation is defined by the expression of CD206, also known as mannose receptor, which is a useful marker for M2 phenotypes [46]. However, in the presence of persistent injury, M2 macrophages adopt a pathologic phenotype, secreting large amounts of pro-fibrotic mediators, including TGF-β and IL-13, which become central to fibrosis progression via the chronic activation of HSCs and collagen accumulation [47,48,49]. Crucially, this is one of the most significant impediments in elucidating liver disease immunopathology, as many cells, not only macrophages, exhibit this dichotomy.
One cell type which does not seem to contribute to the paradox of fibrotic exacerbation versus attenuation is the predominant innate lymphoid cells of the liver: natural killer (NK) cells. These large granular lymphocytes are vital in liver antiviral and anti-tumour immunity, fuelling a severe cytotoxic response [50,51]. Additionally, NK cells are important components of the sinusoidal cellular network which governs fibrosis. Following liver injury, NK cells are suggested to directly kill activated HSCs, diminishing myofibroblast differentiation and ECM deposition. Moreover, the production of IFN-γ, a signature pro-inflammatory cytokine of these cells, induces HSC apoptosis and cell cycle arrest, thus behaving in an anti-fibrotic manner [52]. This behaviour is shown to allow fibrosis reversibility via shifting the immune response from inflammation to resolution, particularly within early to moderate liver fibrogenesis [53]. In fact, NK cells maintain this anti-fibrotic role within advanced fibrosis due to the hallmark IFN-γ production, which is also reported to downregulate TGF-β expression [54]. As such, tuning NK cell effector functions represents an appealing immunotherapeutic strategy for many liver diseases. Direct investigation into NK-cell-based therapies in liver fibrosis settings remains limited, and further in vivo analysis of NK–HSC spatiotemporal interaction is required before the true beneficial extent of NK-cell-based engineering can be established [51]. However, pilot studies investigating the clinical administration of IFN-γ-1b (a recombinant form of human IFN-γ) demonstrated a reduction in advanced fibrosis in selected HCV- and HBV-infected patients, supporting its well-documented anti-fibrotic role and suggesting a favourable biologic rationale in certain patient subgroups [55].
As discussed, this innate cell network can efficiently induce inflammation in response to damaging stimuli. However, the bridging of the innate and adaptive arms of the immune system is of equal importance in liver damage and fibrosis settings. Alongside macrophages and monocytes, dendritic cells (DCs) are a proficient population of heterogenous APCs. While hepatic-resident DCs exist, their distribution is sparse, primarily being found within portal regions and, sporadically, the parenchyma. Despite their low frequency, DCs can produce copious amounts of pro-inflammatory cytokines, including TNF-α and IL-1 derivatives, and possess a remarkable migratory capacity, primarily distinguishing them from macrophages. The migration of DCs out of the liver may be critically linked to their significant MMP production, suggesting a potential influence on fibrogenesis [56]. Moreover, as professional APCs, DCs possess the ability to process and present antigens to adaptive lymphocytes. After migration to secondary lymphoid organs, antigen presentation to naïve T cells enables proliferation and differentiation into CD4+ T helper (Th) and CD8+ T cytotoxic (Tc) cell subsets [41]. DCs can also engage B lymphocyte function via cell–cell contact. Consequently, these immunological functions of DCs enable a superior promotion of adaptive immune responses. This distinguishes them from KCs and is postulated to provide hepatic DCs with an indirect capability to regulate fibrosis, potentially behaving as a central axis in conjunction with KCs for the elicitation of tolerogenic or inflammatory responses [57]. However, the role of hepatic DCs in liver fibrogenesis is less clear than that of KCs and infiltrating monocytes. Depending on fibrosis aetiology, DCs could play antagonistic roles in liver fibrogenesis; in ALD, alcohol intake is suggested to impair the proficiency of DCs, resulting in diminished adaptive cell proliferation [58]. NAFLD ablation studies further reported conflicting evidence in the role of DCs throughout disease evolution, depending on the experimental setting [59]. Evidently, DCs could elicit exogenous signalling manipulation due to their innate–adaptive mediatory ability. Understanding whether these cells should be targeted for treating fibrosis will be a key area of future research, particularly the relationship of different DC subsets with MMPs and the suggested role that DCs may regulate the number and activity of cells which control fibrosis development, including NK cells and CD8+ cells [56].
2.2. Adaptive Immune Cells
DCs are essential regulators for the maintenance of a tolerogenic hepatic microenvironment. This ability lies in their capacity to induce specific T cell subsets and B cell subsets, establishing the lymphocyte population via highly specialised human leukocyte antigen (HLA) signalling and cytokine secretion [60]. The role of B cells within liver fibrosis remains relatively obscure in comparison to T cells. Being implicated mainly within infection- or autoimmune-induced liver fibrosis, B cell function is thought to centre around autoantibody production and the ability to activate HSCs for myofibroblast differentiation, the central mechanism of liver fibrogenesis [61,62]. Similarly, CD8+ Tc cells are vital for initiating killing mechanisms against malignancies and cells infected with intracellular pathogens. While the involvement of CD8+ Tc cells in liver fibrosis is also incompletely understood, models of acute liver injury have demonstrated their potential role in HSC activation [63]. Comparatively, memory populations of tissue-resident CD8+ T cells have been shown to promote fibrosis resolution via HSC apoptosis under NASH, demonstrating a disparate role. Further studies which identify antigens contributing to CD8+ T memory cell development, likely from gut-derived and intrinsic molecules, under these conditions are warranted. However, the manipulation of this antigenic environment could stimulate pro-resolving T cell receptor (TCR) clonotypes [64]. Additionally, experimental models of NAFLD have conveyed the potential immunoregulatory role of CD8+ Tc cells during liver fibrosis via the production of IL-10 [65,66]. While IL-10 has not been categorised as anti-fibrotic, several in vivo reports have suggested the regulatory cytokine may play a role in protecting the liver against fibrogenesis assault, and it thus presents an enticing anti-fibrotic therapeutic angle [67].
Much like macrophages, CD4+ Th cells express a fundamental dichotomy which instigates a reciprocal antagonistic relationship for homeostatic maintenance. Depending on the cytokine milieu at the time of activation, naïve CD4+ T cells have the potential to differentiate into one of two principal effector subtypes: Th1 or Th2 cells. The former are denoted as pro-inflammatory, relying on the expression of IL-12 and IFN-γ for differentiation, and implicate M1 macrophages in type 1 pro-inflammatory immune responses. Conversely, Th2 cells mirror anti-inflammatory M2 macrophages, orchestrating type 2 resolution-like responses [68] with the production of the signature cytokines IL-4, IL-5, and IL-13 [48,49]. While both phenotypes have been implicated in liver fibrosis immunopathology, it is acknowledged that dysregulated type 2 responses cultivate fibrogenesis in the presence of persistent injury to initiate chronic wound healing [69,70,71]. However, resembling KCs, the Th cell population is not simple, and the understanding of CD4+ Th cell lineages has significantly developed; decades of experimental investigation has identified at least seven CD4+ Th cell subsets (Figure 3). In addition, these lymphocytes have the potential to switch phenotypes, and even adopt intermediary states, depending on the cytokine environment and expression of specific lineage-defining transcription factors [72,73]. This is feasible due to the plasticity that the majority of CD4+ T cells retain after differentiation. As such, this ability offers an opportunity to better understand how subsets interact with one another and with the microenvironment, which could be therapeutically manipulated in endless disease settings. The domain of T cell therapy has rapidly expanded in recent years, particularly in relation to cancer and autoimmune disease treatment, with an emphasis on cell engineering and cytokine co-operative approaches [74]. However, the potential for therapeutic use for liver fibrosis remains comparatively unexplored. Additionally, the identification of the Th17 phenotype has led to the recent establishment of type 3 immunity: an effector response driven by cells with an enhanced ability to produce the signature cytokines IL-17A, IL-17F, and IL-22 [75,76]. These cytokines are reported to have pro-fibrogenic roles and are markers of liver fibrosis, with important producers in situ being Th17 and Th22 cells [75,76]. The function of Th17 cells has also been extensively reported in promoting distinctive hepatocellular injuries and HSC activation via TGF-β signalling, thus indirectly and directly promoting liver fibrogenesis [77,78].
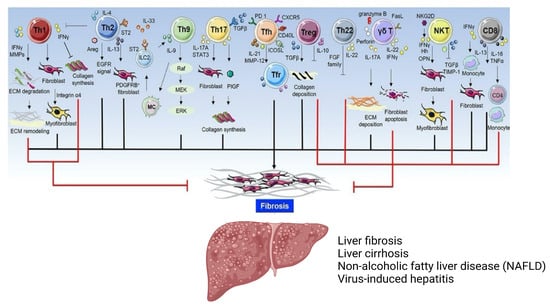
Figure 3.
General overview of T cell subsets within liver disease.
The T cell repertoire is vast, each with unique effector functions. While the immunopathology of each fibrotic disease is distinctive, convergent roles exist due to the biological mechanisms of each cell subset, including their cytokine profiles and ability to interact other cell types, such as fibroblasts. Some cells may express a dual nature, with the capacity to inhibit and exacerbate fibrogenic processes [49].
Presently, a T cell subset which has gained investigational traction within a hepatic fibrosis setting is that of regulatory T cells (Tregs), a specialised population with the capacity to suppress immune responses. The role of these cells within liver fibrosis remains elusive and contentious, perhaps due to the complexity in defining specific Treg phenotypes [79,80]. Delineating the potential role of Tregs within liver fibrosis may provide a deeper insight into the mechanisms governing wound-healing responses. Therefore, this review will examine the current evidence of Treg function within liver fibrosis, with an exploration of prospective therapeutic agents which may manipulate Treg mechanisms for fibrotic resolution.
3. Regulatory T Cells
As the name alludes to, Tregs have a central role in regulating the immune response, namely the initiation and maintenance of peripheral tolerance and homeostasis; hence, they are vital within liver immunology. Identified in 1995 as a specialised subset of CD4+ Th cells, characterised by their high expression of the IL-2 receptor α-chain (CD25) [81], the understanding of Treg biology has exponentially grown in the following decades. Significantly, the identification of transcriptional regulatory forkhead box P3 (FoxP3) as the focal transcription factor defining Treg lineages was a breakthrough facilitating subset characterisation [82]. The purification of “real” human Tregs, as opposed to other effector T cells which may adopt these markers after activation, is also facilitated by the downregulated expression of the IL-7 receptor α-chain (CD127) with weak positive or negative CD25 expression, showing a reverse correlation [83,84]. In addition, the production of pleiotropic regulatory cytokines, namely IL-10, TGF-β, and IL-35, facilitates the suppressive functions of Tregs (Figure 4). These signature cytokines can inhibit the proliferation of naïve lymphocytes, as well as directly impede antigen presentation for the generation of tolerogenic DCs in the periphery [85,86]. Despite these defining features, the ability to functionally identify specific markers of CD4+ Treg phenotypes has remained challenging, as no singular marker is constitutive to this heterogenous population of cells, including that aforementioned [83].
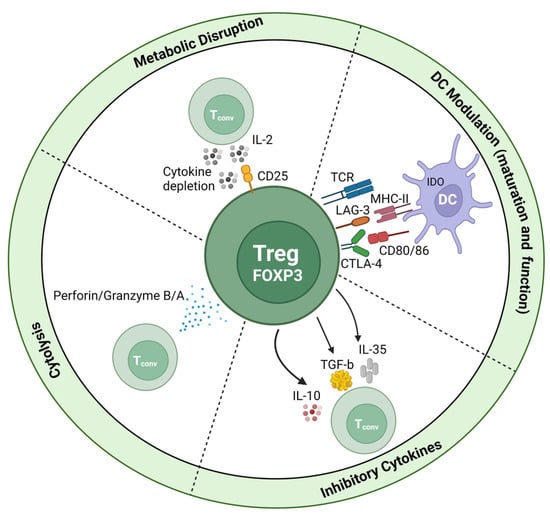
Figure 4.
Mechanisms of FoxP3+ Treg suppression.
Tregs have the ability to suppress immune responses in an antigen-dependent manner, such as via T cell receptors (TCRs) or in an antigen-independent manner (also known as bystander suppression), such as cytokine secretion. An interesting function of Tregs which are CD25hi is their ability to behave as IL-2 sinks; by soaking up free IL-2 within the microenvironment, they can dampen local pro-inflammatory signalling [87].
Ontogenetically, Tregs may be differentiated into naïve CD4+ T cells in the thymus, being termed “natural” Tregs (nTregs), or outside the thymus (such as in secondary lymphoid organs or in cell culture) whereby they are denoted as “inducible” Tregs (iTregs) [88,89]. The former may be promoted when thymocytes have potent TCR signalling in conjunction with CD28 co-stimulation without instigating negative selection, committing the progenitor cell to the nTreg lineage. Conversely, iTregs are produced under particular antigenic conditions from mature T cells, including TGF-β and IL-2 in the cytokine microenvironment, alongside weak TCR signalling [90,91]. It should be emphasised that, while the markers denoting both nTreg and iTreg lineages have been documented within mice studies, the surface markers within humans are yet to be revealed. FoxP3 transiency has been denoted an effective marker for different human Treg populations, as FoxP3 expression is relatively unstable and is suggested to enable self- and non-self-discrimination [92]. However, two phenotypes belonging to the CD4+ iTreg pool lack FoxP3 expression: type 1 regulatory T (Tr1) cells and Th3 cells, which secrete high levels of IL-10 and TGF-β, respectively [93]. Further, the classification of CD8+ Tregs presents another perplexity in defining lineage markers and functionality [94]. This evidently highlights the malleability of the Treg pool, almost certainly to counteract the plethora of effector agents within the immune response.
Concerning immune-mediated liver injury, our understanding of the intrahepatic Treg pool is still developing. The liver participates in specific physiological processes, including detoxification and haemostasis; hence, resident Treg metabolism and function will be specialised in comparison to circulatory Tregs [95]. While the majority of Tregs are produced within the thymus, the liver appears to be a substantial source for peripheral iTregs, with hepatic Treg generation being linked to the organ’s aptitude to instigate peripheral immune tolerance [96]. Traditional effector markers, including FoxP3, CTLA-4 (cytotoxic T-lymphocyte antigen-4), and IL-10, are essential for denoting Treg function and activation states. However, the subclassification of liver- and microenvironment-specific phenotypes of tissue-resident Tregs and how these integrate local signalling has channelled the possibility of modulating Treg phenotypes at a more precise scale. Under homeostatic conditions, hepatic Tregs have been reported as predominantly effector memory Tregs (mTregs) with minimal IL-2 in the microenvironment, dampening their immunosuppressive function [97]. The division of Tregs into memory and resting (rTreg) categories was first defined in a mice model; however, human orthologues have been demarcated by the variable expression of CD45 and FoxP3. Human rTregs are characterised by CD45RA+FoxP3low, while human mTregs have a CD45RA-FoxP3hi phenotype with high expression of CD45RO, the memory isoform of CD45 (Figure 5) [98,99,100,101]. Additionally, the intrahepatic microenvironment is highly enriched with hormones, metabolites, and cytokines promoting Treg induction. For instance, the pro-inflammatory cytokines produced during liver inflammation, such as IFN-γ and IL-1β [102], can induce a hypoxic environment which subsequently stabilises stress proteins, particularly hypoxia-inducible factor 1α (HIF-1α), considered to be a master regulator of this process. This transcription factor is expressed in naïve CD4+ T cells, and upon activation, can incite FoxP3 expression to prompt an iTreg phenotype [97]. The activation of the HIF-1α pathway is also suggested to exert control over the Treg/Th17 ratio, alongside many other significant signalling dynamics, and may bolster the immunosuppressive capacity of Tregs [95,103]. While hypoxic environments have been shown to promote liver fibrosis advancement, this ability to upregulate immunosuppressive mechanisms may be an interesting molecular target, as hypoxia has been shown to attenuate liver fibrosis and promote regeneration in recent in vivo models [104]. Notably, the liver–gut axis has also been suggested to influence and maintain Treg differentiation within the liver, such as in the development of NASH. In addition to an inverted Treg/Th17 balance, NASH patients display a “leaky gut”, increasing exposure to PAMPs and DAMPs, which can be delivered directly to the liver via the portal vein. Significant data suggest that altered DAMP profiles can directly impact FoxP3 expression, and accordingly iTreg generation due to a critical mechanism prompted by TLR signalling. In particular, the ligation of TLR2 heterodimers has been shown to promote Treg proliferation whilst simultaneously reducing the suppressive function, likely via downregulated FoxP3 expression [105]. Comparatively, exposure to many other inflammatory conditions can be detrimental to Treg survival [106]. This demonstrates the unclear role of Tregs in hepatic fibrogenesis, suggesting it may be contextual to the specific injury perpetuating the liver, potentially alongside the predominant Treg phenotype engaging in the immunopathogenesis of the disease. Both factors will be taken into consideration when deliberating the current evidence presented on the role of Tregs in liver fibrogenesis.
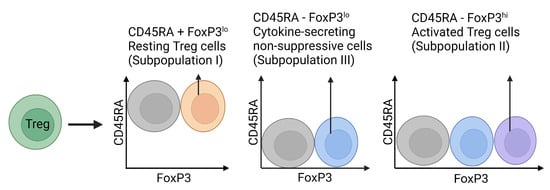
Figure 5.
Classification of human Treg subpopulations.
FoxP3 and CD45RA expression have been suggested as important features when defining the activation state and suppressive capacity of human Treg populations. While populations exist without FoxP3 and CD45RA expression, these three distinctive subpopulations have been widely observed in regulatory T cell heterogeneity and function [101].
3.1. Tregs as Potential Inducers of Liver Fibrosis
Due to the tolerogenic nature of Tregs, it could be expected they would exert an attenuative role in liver fibrogenesis. Of the liver diseases, perhaps the most well discussed in respect to the role of Tregs is viral hepatitis, likely due to the global burden [107]. It is known that as the body undergoes many types of injury, the expansion of Treg populations is initiated [108,109,110]. Expectedly, patients chronically infected with HBV are reported to have increased numbers of infiltrating CD4+CD25+FoxP3+ Tregs, displaying a positive correlation with HBV DNA in the serum. It may be inferred that the proliferation of Tregs is directly linked to the rate of viral replication. This was concurrent with the infection severity, showing increasing Treg infiltration into the liver, which may be indicative of an advanced fibrotic grade due to projected disease progression [111]. As this study is consistent with other reports, the circulating Tregs may elicit a viral persistence mechanism by potentially modulating HBV-specific immune responses [112,113,114]. Accordingly, these data demonstrate the potential pathogenic role of Tregs within HBV infection and indirect role in hepatic fibrosis progression. While the underlying cause of increased Tregs in response to HBV is yet to be clarified, it can be postulated that continual exposure to signature pro-fibrotic cytokines and HBV antigens elicits iTreg generation. For example, the presence of hepatitis B envelope antigen (HBeAg) during chronic HBV infection is shown to positively correlate with the number of peripheral iTregs via transforming CD4+CD25- T cells into CD4+CD25+FoxP3+ Tregs, alongside an augmented ability to produce TGF-β in vitro [115]. As activated HSCs are the primary source of free TGF-β in the liver, their continual stimulation sustains a pro-fibrogenic milieu at the onset of hepatic fibrosis [116,117]. Consequently, it could be hypothesised to cause a Treg surplus, which may become detrimental and contribute to end-stage cirrhosis during chronic wound healing. In this context, the suppression of Treg proliferation and/or activity may offer a desirable strategy for subjugating the underlying liver disease and indirectly perturbing liver fibrosis. Several Treg ablation methods have been demonstrated within various experimental investigations; however, as a therapeutic technique, which delivery mechanisms would enable safe and efficacious treatment, where, and when still require vigorous debate.
In contrast, the frequency of Tregs during HCV infection has remained controversial. Similar to chronic HBV infections, some data report an increase in peripheral Tregs [118,119] as the severity of HCV escalates, as well as in the liver [120,121]. Intriguingly, several chronic HCV patients were shown to harbour CD4+CD25+ Tregs with elevated suppressive activity, which could inhibit the differentiation of CD8+ Tc cells and the production of IFN-γ [122]. Further, while the cytokine profiles of the Treg phenotypes were not entirely clarified within these studies, one report described the expansion of intrahepatic IL-8-producing CD4+FoxP3+ Tregs during chronic HCV infection [123]. The major pro-fibrotic cytokines encompass TGF-β, IL-13, as well as IL-33 in the case of chronic hepatocellular injury [124]. However, IL-8—a principal chemokine facilitating immune cell infiltration into inflammatory sites—has been reported to enable mesenchymal stem cell (MSC) differentiation into fibroblasts for collagen production in pulmonary fibrosis [125]; hence, it has been identified as a non-invasive biomarker for liver fibrosis. During chronic liver disease, IL-8 has also been demonstrated to become upregulated intrahepatically by parenchymal and non-parenchymal liver cells [126]. Thus, this study reports an additional source of IL-8 during liver fibrosis immunopathology. The IL-8-producing Treg phenotype was, coincidingly, proposed to induce pro-fibrogenic markers in HSCs, including TIMP1, MMP2 and α-SMA, signifying an activated state for established collagen synthesis [123]. TIMP-1 and α-SMA are serum biomarkers for liver fibrosis [127], although the role of MMP2 is controversial. Depending on the liver disease aetiology, both pro-fibrotic and anti-fibrotic roles have been suggested [128]. Within this report, it is possible that MMP2 behaves in a pro-fibrotic manner. Due to IL-8 being a powerful chemoattractant, this Treg phenotype could increase the infiltration of circulating inflammatory cells into the hepatic environment, demonstrated in vitro via the recruitment of neutrophils [129]. These inflammatory cells may contribute to liver fibrogenesis depending on the cytokine milieu, in particular TGF-β, which was also upregulated in response to IL-8 production [123,125]. Accordingly, the IL-8-producing Treg phenotype could be perceived as a functional “pro-inflammatory” subpopulation [130], although not as robust as Th1 or Th17 cell induction. Indeed, high levels of IL-8 are associated with liver fibrosis progression; a report demonstrated that elevated intrahepatic IL-8 was directly associated with increased neutrophil infiltration under primary biliary cirrhosis, and with hepatic macrophage infiltration in non-cholestatic cirrhosis [126]. Whether this presents an angle for therapeutic intervention is yet to be discussed despite the concurrent association of IL-8 with other fibrotic diseases, including idiopathic pulmonary fibrosis and cardiac fibrosis [125,131]. Fascinatingly, one study reported the use of intravenous transfusion of epithelial cells transduced with IL-8 receptors as able to significantly inhibit pro-inflammatory mediator production and thus the infiltration of innate inflammatory cells in post-myocardial infarction hearts of rats. By mimicking neutrophil behaviour, the application of these cells resulted in an effectively increased viable myocardium and reduced fibrosis by inhibiting IL-8 activated pro-inflammatory responses (Table 1) [132]. Whether this experimental design could be extrapolated to hepatic epithelial cells would be an interesting area of research. However, the source of IL-8 production appears to be multifaceted, and the chemokine may recruit immune phenotypes which aid in fibrosis resolution. Additionally, while these data are appealing, IL-8-producing Tregs are suggested to be in small quantities under homeostatic conditions; hence, further investigation into whether additional IL-8 CD4+FoxP3+ Treg infiltration or differentiation occurs and the extent of their role within liver fibrogenesis is required [133].

Table 1.
Experimental liver fibrosis treatments including in vivo and human clinical trials.
Nonetheless, HCV and HBV are not the only infections which can elicit liver fibrosis. Many parasites tend to express liver tropism during certain stages of their life cycle, causing hepatocellular damage and fibrosis [147,148]. One such parasite is Clonorchis sinensis, commonly referred to as Chinese liver fluke, which infects mammals to instigate clonorchiasis and biliary fibrosis [149]. As with many parasitic infections, pro-fibrotic type 2 immune responses are highly upregulated for host protection [48,49,150]. Therefore, the expansion of Treg populations to control the anti-parasitic response would be expected. Indeed, an elegant study investigating hepatic CD4+ Th cell profiles of different mice strains reported their potential role during biliary fibrosis in response to C. sinensis [151]. A dramatic increase in hepatic Th2 and CD4+CD25+FoxP3+ Treg cells, alongside their respective cytokines, including IL-4 and TGF-β, was seen in BALB/c and FVB mice. Crucially, upregulation was positively correlated with hepatic hydroxyproline content, an essential component of all collagen types for stability and thus a tool for identifying collagen metabolism [151,152]. The most extreme fibrotic changes resemblant of cirrhosis were identified in FVB mice, followed by severe fibrosis in BALB/c mice in contrast to the C57BL/6 and control strains. While the study did not identify the underlying mechanisms of the Th cell subsets, the data strongly suggest potential roles in the formation of biliary fibrosis, supported by the production of pro-fibrogenic cytokines by both subsets. Comparatively, a slight increase in hepatic Th1 and Th17 populations was reported; however, it was not suggested to be significant between infected and non-infected mice [151]. Interestingly, another study investigating C. sinensis infection in mice identified a significant increase in the hepatic Treg/Th17 ratio as infection developed, causing an imbalance. This shifted axis was shown to be positively correlated with increased inflammatory cell infiltration and collagen deposition, the latter identified via hydroxyproline, during progressive infection. While the exact role of Tregs was not clarified in this study, it is suggested to be centred around the induction of peripheral tolerance to C. sinensis and could contribute to fibrosis progression. Thus, it is curious as to why the prior study did not identify a significant increase in Th17 cells between infected and non-infected mice when both studies used BALB/c strains. This could be due to differences in experimental setup, such as the number of mice used and length of study; a more significant increase in Th17 cells and disruption to Treg/Th17 ratios may occur at more advanced stages of infection, as demonstrated by the latter study [153].
Aside from infection models, many in vivo experiments artificially induce liver fibrosis via physical intercession, such as bile duct ligation (BDL), or using hepatoxins, such as carbon tetrachloride (CCl4). Principally, CCl4-induced liver fibrosis via single or repeated application is one of the most common experimental models [154], as it becomes metabolised into radicals by liver cytochrome P450 enzymes which prompt fibrosis [155]. When using this method in a cirrhosis mice model, one research group reported hepatic fibre degradation was inhibited by the action of CD4+CD25+FoxP3+ Tregs to enable fibrosis persistence. It was demonstrated that Tregs were able to alter the TIMP/MMP balance within the liver microenvironment, thus determining the proteolytic activity controlling ECM turnover. Namely, Treg depletion showed increased ratios of TIMP1 with MMP9 and MMP13, alongside depleted hepatic TGF-β levels [156]. MMP13 has been established as a key collagenase for fibrosis resolution in experimental rat models, with investigation into its application as an antifibrotic therapy; experimental administration of hyaluronic-acid-shielded plasmids encoding MMP13 DNA has been shown to enhance hepatic MMP13 expression and reduced collagen deposition, highlighting its potential as a genetic antifibrotic strategy [134,135].
Conversely, the role of MMP9 is contentious within liver fibrosis amid reports of both anti-fibrotic and pro-fibrotic characteristics depending on the animal model used and cell origin (Figure 6) [30]. However, in combination with reduced TGF-β and TIMP-1 expression, this ratio is indicative of a pro-fibrogenic response. Interestingly, Treg depletion also caused enhanced expression of MMP2 and MMP14. Several reports have suggested the resolving nature of both MMPs within various stages of liver fibrosis to promote ECM degradation, although there remains some controversy, particularly for MMP2 [128]. This expression of MMP2 after Treg ablation is in direct contrast with a prior study, which correlated MMP2 upregulation with an increase in IL-8 CD4+FoxP3+ Treg cells [123]. There are likely many factors regulating the fibrotic activity of MMPs, including the cytokine milieu [128,157], and it may be possible that the secretion of MMP2 is specific to a certain Treg phenotype, or not directly linked to Treg expansion at all. Considering this, the expression profile after Treg depletion may be indicative of fibre degradation, supporting the notion that Tregs could prevent fibrosis regression. Additionally, the authors identified that Treg ablation resulted in increased levels of NK cells and M1 KCs [156]. This is reinforced by preceding work on a CCl4-induced liver fibrosis model; Tregs were demonstrated to suppress the activity of NK cells and M1 KCs [158], both suggested to be anti-fibrotic in nature due to their distinctive IFN-γ expression [42,52,159]. Thus, the conduction of chronic inflammation is seemingly favoured by the action of Tregs in this study, enabling fibrosis persistence. The immune regulation of NK cells during liver fibrosis is also supported in the context of chronic HCV infection; intrahepatic accumulation of Tregs was reported to modify the interaction between NK cells and HSCs via cell-contact-dependent inhibition and the downregulation of HLA-I in HSCs, an NK-cell-activating receptor ligand. The mechanisms mediating these responses involved the secretion of CTLA-4, a co-inhibitory receptor for T cells indicating an active state, and TGF-β and IL-8, respectively [160]. Blockade of NK cell inhibitors has been suggested as a potential immunotherapeutic to protect against fibrosis; clinical trials using tremelimumab, an anti-CTLA-4 monoclonal antibody, to treat HCC are currently underway with promising results. By inhibiting the downregulatory reaction of CTLA-4 binding to its ligand, B7, T cell effector mechanisms are elicited alongside NK cell activation [3]. However, the microenvironment and cellular interactions of HCC differ greatly at earlier and later stages of fibrosis due to underlying liver injury (Figure 2); hence, further exploration into the antagonistic relationship between Treg and NK cell signalling is required before this type of broad spectrum therapeutic could be clinically applied. Nevertheless, these data advocate for Treg contribution of fibrogenesis by protecting HSCs from NK-cell-induced apoptosis, a key mechanism for a reduction in liver fibrosis.
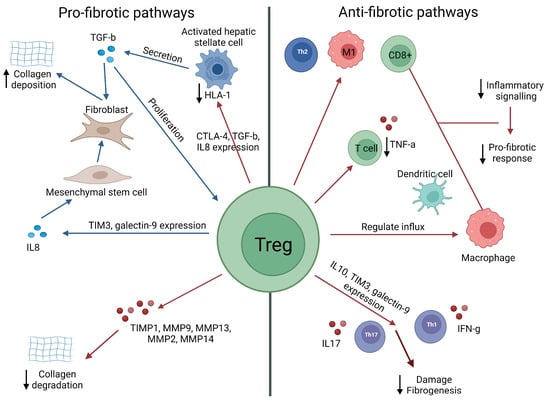
Figure 6.
Schematic representation of how Tregs could either promote or suppress liver fibrosis.
Within the liver microenvironment, Tregs can induce fibrotic responses mostly via a direct impact on fibroblast/stellate cells and mesenchymal stem cells, leading to increased collagen deposition (e.g., in response to high levels of TGF-b), or the suppression of collagen degradation (e.g., via the suppression of MMPs). Paradoxically, Tregs can also suppress inflammatory signalling and profibrotic responses via direct suppression of effector T cells, inducing a regulatory phenotype in antigen-presenting cells (e.g., via the production of IL-10 or via direct cell–cell contact). In the figure, red arrows = inhibition/downregulation, and blue arrows = expansion/upregulation [116,117,123,126,161,162,163].
3.2. Potential Role of Tregs in the Attenuation of Liver Fibrosis
Despite the evidence proposing an aggravative role of Tregs in pathogenic liver fibrosis, the notion that Tregs may ameliorate fibroplasia is also supported by various studies. Concerning infectious liver disease, a schistosomiasis mice model reported that CD4+CD25+ Tregs alongside innate APCs were able to protect the liver from damage and fibrosis progression due to elevated secretion of IL-10 [161]. Similarly, one report identifying the increased frequency of infiltrating CD4+FoxP3+ Tregs in the livers of chronic HCV patients alluded that the cells may limit the extent of hepatic fibrosis also due to the production of IL-10. These cells expressed a highly activated effector memory phenotype, as indicated by the expression of CTLA-4 and CD45RO (the memory isoform of CD45RA) [164]. Comparatively to some chronic HBV infection studies in humans, [111,112,113] this study identified a significant inverse correlation between the fibrosis score and intrahepatic Tregs; the CD4+FoxP3+ mTregs were only found in HCV-infected livers with limited fibrosis and did not correlate with the load of HCV RNA or alanine transaminase (ALT), a fundamental marker of liver injury [164,165]. Taken together, these data suggest the protective role of IL-10-producing Tregs against infection-induced fibrosis.
An experimental model of acute HBV infection also supported this protective function of Tregs, suggesting recruitment of CD4+FoxP3+ nTregs to the liver can limit hepatocellular damage at the expense of viral persistence. The immunosuppressive mechanism controlling this was proposed to be the regulation of DC and macrophage influx into the liver during early infection, as well as weakening TNF-α-producing T cell responses, particularly CD8+ Tc cells, to limit pro-inflammatory cytokine secretion [166]. This HBV-specific damage is distinctive to that of a chronic wound-healing response; however, limiting this initial injury could delay the initiation of early fibrogenesis in response. The relationship of mTregs with HBV chronicity progression was further delineated in a comprehensive investigation. Different infection stages in humans were analysed; only levels of mTregs with a robust suppressive ability were shown to infiltrate the hepatic environment during advanced inflammatory stages [100], comparable to a prior study [111]. In contrast to HCV infection [164], these cells positively correlated with serum ALT levels, and consequently, the authors suggested mTregs simply regulate ongoing HCV-induced liver injury rather than directly exacerbating hepatocellular damage. However, the authors did not histochemically analyse the extent of hepatic fibrosis within the different infection stages, most likely due to the invasive nature of biopsy; thus, it can only be assumed that fibrosis would be worse at more advanced stages based on the understanding of viral hepatitis infection [100]. Consequently, a correlation between Treg infiltration and the extent of fibrosis cannot be definitively determined within this study. Nonetheless, what is noteworthy is the identification of the galectin-9/Tim-3 interaction, which enables mTreg expansion [100], reported to be the mechanism which also elicited the differentiation of IL-8-producing CD4+FoxP3 Tregs [123].
Galectin-9 is a tandem lectin highly expressed within the liver and is the natural ligand of Tim-3 receptor (T cell immunoglobulin and mucin domain containing protein 3). The galectin-9-Tim-3 signalosome is associated with cell renewal, but in the case of hepatocellular damage, displays a dichotomous role depending on the injury. For example, in viral hepatitis and liver ischemia, galectin-9 is reported to enable viral persistence and attenuate hepatocyte damage, correspondingly. However, during autoimmune hepatitis, galectin-9+ Tregs can control the disease severity by regulating IL-17- and IFN-γ-producing cells, such as the Th17 and Th1 subsets [162,163,167]. Additionally, Tim-3+ Treg cells exist in small quantities; however, the enhancement of Tregs by this receptor is strongly associated with increased IL-10 expression, which may be indicative of anti-fibrotic properties and the suppression of Th17 cells [168]. Evidently, more research into the galectin-9/Tim-3 relationship with Tregs needs to be accomplished before its impact on injury-specific liver fibrosis can be fully established. Despite this paradigm, it again highlights the role of the Treg/Th17 ratio and how this may impact fibrosis. The tentative relationship between these two cell types has been suggested as both synergistic and antagonistic, much like that of Th1 and Th2 cells [169]. Defining this further in the case of liver diseases, an imbalance usually denotes an increase in Th17 cells and a decrease in Tregs, skewing to a pro-inflammatory environment during the onset of injury. Several studies have reported this, with an increase in intrahepatic Treg populations as liver damage becomes more advanced, progressing to cirrhosis. However, rather than Tregs being associated with direct fibrotic damage, their expansion is suggested to arbitrate inhibitory responses towards upregulated pro-fibrotic Th17 cells [153,169,170].
The effect of downregulated Tregs and imbalanced Treg/Th17 ratios has also been reported within retrospective studies and mice models of NAFLD; amplified populations of peripheral and intrahepatic Th17 cells in conjunction with diminished Treg populations enable the progression of liver disease, encompassing fibrosis. Thus, it can be inferred that the expansion of Tregs at later stages of liver injury is a suppressive response to regulate the level of Th17 cells, with fewer rTregs circulating in the periphery due to their activation into CD4+CD25hiFoxP3+ Treg phenotypes [136,171]. Supporting this notion is the plasticity of Tregs and Th17 cells within the idea of type 3 inflammatory responses. One study demonstrated that dysregulation of the Treg/Th17 ratio enabled increased production of signature type 3 cytokines, which in turn advanced fibrosis in vitro and in vivo via the enhancement of TGF-β signalling in human liver disease aetiologies and CCl4-induced liver fibrosis in mice. Concordant to the preceding results, the authors noted a positive correlation between the frequency of CD4+FoxP3hi Tregs and increased serum ALT levels as fibrosis became more advanced. However, their cellular distribution in fibrotic lesions was not consistent, unlike the unscarred parenchyma within cirrhotic livers [76]. One explanation for this observed imbalance is that of CD4+ Th cell transdifferentiation. FoxP3+ differentiation of Tregs is not a stagnant lineage, and the development of an IL-17-producing phenotype of Tregs or a phenotypic switch to a true Th17 subset has been widely reported in the presence of IL-6, which can aggravate fibrosis [172]. Considering this, it could be assumed that the transdifferentiation of Tregs towards a more pro-inflammatory Th17 phenotype may contribute to Treg depletion during early liver injury, thus promoting fibrosis. As liver chronicity and fibrosis develops, the suppressive CD4+ Treg population would repopulate, potentially via the transdifferentiation of Th17 subsets into particular Treg phenotypes, to control the ongoing damage. However, more research needs to be undertaken before this concept can be definitively supported [75,76,172].
As seen, experimental models of Tregs in liver damage have additionally provided evidence for their potential attenuative roles in the development of fibrosis, conflicting with other reports [156,160]. Recently, a CCl4-induced in vivo mice model of liver inflammation identified that CD4+FoxP3+ Tregs inhibit fibrosis via the regulation of chronic inflammatory signatures in the liver, particularly the infiltration of Th2 cells and M1-type monocytes and macrophages, as well as CD8+ Tc cells. Additionally, prior expansion of Tregs was instigated by the presence of IL-33 release, potentially from damaged hepatocytes, and that depletion of Tregs resulted in higher serum ALT and collagen deposition in comparison to controls. While this study modelled acute liver injury, the elevated expression of tissue fibrosis markers in response to Treg ablation, including α-SMA, strongly indicate a suppressive role against liver fibrosis progression [155]. Coincidingly, hepatic CD4+CD25+FoxP3+ Tregs in an acute cholestasis model using BDL demonstrated the ability to modulate liver inflammation and fibrosis. Protection against fibrogenesis was showed via Treg depletion, which resulted in increased IL-6 production and a reduction in IL-10 by effector T cells in the intrahepatic environment, leading to exacerbated fibrosis [173]. While the precise T cell subset for cytokine production was not defined, the importance of IL-10 production against liver fibrogenesis has been described, suggesting an antagonistic role against TGF-β [137,174,175,176], supporting the deductions stated earlier regarding protective IL-10-producing Tregs. In agreement with this, a similar BDL cholestasis model demonstrated expanded FoxP3+ Tregs in response to injury and suppression of pro-fibrogenic CD8+ Tc cell and Th17 cell activity, conforming with the severity of fibrosis after Treg ablation. This, clearly, further supports the notion of Treg modulation of the Treg/Th17 ratio and applicable cytokines, which may indirectly influence myofibroblast function during hepatic fibrosis regardless of liver disease aetiology.
3.3. Tregs: Pro-Fibrotic or Protective?
A closer look at the literature highlights the complex role of Tregs in the pathophysiology of liver fibrosis. Comparing different liver disease aetiologies, Treg expansion has shown to be variable in association with fibrosis progression, particularly within viral hepatitis settings. Indeed, most human investigations into the role of Tregs during liver fibrosis has been completed within chronic HCV and HBV patients. Consequently, differences in Treg expansion are most evident within these settings; however, several experimental models have enabled a better understanding of how Tregs may behave during liver fibrosis. Potential exacerbation of fibrosis was demonstrated via the downregulation of protective NK cells and the inhibition of IFN-γ, both anti-fibrotic in nature [160]. This is an expected function of Tregs due to their characteristic suppression of pro-inflammatory mechanisms to control the immune response. Additionally, the discovery of an IL-8+ CD4+FoxP3+ Treg phenotype, which may elicit inflammatory cell infiltration, suggests a pro-fibrotic mechanism due to increased association with collagen synthesis markers [123]. Interestingly, these studies were completed by the same research group, raising the issue of potential bias towards the pro-fibrogenic role of Tregs. More investigation is required before these mechanisms can be directly associated with liver fibrosis progression, although it is indicative of a pathogenic role in chronic injury settings. Associations with the TIMP/MMP balance also suggest Tregs can skew a pro-fibrogenic environment. However, disparities were observed between the two studies suggesting this relationship; one study reported an upregulation of MMP2 and decreased fibrosis in response to CD4+CD25+FoxP3+ Treg ablation [156], while another study demonstrated a positive correlation between IL-8+ CD4+FoxP3+ Treg function with MMP2 induction and fibrogenesis [123]. These differences may be due to the differing Treg phenotypes investigated or MMP2 upregulation not being related to Treg expansion; combined with the tentative role of MMP2 within fibroplasia, this relationship remains inconclusive.
Emergent research has indicated the importance of the Treg/Th17 relationship, mirroring that of the Th1/Th2 dichotomy. Understanding this, it could be suggested Th17 cells would aggravate pro-inflammatory damage during liver injury and Tregs would control their response, thus inducing pro-fibrotic regulatory mechanisms, corresponding to Th1 and Th2 subset roles. However, Th17 cells are largely pro-fibrotic via the induction of type 3 immune responses, driving both pro-inflammatory damage and liver fibrogenesis. In response, Treg cells also become progressively upregulated [76]. While the increase in both subsets may be pathologic within the liver, enabling fibrosis progression [153], it is more likely that Tregs expand to dampen the Th17 response, rather than as a direct cause of fibrosis exacerbation. Moreover, although increasing levels of pro-fibrotic TGF-β were associated with hepatic Treg aggregation, TGF-β is likely to exist as a membrane-bound form on Tregs, requiring cell–cell contact to elicit suppression [177]. Additionally, HSCs and Th17 cells have also been reported to be critical sources of free TGF-β in vitro, behaving in an autocrine manner for cell proliferation [116,178]. Hence, it is questionable whether Tregs constitute a significant source of TGF-β to directly progress the pro-fibrotic activation of HSCs in situ. Knowing this, it may be proposed that the Treg/Th17 balance has an inverse role to that of the Th1/Th2 ratio during liver fibrosis; however, more research would need to be conducted before a clear verdict could be formulated.
Furthermore, several reports suggested the protective role of CD4+ Tregs via IL-10 production during acute liver injury [137,161]. During chronic injury settings, IL-10 was suggested to be produced by a CD4+CD25+FoxP3+ mTreg subset [164]. Although this evidence was primarily presented in experimental rodent models, which cannot definitively replicate human liver fibrosis due to the timespan limitations and thus differing immunopathology, it certainly highlights the importance of IL-10-producing Tregs, perhaps the Tr1 phenotype (although this was not defined within the studies), and how they employ an anti-fibrotic role. This indicates different Treg phenotypes may become upregulated as injury persists, thus prompting distinctive mechanisms to influence distinct fibrotic immunopathologies. It may also explain the expansion of an IL-8+ Treg population and support the notion of a Treg/Th17 hybrid phenotype during a more pro-inflammatory phase of liver fibrosis, due to the plastic nature of CD4+ Tregs.
Accumulating these data, it is plausible that the role of Tregs is distinctive for individual patients and depending on liver disease aetiology. Indeed, the cellular interactions and hepatic microenvironment can govern the induced Treg phenotype in a complex mechanism which is yet to be fully explicated. The role of Tregs may not be entirely opposed to that of pro-fibrotic Th17 and Th2 cells, with the induction of bystander suppression, which inadvertently downregulates anti-fibrotic mediators. Additionally, there appears to be a difference between Treg mechanisms in acute versus chronic injury. This insinuates there may be an evolution of Treg function as hepatocellular damage changes to indirectly influence fibrosis depending on the disease setting; however, this evidence is mainly experimental. Conversely, it is probable that Tregs are inadvertently correlated with increasing fibrosis; their expansion is affiliated with the downregulation of self-harming, pro-fibrotic mechanisms (such as Th17 or Th2 cells), rather than actively, or directly, participating in fibrosis progression. From this, CD4+ Treg phenotypes expressing FoxP3 and/or CD25 appear to be double-edged swords, with the potential to elicit pro-fibrotic bystander suppression while managing the on-going fibrosis pathogenesis. Evidently, more research into different Treg phenotypes and how they interact with the hepatic network is required. However, this review explicates an essential preliminary understanding of their role within liver fibrosis settings.
4. Immunotherapy of Liver Fibrosis
The body of literature surrounding the potential for liver fibrosis resolution is robust (Figure 7) [55]. In the treatment of hepatic fibrosis, targeted therapies to modify or constrain pro-fibrotic crosstalk is the future frontier of disease management. The current research focus for many anti-fibrotic therapeutics centres around the control of HSC function, impeding signalling pathways which can activate these cells to reduce matrix production, and in turn promoting ECM degradation to reduce the overall scar tissue formation [55]. Alas, no medications have been approved or yet accomplish this task under liver fibrosis conditions. In the case of idiopathic pulmonary fibrosis (IPF), two anti-fibrotic therapies have been approved to reduce the event of acute respiratory regression and slow deterioration of lung function: pirfenidone and nintedanib. While the exact mechanism of the former remains unknown, nintedanib is a potent tyrosine protein kinase inhibitor which targets key pro-fibrotic agonists within IPF immunopathology, such as platelet-derived growth factor receptor (PDGFR) [179,180]. PDGFR has also been shown to positively correlate with liver fibrosis progression [181], and its ligand (PDGF) is produced in high quantities by M2a wound-healing macrophages [45], suggesting that nintedanib may be helpful in the treatment of liver fibrosis.
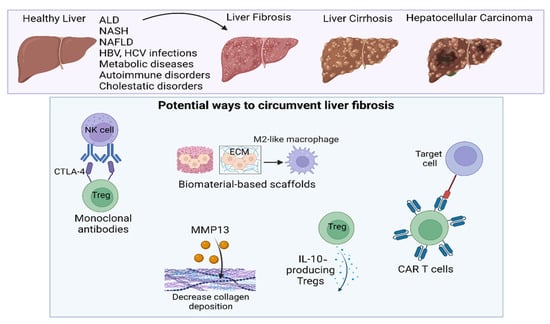
Figure 7.
Potential routes to reduce liver fibrosis.
Therapies aimed at directly targeting liver fibrosis do not currently exist; however, as research advances, some potential routes to attenuate the advancement of liver fibrosis include the use of monoclonal antibodies, immune-instructive scaffolds, IL-10-producing Tregs (HCV patients), decreased collagen deposition via MMP13 production, and CAR T cell therapy.
However, as fibrogenesis is an immune-mediated process, with inflammation able to exacerbate fibrosis progression, the development of anti-fibrotic immunotherapeutics is also of research interest. Pilot studies investigating IFN-γ and IL-10 as therapeutic agents demonstrate high efficacy in the management of liver fibrosis. Under viral hepatitis conditions, IFN-γ-1b (a recombinant form of human IFN-γ) could reduce advanced fibrosis in selected HCV- and HBV-infected patients, supporting its well-documented anti-fibrotic role and suggesting a favourable biologic rationale in certain patient subgroups. Additionally, early human trials of IL-10 treatment within HCV-infected patients exhibited a degree of fibrosis resolution, however, simultaneously exacerbating the HCV viral load [55]. Comparatively, a pro-fibrotic cytokine blockade may also offer beneficial effects on liver fibrosis, such as the inhibition of the TGF-β signalling pathway. Promising research into the in vivo delivery of small interfering RNA (siRNA) to inhibit TGF-β expression has been shown to decrease HSC production of type I collagen and α-SMA, administering an anti-fibrotic effect in CCl4-induced livers [182]. However, due to the complexity of the hepatic microenvironment, successful cytokine therapy remains a challenge. Many cytokines involved in liver fibrogenesis, including TGF-β, are pleiotropic, exerting differential effects depending on the cell they were secreted from, the location, neighbouring cytokines, and even the fibrotic stage and cause of liver injury. Silencing or amplification of one particular cytokine could be detrimental to the liver disease, thus exacerbating fibrogenesis. Hence, clarifying the extent of cytokine function and interaction during liver fibrosis is imperative.
As primary cellular targets identified within liver fibrosis, research into macrophages and monocytes is extensive. In experimental mice models of acute liver injury, the application of M2 macrophages as a monotherapy, and in combination with MSC transplantation, was able to rapidly lessen the effect of inflammatory mediators and hepatocellular necrosis, as well as diminish fibrotic lesions. This improvement in fibrotic sequelae is likely caused by increasing the number of highly phagocytic macrophages, potentially IL-10-stimulated M2c phenotypes [183], circulating in the liver microenvironment, which reduced scarring debris for tissue regeneration [138,139]. A more recent study investigated the effect of MSCs’ anti-fibrotic properties, finding that TNF-stimulated gene 6 (TSG-6) is an integral cytokine for this behaviour. Using calcium phosphate nanoparticles to deliver TSG-6 to CCl4-treated mice livers, MSCs showed elevated expression of TSG-6, leading to attenuated liver fibrosis via the modulation of M2 macrophages and increased production of MMP12, demonstrating a protective role. Additionally, an inhibitory feedback loop exerted by TNF-α, IL-6, and IL-1β on MMP12 expression was highlighted, which was interrupted by TSG-6 intervention. The evolution of liver fibrosis may be delineated by feedback loops such as this and presents an exciting avenue for future research. Despite these reports, there are still significant limitations when extrapolating this to human liver fibrosis. The advancement of fibrosis within mice models is rapid due to the experimental limitations, whereas human fibrosis is a long-term progression. As such, the cellular mechanisms governing fibrosis development will be different in situ, with pro-fibrotic M2 phenotypes suggested to play a central role within chronic wound-healing pathogenesis. Indeed, several studies investigating the effect of inhibiting TGF-β production using M2 macrophages have reported favourable clinical outcomes in reversing hepatocellular senescence and prompting fibrosis resolution [140,184]. Evidently, the role of macrophages within liver fibrosis is contextual to the induced phenotype and disease progression (acute versus chronic advancement). Elucidating the function of specific phenotypes within pathogenesis would enable improvements into cellular reprogramming therapies for fibrosis treatment.
4.1. Clinical Potential of CAR T Cells
Emerging research into bioengineered immune cells as a clinical intervention represents the forefront of an “immunotherapy revolution” [185]. Perhaps the most well known and exciting of these developments is the adoptive transfer of chimeric antigen receptor (CAR) T cell therapy. CARs are a type of artificial receptor; different generations can be bioengineered onto a T cell to execute the lymphocyte effector function against a specific antigen target. Clinical studies comparing the efficacy of CAR generations for one specific antigen are yet to be completed; therefore, it remains unclear which generation is the most potent, and safe, for patient application [186]. Currently, T cell therapeutics are mainly focused on treating cancer, including HCC, and inducing tolerance after liver transplantation [187]. Although HCC occurrence is increasing parallel to a global upsurge in liver diseases [4,7,9], hepatic fibrosis does not always instigate oncogenesis. Additionally, one of the major difficulties in developing CAR T cell therapy for hepatic diseases and fibrosis is the lack of suitable targets due to the complex liver microenvironment [188].
Nevertheless, emerging studies have demonstrated the benefits of T cell therapy and the exciting prospect they represent for liver fibrosis treatment. Recently, one research group investigated the clinical potential of novel CAR T cells, with an effector memory phenotype (CD45RA-), directed towards the urokinase-type plasminogen activator receptor (uPAR) in different disease models in vivo (Figure 8) [141]. uPAR is a cell surface protein which becomes upregulated during inflammation and cellular senescence pathways. In fact, it has recently been identified as a serum fibrosis marker within HBV, HCV, and NAFLD patients, positively correlating with the fibrotic stage [189,190]. Mice with CCl4-induced liver fibrosis or diet-induced NASH were treated with uPAR-specific CAR T cells, which congregated around hepatic scar tissue. Therapeutically, the CAR T cells efficiently eliminated senescent HSCs and instigated fibrosis resolution, which was sustained long-term with no reported toxicity in follow-up studies. This improved liver function in both aetiologies, therefore suggesting an extensive therapeutic potential for human implementation [141].
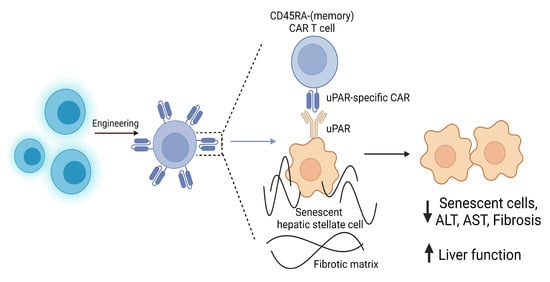
Figure 8.
The use of senolytic CAR T cells in liver fibrosis.
Cell senescence contributes to a range of chronic tissue pathologies, including liver fibrosis. Novel CAR T cells, with an effector memory phenotype (CD45RA-) directed towards the urokinase-type plasminogen activator receptor (uPAR) expressed on senescent hepatic stellate cells, have been shown to eliminate pro-inflammatory senescent HSCs, reduce fibrosis, and thus improve liver function. These senolytic CAR T cells have been shown in vivo to be effective against liver fibrosis of different etiologies, therefore offering therapeutic potential for human implementation [141].
Research into CAR T cell therapy for hepatic fibrosis currently remains few and far between as the treatment is complex and expensive. However, investigation into other tissue fibrosis models has shown promising results, which may further support the use of CAR T cells in the treatment of fibrogenesis throughout the body. Importantly, cardiac fibrosis resolution has responded successfully to CAR T cells in vivo. Fibroblast activation protein (FAP) on the surface of cardiac fibroblasts has been identified as a potential endogenous target. CAR T cells directed towards this protein in an in vivo mice model of heart disease demonstrated effective resolution of fibrosis and restored heart function post-injury, with no reported toxicity [142]. Several other reports using FAP-specific CAR T cell generation supported their expedient potential within various in vivo malignancies, inhibiting stromal fibroblasts and consequently hindering tumour growth and fibrosis [191,192]. Evidently, anti-FAP therapy may have a broad immunotherapeutic application, due to FAP expression being upregulated in several fibrosis-associated pathologies, including liver fibrosis [193,194,195].
4.2. Treg Manipulation as a Next-Generation Therapy
While it is currently not an approved clinical practice, the development of regulatory CAR T cells (CAR-Tregs) is an exciting branch of adoptive transfer therapy. Preceding this, the efficacy and safety of autologous polyclonal Treg therapy has been demonstrated largely within autoimmune conditions, such as autoimmune hepatitis [143], and under organ transplantation conditions, including with the liver, to prevent acute rejection and enhance tolerance [144]. In vivo expansion of polyclonal Tregs from naïve CD4+ T cells can be achieved via immunoregulatory cytokine and monoclonal antibody (mAb) application; several mice models of chronic inflammatory diseases, including multiple sclerosis and rheumatoid arthritis, showed delayed disease progression after treatment. In particular, the application of anti-CD3 mAb therapy, which interferes with T cell activation, was reported to improve immunoregulation by inducing TGF-β-secreting Th3 cells [87]. However, in the context of liver diseases, this type of therapy could be debilitating due to the pathogenic role of TGF-β in fibrogenesis. Techniques which can control TGF-β signalling would be more beneficial in liver fibrosis settings. Interestingly, there are preliminary data to suggest the TGF-β/SMAD signalling pathway is a regulator of the Treg/Th17 balance within the liver [196,197,198]; however, deeper investigation into this function would be required before the establishment of therapeutic agents to control it. Additionally, DC interaction is a key influencer of T cell phenotype, and there is research investigating DC-based therapeutics to control Treg expansion. A study investigating the clinical effect of IL-10 gene-enhanced DCs was also able to expand the Treg population and downregulate peripheral CD4+ T cell populations, potentially Th17 cells. The induction of a tolerogenic microenvironment post-treatment was shown to provide protection against CCl4-induced liver fibrosis in mice [137]. Notably, the advent of synthetic APCs fabricated using biocompatible platforms for T cell priming is an exciting research area. The induction of iTregs with a potent suppressive function was demonstrated using polymer-based synthetic APCs in the presence of TGF-β in vivo [145]. Evidently, the clinical implications of this type of therapy could be detrimental in liver fibrosis settings as the exact role of Treg cells and the extent of TGF-β pathology remains unclear. However, it presents the opportunity to develop a more cost-effective and less invasive “off-the-shelf” therapeutic in comparison to adoptive cell transfer. Alternatively, the repurposing of well-known immunosuppressive drugs has shown promising results for treating liver fibrosis via Treg manipulation, such as rapamycin. This macrolide targets the mTOR (mechanistic target of rapamycin) pathway, a central regulator for cell growth and proliferation, and is closely linked to TLR signalling for innate cell activation [199]. In a CCl4 mice model, rapamycin has been shown to directly alleviate hepatic fibrosis sequelae by upregulating Treg suppressive activity on HSC activation and correcting the Th17/Treg imbalance [146]. These studies further support the significance of Th17/Treg balance within liver diseases, suggesting its importance as a clinical target for fibrosis treatment.
Improving upon this, genetic engineering of Tregs via altering the antigen specificity, either via TCR or CAR modification, has demonstrated beneficial therapeutic effects in vivo. By generating antigen-specific Tregs, the suppression potency towards a targeted pathway or constituent is increased in comparison to treatment with polyclonal Tregs. Additionally, when comparing the efficacy of CAR engineering to TCR modification, CAR-Tregs display an advantage as their antigen repertoire is not HLA-restricted like that of TCRs [200]. Refining the antigen specificity would also prevent the off-target bystander suppression of immune responses, which could exacerbate the original liver disease and thus advance fibroplasia [87]. Preclinical studies of CAR-Tregs directed towards HLA-A2 (an alloantigen commonly mismatched in tissue graft and solid organ transplantation) have successfully induced tolerance and prevented xenogeneic-graft-versus-host disease and rejection in mice [200]. Therefore, the ex vivo expansion of CAR-Tregs is a promising next-generation therapy which could reverse advanced liver fibrosis if appropriate targets are identified.
5. Conclusions and Future Perspectives
Liver immunopathology is central to the manifestation of fibrosis for many liver disease aetiologies. Emergent research has increasingly elucidated the hepatic cellular network, revealing intricate crosstalk between non-immune, innate, and adaptive immune mechanisms. The infiltration of circulating immune cells and the dysregulation of resident liver cells, including KCs, can fabricate a pro-fibrogenic microenvironment via HSC activation and ECM protein deposition, thus driving scar formation [23]. Conversely, cells such as NK cells can elicit fibrosis regression via upregulated HSC apoptosis [52,54]. Despite this, many immune cells display contradictory roles contextual to the phenotype during liver fibrosis pathology, highlighting the complex nature of this condition. Perhaps the most ambiguous cell type within liver fibrosis is Tregs, with few studies being conducted on them compared to innate constituents, such as macrophages. As such, whether Tregs can exacerbate or convalesce liver fibrosis remains a complicated discussion. Theoretically, cells which exert regulatory and anti-inflammatory properties should avert self-harm. However, the very nature of a chronic wound-healing response is the dysregulation of the body’s own protective mechanisms [69]. Therefore, it may be speculated that Tregs could contribute pathogenically to fibrosis. Although research into their specific effector functions and how these may be implicated within fibrogenesis is incomplete, several key studies have reported significant correlations between Treg proliferation and fibrosis; thus, deductions can be made based on the presented data.
The significance of elucidating Treg function during liver fibrogenesis is not inconsequential; by exposing the complex crosstalk within the hepatic network, novel therapeutic targets can be unveiled for better treatment. While preclinical trials into cytokine- [55] and macrophage-based [138,139] therapies have shown a capacity for fibrosis regression, many cellular and inflammatory mediators exert pleiotropic effects, making their therapeutic application difficult. Hence, continued research into specific cell phenotypes and their roles within fibrogenesis is essential for therapy development. Additionally, the repurposing of IPF-approved drugs, such as nintedanib [180], for application within liver fibrosis settings may also be a point of research interest due to converging targets within both immunopathologies. However, the use of broad-spectrum immunosuppressant therapies presents the issue of unfavourable side effects, due to their unspecific targeting within various tissues. As such, the need for accurate, personalised therapy is ever-growing.
Tregs are one of the body’s own mechanisms for controlling the immune response; hence, the ability to beneficially manipulate their suppressive functions to subvert fibrosis progression represents an exciting development for anti-fibrotic therapy. Advancements into new-generation CARs and CAR-Tregs promise personalised therapy by eliminating immunogenic HLA-restriction and optimising the antigen specificity within target tissues. However, despite this prospect, the transmission of CAR-Treg research into a clinical-grade cell therapy faces significant challenges. Namely, the manufacture of safe and potent CAR constructs for optimal Treg function, without inducing debilitating immunogenicity, is of particular importance [186]. Clearly, this type of next-generation therapy currently remains beyond the bounds of clinical application due to an incomplete understanding of Treg phenotypes and their functions. By increasing this knowledge, the CAR-Treg field can be advanced for their use within the medical field, and prospectively, liver fibrosis settings. One of the key limitations in the study of Treg function is the complication of definable surface markers. As stated earlier, the presence of absence of CD25 or FoxP3 expression does not necessarily indicate regulatory function, and several other activated lymphoid and myeloid populations can also display CD25 and FoxP3 [201]. Indeed, the use of anti-CD25 monoclonal antibodies, a commonly used method to deplete Tregs, can knock down other effector cell populations [83]. As such, this can create discrepancies between data and can offer an explanation as to why different populations of Tregs behave differently in the same disease setting, as is the case in liver fibrosis. Future research into defining more reliable markers will therefore provide greater certainty when identifying Treg phenotypes and allow the development of new strategies to target Tregs for depletion or isolation, as well as elucidating their role within liver fibrosis.
Author Contributions
K.O.L. and S.M. wrote the review. C.E.T., L.E.F. and A.M.G. supervised, reviewed, and edited the article. All authors have read and agreed to the published version of the manuscript.
Funding
A.M.G., C.E.T., and L.E.F. were funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement 760921 (PANBioRA).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the studies of the author.
Data Availability Statement
Not applicable.
Acknowledgments
All figures were created using BioRender.com. Accessed on 11 July 2023.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pan, X.; Ma, X.; Jiang, Y.; Wen, J.; Yang, L.; Chen, D.; Cao, X.; Peng, C. A Comprehensive Review of Natural Products against Liver Fibrosis: Flavonoids, Quinones, Lignans, Phenols, and Acids. Evid.-Based Complement. Altern. Med. 2020, 2020, 7171498. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Keenan, B.P.; Fong, L.; Kelley, R.K. Immunotherapy in hepatocellular carcinoma: The complex interface between inflammation, fibrosis, and the immune response. J. Immunother. Cancer 2019, 7, 267. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, 2–6. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Sharma, A.; Nagalli, S. Chronic Liver Disease; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Xia, M.-F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef]
- Fattahi, M.R.; Niknam, R.; Safarpour, A.; Sepehrimanesh, M.; Lotfi, M. The Prevalence of Metabolic Syndrome in Non-alcoholic Fatty Liver Disease; A Population-Based Study. Middle East J. Dig. Dis. 2016, 8, 131–137. [Google Scholar] [CrossRef]
- Elferink, R.O. Cholestasis. Gut 2003, 52, ii42. [Google Scholar] [CrossRef]
- Nallagangula, K.S.; Nagaraj, S.K.; Venkataswamy, L.; Chandrappa, M. Liver fibrosis: A compilation on the biomarkers status and their significance during disease progression. Future Sci. OA 2017, 4, FSO250. [Google Scholar] [CrossRef]
- Yeom, S.K.; Lee, C.H.; Cha, S.H.; Park, C.M. Prediction of liver cirrhosis, using diagnostic imaging tools. World J. Hepatol. 2015, 7, 2069–2079. [Google Scholar] [CrossRef]
- Ginès, P.; Graupera, I.; Lammert, F.; Angeli, P.; Caballeria, L.; Krag, A.; Guha, I.N.; Murad, S.D.; Castera, L. Screening for liver fibrosis in the general population: A call for action. Lancet Gastroenterol. Hepatol. 2016, 1, 256–260. [Google Scholar] [CrossRef]
- Lucero, C.; Brown, R.S., Jr. Noninvasive Measures of Liver Fibrosis and Severity of Liver Disease. Gastroenterol. Hepatol. 2016, 12, 33–40. [Google Scholar]
- Wang, X.; Wu, B. Critical issues in the diagnosis and treatment of liver cirrhosis. Gastroenterol. Rep. 2019, 7, 227–230. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef]
- Bessone, F. Non-steroidal anti-inflammatory drugs: What is the actual risk of liver damage? World J. Gastroenterol. 2010, 16, 5651–5661. [Google Scholar] [CrossRef]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef]
- da Silva Morais, A.; Vieira, S.; Zhao, X.; Mao, Z.; Gao, C.; Oliveira, J.M.; Reis, R.L. Advanced Biomaterials and Processing Methods for Liver Regeneration: State-of-the-Art and Future Trends. Adv. Healthc. Mater. 2020, 9, 1901435. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, Z.; Wang, F.-S. Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell. Mol. Immunol. 2012, 9, 296–301. [Google Scholar] [CrossRef]
- Yang, L.; Seki, E. Toll-Like Receptors in Liver Fibrosis: Cellular Crosstalk and Mechanisms. Front. Physiol. 2012, 3, 138. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Dobie, R.; Henderson, N.C. Homing in on the hepatic scar: Recent advances in cell-specific targeting of liver fibrosis. F1000Research 2016, 5, 1749. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7, 921. [Google Scholar] [CrossRef]
- Xu, J.; Liu, X.; Koyama, Y.; Wang, P.; Lan, T.; Kim, I.-G.; Kim, I.H.; Ma, H.-Y.; Kisseleva, T. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol. 2014, 5, 167. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, B.G.; Pestana, R.C.; Abugabal, Y.I.; Krishnan, S.; Chen, J.; Hassan, M.M.; Wolff, R.A.; Rashid, A.; Amin, H.M.; Kaseb, A.O. Origin and role of hepatic myofibroblasts in hepatocellular carcinoma. Oncotarget 2020, 11, 1186–1201. [Google Scholar] [CrossRef]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef]
- Naim, A.; Pan, Q.; Baig, M.S. Matrix Metalloproteinases (MMPs) in Liver Diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef]
- Roeb, E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018, 68–69, 463–473. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Dou, L.; Ono, Y.; Chen, Y.F.; Thomson, A.W.; Chen, X.P. Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin. Liver Dis. 2018, 38, 170–180. [Google Scholar] [CrossRef]
- Bartneck, M.; Wang, J. Therapeutic Targeting of Neutrophil Granulocytes in Inflammatory Liver Disease. Front. Immunol. 2019, 10, 2257. [Google Scholar] [CrossRef]
- Pulli, B.; Ali, M.; Iwamoto, Y.; Zeller, M.W.G.; Schob, S.; Linnoila, J.J.; Chen, J.W. Myeloperoxidase-Hepatocyte-Stellate Cell Cross Talk Promotes Hepatocyte Injury and Fibrosis in Experimental Nonalcoholic Steatohepatitis. Antioxid. Redox Signal. 2015, 23, 1255–1269. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef]
- Cardoso, C.C.; Matiollo, C.; Pereira, C.H.J.; Fonseca, J.S.; Alves, H.E.L.; da Silva, O.M.; de Souza Menegassi, V.; dos Santos, C.R.; de Moraes, A.C.R.; de Lucca Schiavon, L.; et al. Patterns of dendritic cell and monocyte subsets are associated with disease severity and mortality in liver cirrhosis patients. Sci. Rep. 2021, 11, 5923. [Google Scholar] [CrossRef]
- Ploeger, D.T.; Hosper, N.A.; Schipper, M.; Koerts, J.A.; de Rond, S.; Bank, R.A. Cell plasticity in wound healing: Paracrine factors of M1/M2 polarized macrophages influence the phenotypical state of dermal fibroblasts. Cell Commun. Signal. 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-Y.; Li, X.-F.; Meng, X.-M.; Huang, C.; Zhang, L.; Li, J. Macrophage Phenotype in Liver Injury and Repair. Scand. J. Immunol. 2017, 85, 166–174. [Google Scholar] [CrossRef]
- Witherel, C.E.; Abebayehu, D.; Barker, T.H.; Spiller, K.L. Macrophage and Fibroblast Interactions in Biomaterial-Mediated Fibrosis. Adv. Healthc. Mater. 2019, 8, 1801451. [Google Scholar] [CrossRef]
- Li, M.; Hou, Q.; Zhong, L.; Zhao, Y.; Fu, X. Macrophage Related Chronic Inflammation in Non-Healing Wounds. Front. Immunol. 2021, 12, 681710. [Google Scholar] [CrossRef]
- Chung, L.; Maestas, D.R.; Housseau, F.; Elisseeff, J.H. Key players in the immune response to biomaterial scaffolds for regenerative medicine. Adv. Drug Deliv. Rev. 2017, 114, 184–192. [Google Scholar] [CrossRef]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef]
- Liu, Y.; Munker, S.; Müllenbach, R.; Weng, H.-L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S.; Park, O. Liver natural killer and natural killer T cells: Immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 2009, 86, 513–528. [Google Scholar] [CrossRef]
- Fasbender, F.; Widera, A.; Hengstler, J.G.; Watzl, C. Natural Killer Cells and Liver Fibrosis. Front. Immunol. 2016, 7, 19. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S.; Jeong, W.-I.L. Activation of natural killer cells inhibits liver fibrosis: A novel strategy to treat liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 2007, 1, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol. Res. 2020, 155, 104720. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Mertens, P.R.; Gressner, A.M.; Dooley, S. IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J. Hepatol. 2007, 46, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C. Current and future anti-fibrotic therapies for chronic liver disease. Clin. Liver Dis. 2008, 12, 939–962. [Google Scholar] [CrossRef]
- Rahman, A.H.; Aloman, C. Dendritic cells and liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Almeda-Valdes, P.; Aguilar Olivos, N.E.; Barranco-Fragoso, B.; Uribe, M.; Méndez-Sánchez, N. The Role of Dendritic Cells in Fibrosis Progression in Nonalcoholic Fatty Liver Disease. BioMed Res. Int. 2015, 2015, 768071. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.-Y.; Wang, N.; Feng, Y.; Wang, X.; Feng, Y. Recent Insights Into the Role of Immune Cells in Alcoholic Liver Disease. Front. Immunol. 2019, 10, 1328. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Valencia-Rodríguez, A.; Coronel-Castillo, C.; Vera-Barajas, A.; Contreras-Carmona, J.; Ponciano-Rodríguez, G.; Zamora-Valdés, D. The cellular pathways of liver fibrosis in non-alcoholic steatohepatitis. Ann. Transl. Med. 2020, 8, 400. [Google Scholar] [CrossRef]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef]
- Bhogal, R.K.; Bona, C.A. B cells: No longer bystanders in liver fibrosis. J. Clin. Investig. 2005, 115, 2962–2965. [Google Scholar] [CrossRef]
- Faggioli, F.; Palagano, E.; Di Tommaso, L.; Donadon, M.; Marrella, V.; Recordati, C.; Mantero, S.; Villa, A.; Vezzoni, P.; Cassani, B. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 2018, 67, 1970–1985. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Ricci, O.E.; Paoletti, F.; Surrenti, C. Immune mechanisms for hepatic fibrogenesis. T-lymphocyte-mediated stimulation of fibroblast collagen production in chronic active hepatitis. Liver 1985, 5, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Koda, Y.; Teratani, T.; Chu, P.-S.; Hagihara, Y.; Mikami, Y.; Harada, Y.; Tsujikawa, H.; Miyamoto, K.; Suzuki, T.; Taniki, N.; et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 2021, 12, 4474. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, P.; Wu, Y.; Wang, L. Metabolic tissue-resident CD8+ T cells: A key player in obesity-related diseases. Obes. Rev. 2021, 22, 13133. [Google Scholar] [CrossRef] [PubMed]
- Breuer, D.A.; Pacheco, M.C.; Washington, M.K.; Montgomery, S.A.; Hasty, A.H.; Kennedy, A.J. CD8+ T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, 211–224. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Wang, X.-Z. Interleukin-10 and chronic liver disease. World J. Gastroenterol. 2006, 12, 1681–1685. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Li, H.; You, H.; Fan, X.; Jia, J. Hepatic macrophages in liver fibrosis: Pathogenesis and potential therapeutic targets. BMJ Open Gastroenterol. 2016, 3, e000079. [Google Scholar] [CrossRef]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 723–728. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.; Gottschalk, S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef]
- Rainard, P.; Cunha, P.; Martins, R.P.; Gilbert, F.B.; Germon, P.; Foucras, G. Type 3 immunity: A perspective for the defense of the mammary gland against infections. Vet. Res. 2020, 51, 129. [Google Scholar] [CrossRef] [PubMed]
- Fabre, T.; Molina, M.F.; Soucy, G.; Goulet, J.-P.; Willems, B.; Villeneuve, J.-P.; Bilodeau, M.; Shoukry, N.H. Type 3 cytokines IL-17A and IL-22 drive TGF-β–dependent liver fibrosis. Sci. Immunol. 2018, 3, 7754. [Google Scholar] [CrossRef] [PubMed]
- Shoukry, N.H.; Fabre, T.; Molina, M.F.; Soucy, G.; Willems, B.; Villeneuve, J.-P.; Bilodeau, M. Th17 Cytokines Drive Liver Fibrosis Progression by Regulating TGF-β Signaling through Activation of MAPKs. J. Immunol. 2017, 198, 197.12. [Google Scholar] [CrossRef]
- Lafdil, F.; Miller, A.M.; Ki, S.H.; Gao, B. Th17 cells and their associated cytokines in liver diseases. Cell. Mol. Immunol. 2010, 7, 250–254. [Google Scholar] [CrossRef]
- Mason, G.M.; Lowe, K.; Melchiotti, R.; Ellis, R.; de Rinaldis, E.; Peakman, M.; Heck, S.; Lombardi, G.; Tree, T.I.M. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J. Immunol. 2015, 195, 2030. [Google Scholar] [CrossRef]
- Wan, M.; Han, J.; Ding, L.; Hu, F.; Gao, P. Novel Immune Subsets and Related Cytokines: Emerging Players in the Progression of Liver Fibrosis. Front. Med. 2021, 8, 604894. [Google Scholar] [CrossRef]
- Kitz, A.; Singer, E.; Hafler, D. Regulatory T Cells: From Discovery to Autoimmunity. Cold Spring Harb. Perspect. Med. 2018, 8, a029041. [Google Scholar] [CrossRef]
- Devaud, C.; Darcy, P.K.; Kershaw, M.H. Foxp3 expression in T regulatory cells and other cell lineages. Cancer Immunol. Immunother. 2014, 63, 869–876. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; de St. Groth, B.F.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Wallet, M.A.; Sen, P.; Tisch, R. Immunoregulation of dendritic cells. Clin. Med. Res. 2005, 3, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, 22236. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.; Biswas, M. Regulatory T cell therapy: Current and future design perspectives. Cell. Immunol. 2020, 356, 104193. [Google Scholar] [CrossRef]
- Curotto de Lafaille, M.A.; Lafaille, J.J. Natural and Adaptive Foxp3+ Regulatory T Cells: More of the Same or a Division of Labor? Immunity 2009, 30, 626–635. [Google Scholar] [CrossRef]
- Lin, X.; Chen, M.; Liu, Y.; Guo, Z.; He, X.; Brand, D.; Zheng, S.G. Advances in distinguishing natural from induced Foxp3(+) regulatory T cells. Int. J. Clin. Exp. Pathol. 2013, 6, 116–123. [Google Scholar]
- Freudenberg, K.; Lindner, N.; Dohnke, S.; Garbe, A.I.; Schallenberg, S.; Kretschmer, K. Critical Role of TGF-β and IL-2 Receptor Signaling in Foxp3 Induction by an Inhibitor of DNA Methylation. Front. Immunol. 2018, 9, 125. [Google Scholar] [CrossRef]
- Tischner, D.; Wiegers, G.J.; Fiegl, H.; Drach, M.; Villunger, A. Mutual antagonism of TGF-beta and Interleukin-2 in cell survival and lineage commitment of induced regulatory T cells. Cell Death Differ. 2012, 19, 1277–1287. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X. Foxp3 Instability Helps tTregs Distinguish Self and Non-self. Front. Immunol. 2019, 10, 2226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liao, X.; Kang, Y. Tregs: Where We Are and What Comes Next? Front. Immunol. 2017, 8, 1578. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ma, X.; Gong, R.; Zhu, J.; Wei, L.; Yao, J. Recent advances in CD8+ regulatory T cell research. Oncol. Lett. 2018, 15, 8187–8194. [Google Scholar] [CrossRef]
- Wawman, R.E.; Bartlett, H.; Oo, Y.H. Regulatory T Cell Metabolism in the Hepatic Microenvironment. Front. Immunol. 2018, 8, 1889. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Lohse, A.W. Immune tolerance: What is unique about the liver. J. Autoimmun. 2010, 34, 1–6. [Google Scholar] [CrossRef]
- Jeffery, H.C.; Braitch, M.K.; Brown, S.; Oo, Y.H. Clinical Potential of Regulatory T Cell Therapy in Liver Diseases: An Overview and Current Perspectives. Front. Immunol. 2016, 7, 334. [Google Scholar] [CrossRef]
- Devi, M.; Vijayalakshmi, D.; Dhivya, K.; Janane, M. Memory T Cells (CD45RO) Role and Evaluation in Pathogenesis of Lichen Planus and Lichenoid Mucositis. J. Clin. Diagn. Res. 2017, 11, 84–86. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Way, S.S.; Abbas, A.K. Regulatory T cell memory. Nat. Rev. Immunol. 2016, 16, 90–101. [Google Scholar] [CrossRef]
- Hu, C.-C.; Jeng, W.-J.; Chen, Y.-C.; Fang, J.-H.; Huang, C.-H.; Teng, W.; Hsieh, Y.-C.; Lin, Y.-C.; Chien, R.-N.; Sheen, I.S.; et al. Memory Regulatory T cells Increase Only In Inflammatory Phase of Chronic Hepatitis B Infection and Related to Galectin-9/Tim-3 interaction. Sci. Rep. 2017, 7, 15280. [Google Scholar] [CrossRef]
- Jung, M.K.; Shin, E.-C. Regulatory T Cells in Hepatitis B and C Virus Infections. Immune Netw. 2016, 16, 330–336. [Google Scholar] [CrossRef]
- Scharte, M.; Han, X.; Bertges, D.J.; Fink, M.P.; Delude, R.L. Cytokines induce HIF-1 DNA binding and the expression of HIF-1-dependent genes in cultured rat enterocytes. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 284, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Colgan, S.P.; Eltzschig, H.K. Hypoxia-inducible factors as molecular targets for liver diseases. J. Mol. Med. 2016, 94, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef] [PubMed]
- Atif, M.; Warner, S.; Oo, Y.H. Linking the gut and liver: Crosstalk between regulatory T cells and mucosa-associated invariant T cells. Hepatol. Int. 2018, 12, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Jeffery, H.C.; Hunter, S.; Bhogal, R.; Birtwistle, J.; Braitch, M.K.; Roberts, S.; Ming, M.; Hannah, J.; Thomas, C.; et al. Human intrahepatic regulatory T cells are functional, require IL-2 from effector cells for survival, and are susceptible to Fas ligand-mediated apoptosis. Hepatology 2016, 64, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Z.-q.; Zhang, L.; Zheng, H.; Zhou, M.-g.; Liu, D.-w. Burden of viral hepatitis caused by specific aetiologies in China, 1990–2016: Findings from the GBD 2016. BMC Public Health 2020, 20, 1461. [Google Scholar] [CrossRef] [PubMed]
- Mailer, R.K.W.; Gisterå, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia Induces Differentiation of Regulatory T Cells in the Liver. Circ. Res. 2017, 120, 1740–1753. [Google Scholar] [CrossRef]
- Zhai, N.; Chi, X.; Li, T.; Song, H.; Li, H.; Jin, X.; Crispe, I.N.; Su, L.; Niu, J.; Tu, Z. Hepatitis C virus core protein triggers expansion and activation of CD4+CD25+ regulatory T cells in chronic hepatitis C patients. Cell. Mol. Immunol. 2015, 12, 743–749. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Feng, K.; Fan, D.; Xue, W.; Lu, J. ‘Repair’ Treg Cells in Tissue Injury. Cell. Physiol. Biochem. 2017, 43, 2155–2169. [Google Scholar] [CrossRef]
- Xu, D.; Fu, J.; Jin, L.; Zhang, H.; Zhou, C.; Zou, Z.; Zhao, J.-M.; Zhang, B.; Shi, M.; Ding, X.; et al. Circulating and Liver Resident CD4+CD25+ Regulatory T Cells Actively Influence the Antiviral Immune Response and Disease Progression in Patients with Hepatitis B. J. Immunol. 2006, 177, 739. [Google Scholar] [CrossRef]
- Peng, G.; Li, S.; Wu, W.; Sun, Z.; Chen, Y.; Chen, Z. Circulating CD4+ CD25+ regulatory T cells correlate with chronic hepatitis B infection. Immunology 2008, 123, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Stoop, J.N.; van der Molen, R.G.; Baan, C.C.; van der Laan, L.J.W.; Kuipers, E.J.; Kusters, J.G.; Janssen, H.L.A. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology 2005, 41, 771–778. [Google Scholar] [CrossRef] [PubMed]
- El-Badawy, O.; Sayed, D.; Badary, M.S.; Abd-Alrahman, M.E.; El-Feky, M.A.; Thabit, A.G. Relations of regulatory T cells with hepatitis markers in chronic hepatitis B virus infection. Hum. Immunol. 2012, 73, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Lei, Z.; Wang, X.; Qi, Q.; He, J.; Liu, D.; Wang, X.; Chen, X.; Zhu, J.; Li, Y.; et al. Hepatitis B envelope antigen increases Tregs by converting CD4+CD25- T cells into CD4+CD25+Foxp3+ Tregs. Exp. Ther. Med. 2020, 20, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Mucida, D.; Tyznik, A.J.; Kronenberg, M.; Cheroutre, H. Hepatic stellate cells function as regulatory bystanders. J. Immunol. 2011, 186, 5549–5555. [Google Scholar] [CrossRef] [PubMed]
- Khanam, A.; Chua, J.V.; Kottilil, S. Immunopathology of Chronic Hepatitis B Infection: Role of Innate and Adaptive Immune Response in Disease Progression. Int. J. Mol. Sci. 2021, 22, 5497. [Google Scholar] [CrossRef] [PubMed]
- Ebinuma, H.; Nakamoto, N.; Li, Y.; Price, D.A.; Gostick, E.; Levine, B.L.; Tobias, J.; Kwok, W.W.; Chang, K.-M. Identification and in vitro expansion of functional antigen-specific CD25+ FoxP3+ regulatory T cells in hepatitis C virus infection. J. Virol. 2008, 82, 5043–5053. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ikeda, F.; Stadanlick, J.; Nunes, F.A.; Alter, H.J.; Chang, K.-M. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 2003, 38, 1437–1448. [Google Scholar] [CrossRef]
- Ward, S.M.; Fox, B.C.; Brown, P.J.; Worthington, J.; Fox, S.B.; Chapman, R.W.; Fleming, K.A.; Banham, A.H.; Klenerman, P. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J. Hepatol. 2007, 47, 316–324. [Google Scholar] [CrossRef]
- Rushbrook, S.M.; Ward, S.M.; Unitt, E.; Vowler, S.L.; Lucas, M.; Klenerman, P.; Alexander, G.J.M. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J. Virol. 2005, 79, 7852–7859. [Google Scholar] [CrossRef]
- Boettler, T.; Spangenberg, H.C.; Neumann-Haefelin, C.; Panther, E.; Urbani, S.; Ferrari, C.; Blum, H.E.; von Weizsäcker, F.; Thimme, R. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J. Virol. 2005, 79, 7860–7867. [Google Scholar] [CrossRef] [PubMed]
- Langhans, B.; Krämer, B.; Louis, M.; Nischalke, H.D.; Hüneburg, R.; Staratschek-Jox, A.; Odenthal, M.; Manekeller, S.; Schepke, M.; Kalff, J.; et al. Intrahepatic IL-8 producing Foxp3+CD4+ regulatory T cells and fibrogenesis in chronic hepatitis C. J. Hepatol. 2013, 59, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Kotsiou, O.S.; Gourgoulianis, K.I.; Zarogiannis, S.G. IL-33/ST2 Axis in Organ Fibrosis. Front. Immunol. 2018, 9, 2432. [Google Scholar] [CrossRef] [PubMed]
- She, Y.X.; Yu, Q.Y.; Tang, X.X. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.W.; Seidler, S.; Gassler, N.; Nattermann, J.; Luedde, T.; Trautwein, C.; Tacke, F. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS ONE 2011, 6, 21381. [Google Scholar] [CrossRef]
- Wang, H.; Lafdil, F.; Wang, L.; Yin, S.; Feng, D.; Gao, B. Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: Involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci. 2011, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Himmel, M.E.; Crome, S.Q.; Ivison, S.; Piccirillo, C.; Steiner, T.S.; Levings, M.K. Human CD4+FOXP3+ regulatory T cells produce CXCL8 and recruit neutrophils. Eur. J. Immunol. 2011, 41, 306–312. [Google Scholar] [CrossRef]
- Kryczek, I.; Wang, L.; Wu, K.; Li, W.; Zhao, E.; Cui, T.; Wei, S.; Liu, Y.; Wang, Y.; Vatan, L.; et al. Inflammatory regulatory T cells in the microenvironments of ulcerative colitis and colon carcinoma. OncoImmunology 2016, 5, 1105430. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Frangogiannis, N.G. Chemokines and cardiac fibrosis. Front Biosci (Sch. Ed.) 2009, 1, 391–405. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, W.; Xing, D.; Li, P.; Fu, J.; Gong, K.; Hage, F.G.; Oparil, S.; Chen, Y.-F. Endothelial cells overexpressing IL-8 receptor reduce cardiac remodeling and dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H590–H598. [Google Scholar] [CrossRef]
- Claassen, M.A.A.; Janssen, H.L.A.; de Knegt, R.J.; Boonstra, A. Controversy on the role of FoxP3+ regulatory T cells in fibrogenesis in chronic hepatitis C virus infections. J. Hepatol. 2014, 60, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Kamimura, K.; Kobayashi, Y.; Ohtsuka, M.; Miura, H.; Ohashi, R.; Yokoo, T.; Kanefuji, T.; Suda, T.; Tsuchida, M.; et al. Effective Prevention of Liver Fibrosis by Liver-targeted Hydrodynamic Gene Delivery of Matrix Metalloproteinase-13 in a Rat Liver Fibrosis Model. Mol. Ther. Nucleic Acids 2016, 5, 276. [Google Scholar] [CrossRef]
- Kim, E.-J.; Cho, H.-J.; Park, D.; Kim, J.Y.; Kim, Y.B.; Park, T.G.; Shim, C.-K.; Oh, Y.-K. Antifibrotic effect of MMP13-encoding plasmid DNA delivered using polyethylenimine shielded with hyaluronic acid. Mol. Ther. 2011, 19, 355–361. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wu, L.; Xie, W.; Shao, Y.; Jiang, J.; Zhao, Z.; Yan, M.; Chen, Z.; Cui, D. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. 2017, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tang, X.; Yang, M.; Zhang, S.; Li, S.; Chen, Y.; Liu, M.; Guo, Y.; Lu, M. Interleukin 10 Gene-Modified Bone Marrow-Derived Dendritic Cells Attenuate Liver Fibrosis in Mice by Inducing Regulatory T Cells and Inhibiting the TGF-β/Smad Signaling Pathway. Mediat. Inflamm. 2019, 2019, 4652596. [Google Scholar] [CrossRef]
- Starkey Lewis, P.; Campana, L.; Aleksieva, N.; Cartwright, J.A.; Mackinnon, A.; O’Duibhir, E.; Kendall, T.; Vermeren, M.; Thomson, A.; Gadd, V.; et al. Alternatively activated macrophages promote resolution of necrosis following acute liver injury. J. Hepatol. 2020, 73, 349–360. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tsuchiya, A.; Seino, S.; Kawata, Y.; Kojima, Y.; Ikarashi, S.; Starkey Lewis, P.J.; Lu, W.-Y.; Kikuta, J.; Kawai, H.; et al. Mesenchymal Stem Cells and Induced Bone Marrow-Derived Macrophages Synergistically Improve Liver Fibrosis in Mice. Stem Cells Transl. Med. 2019, 8, 271–284. [Google Scholar] [CrossRef]
- Bird, T.G.; Müller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.-Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, 1230. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Oo, Y.H.; Ackrill, S.; Cole, R.; Jenkins, L.; Anderson, P.; Jeffery, H.C.; Jones, N.; Jeffery, L.E.; Lutz, P.; Wawman, R.E.; et al. Liver homing of clinical grade Tregs after therapeutic infusion in patients with autoimmune hepatitis. JHEP Rep. 2019, 1, 286–296. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Whitehouse, G.; Grageda, N.; Cramp, M.E.; Lim, T.Y.; Romano, M.; Thirkell, S.; Lowe, K.; Fry, L.; Heward, J.; et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am. J. Transpl. 2020, 20, 1125–1136. [Google Scholar] [CrossRef]
- Rhodes, K.R.; Meyer, R.A.; Wang, J.; Tzeng, S.Y.; Green, J.J. Biomimetic tolerogenic artificial antigen presenting cells for regulatory T cell induction. Acta Biomater. 2020, 112, 136–148. [Google Scholar] [CrossRef]
- Gu, L.; Deng, W.S.; Sun, X.F.; Zhou, H.; Xu, Q. Rapamycin ameliorates CCl4-induced liver fibrosis in mice through reciprocal regulation of the Th17/Treg cell balance. Mol. Med. Rep. 2016, 14, 1153–1161. [Google Scholar] [CrossRef][Green Version]
- Anthony, B.; Allen, J.T.; Li, Y.S.; McManus, D.P. Hepatic stellate cells and parasite-induced liver fibrosis. Parasites Vectors 2010, 3, 60. [Google Scholar] [CrossRef]
- Silva Pereira, S.; Trindade, S.; De Niz, M.; Figueiredo, L.M. Tissue tropism in parasitic diseases. Open Biol. 2019, 9, 190036. [Google Scholar] [CrossRef]
- Tang, Z.-L.; Huang, Y.; Yu, X.-B. Current status and perspectives of Clonorchis sinensis and clonorchiasis: Epidemiology, pathogenesis, omics, prevention and control. Infect. Dis. Poverty 2016, 5, 71. [Google Scholar] [CrossRef]
- Allen, J.E.; Sutherland, T.E. Host protective roles of type 2 immunity: Parasite killing and tissue repair, flip sides of the same coin. Semin. Immunol. 2014, 26, 329–340. [Google Scholar] [CrossRef]
- Zhang, B.-B.; Yan, C.; Fang, F.; Du, Y.; Ma, R.; Li, X.-Y.; Yu, Q.; Meng, D.; Tang, R.-X.; Zheng, K.-Y. Increased hepatic Th2 and Treg subsets are associated with biliary fibrosis in different strains of mice caused by Clonorchis sinensis. PLoS ONE 2017, 12, 171005. [Google Scholar] [CrossRef]
- Xu, S.; Gu, M.; Wu, K.; Li, G. Unraveling the Role of Hydroxyproline in Maintaining the Thermal Stability of the Collagen Triple Helix Structure Using Simulation. J. Phys. Chem. B 2019, 123, 7754–7763. [Google Scholar] [CrossRef]
- Yan, C.; Zhang, B.-B.; Hua, H.; Li, B.; Zhang, B.; Yu, Q.; Li, X.-Y.; Liu, Y.; Pan, W.; Liu, X.-Y.; et al. The Dynamics of Treg/Th17 and the Imbalance of Treg/Th17 in Clonorchis sinensis-Infected Mice. PLoS ONE 2015, 10, 143217. [Google Scholar] [CrossRef]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab. Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Ikeno, Y.; Ohara, D.; Takeuchi, Y.; Watanabe, H.; Kondoh, G.; Taura, K.; Uemoto, S.; Hirota, K. Foxp3+ Regulatory T Cells Inhibit CCl4-Induced Liver Inflammation and Fibrosis by Regulating Tissue Cellular Immunity. Front. Immunol. 2020, 11, 584048. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, M.; Liu, X.; Bai, L.; Kong, M.; Chen, Y.; Zheng, S.; Liu, S.; Wan, Y.-J.Y.; Duan, Z.; et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl. Res. 2016, 169, 67–79. [Google Scholar] [CrossRef]
- Zhou, X.; Hovell, C.J.; Pawley, S.; Hutchings, M.I.; Arthur, M.J.; Iredale, J.P.; Benyon, R.C. Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004, 24, 492–501. [Google Scholar] [CrossRef]
- Zhang, X.; Lou, J.; Bai, L.; Chen, Y.; Zheng, S.; Duan, Z. Immune Regulation of Intrahepatic Regulatory T Cells in Fibrotic Livers of Mice. Med. Sci. Monit. 2017, 23, 1009–1016. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S. Natural killer and natural killer T cells in liver fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1061–1069. [Google Scholar] [CrossRef]
- Langhans, B.; Alwan, A.W.; Krämer, B.; Glässner, A.; Lutz, P.; Strassburg, C.P.; Nattermann, J.; Spengler, U. Regulatory CD4+ T cells modulate the interaction between NK cells and hepatic stellate cells by acting on either cell type. J. Hepatol. 2015, 62, 398–404. [Google Scholar] [CrossRef]
- Hesse, M.; Piccirillo, C.A.; Belkaid, Y.; Prufer, J.; Mentink-Kane, M.; Leusink, M.; Cheever, A.W.; Shevach, E.M.; Wynn, T.A. The Pathogenesis of Schistosomiasis Is Controlled by Cooperating IL-10-Producing Innate Effector and Regulatory T Cells. J. Immunol. 2004, 172, 3157. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Rosen, H.R. Galectin-9: Diverse roles in hepatic immune homeostasis and inflammation. Hepatology 2017, 66, 271–279. [Google Scholar] [CrossRef]
- Fujita, K.; Niki, T.; Nomura, T.; Oura, K.; Tadokoro, T.; Sakamoto, T.; Tani, J.; Yoneyama, H.; Morishita, A.; Kuroda, N.; et al. Correlation between serum galectin-9 levels and liver fibrosis. J. Gastroenterol. Hepatol. 2018, 33, 492–499. [Google Scholar] [CrossRef]
- Claassen, M.A.; de Knegt, R.J.; Tilanus, H.W.; Janssen, H.L.; Boonstra, A. Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C infected patients and limit the extent of fibrosis. J. Hepatol. 2010, 52, 315–321. [Google Scholar] [CrossRef]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Stross, L.; Günther, J.; Gasteiger, G.; Asen, T.; Graf, S.; Aichler, M.; Esposito, I.; Busch, D.H.; Knolle, P.; Sparwasser, T.; et al. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology 2012, 56, 873–883. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selnø, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Gonçalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594. [Google Scholar] [CrossRef]
- Banerjee, H.; Nieves-Rosado, H.; Kulkarni, A.; Murter, B.; McGrath, K.V.; Chandran, U.R.; Chang, A.; Szymczak-Workman, A.L.; Vujanovic, L.; Delgoffe, G.M.; et al. Expression of Tim-3 drives phenotypic and functional changes in Treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep. 2021, 36, 109699. [Google Scholar] [CrossRef]
- Lan, Y.-T.; Wang, Z.-l.; Tian, P.; Gong, X.-N.; Fan, Y.-C.; Wang, K. Treg/Th17 imbalance and its clinical significance in patients with hepatitis B-associated liver cirrhosis. Diagn. Pathol. 2019, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, H.; Guo, T. Th17/Treg imbalance is an indicator of liver cirrhosis process and a risk factor for HCC occurrence in HBV patients. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 399–407. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef]
- Katz, S.C.; Ryan, K.; Ahmed, N.; Plitas, G.; Chaudhry, U.I.; Kingham, T.P.; Naheed, S.; Nguyen, C.; Somasundar, P.; Espat, N.J.; et al. Obstructive Jaundice Expands Intrahepatic Regulatory T Cells, Which Impair Liver T Lymphocyte Function but Modulate Liver Cholestasis and Fibrosis. J. Immunol. 2011, 187, 1150. [Google Scholar] [CrossRef]
- Sa’dyah, N.A.C.; Putra, A.; Dirja, B.T.; Hidayah, N.; Yasmine Azzahara, S.; Candra Satria Irawan, R. Suppression of transforming growth factor-β by mesenchymal stem-cells accelerates liver regeneration in liver fibrosis animal model. Universa Med. 2021, 40, 29–35. [Google Scholar] [CrossRef]
- Steen, E.H.; Wang, X.; Balaji, S.; Butte, M.J.; Bollyky, P.L.; Keswani, S.G. The Role of the Anti-Inflammatory Cytokine Interleukin-10 in Tissue Fibrosis. Adv. Wound Care 2020, 9, 184–198. [Google Scholar] [CrossRef]
- Sziksz, E.; Pap, D.; Lippai, R.; Béres, N.J.; Fekete, A.; Szabó, A.J.; Vannay, Á. Fibrosis Related Inflammatory Mediators: Role of the IL-10 Cytokine Family. Mediat. Inflamm. 2015, 2015, 764641. [Google Scholar] [CrossRef]
- Ostroukhova, M.; Qi, Z.; Oriss, T.B.; Dixon-McCarthy, B.; Ray, r.; Ray, A. Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-β. J. Clin. Investig. 2006, 116, 996–1004. [Google Scholar] [CrossRef]
- Oh, S.A.; Li, M.O. TGF-β: Guardian of T Cell Function. J. Immunol. 2013, 191, 3973. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Maher, T.M.; Strek, M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: Time to treat. Respir. Res. 2019, 20, 205. [Google Scholar] [CrossRef]
- Lim, B.J.; Lee, W.-K.; Lee, H.W.; Lee, K.S.; Kim, J.K.; Chang, H.Y.; Lee, J.I. Selective deletion of hepatocyte platelet-derived growth factor receptor α and development of liver fibrosis in mice. Cell Commun. Signal. 2018, 16, 93. [Google Scholar] [CrossRef]
- Zhao, Z.; Lin, C.Y.; Cheng, K. siRNA- and miRNA-based therapeutics for liver fibrosis. Transl. Res. 2019, 214, 17–29. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Wu, H.M.; Kim, T.H.; Kim, A.; Koo, J.H.; Joo, M.S.; Kim, S.G. Liver X Receptor α–Induced Cannabinoid Receptor 2 Inhibits Ubiquitin-Specific Peptidase 4 Through miR-27b, Protecting Hepatocytes From TGF-β. Hepatol. Commun. 2019, 3, 1373–1387. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Johansen, A.K.Z.; Molkentin, J.D. CARdiac Immunotherapy: T Cells Engineered to Treat the Fibrotic Heart. Mol. Ther. 2019, 27, 1869–1871. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Yu, J.; Liu, Z.; Li, C.; Wei, Q.; Zheng, S.; Saeb-Parsy, K.; Xu, X. Regulatory T Cell Therapy Following Liver Transplantation. Liver Transplant. 2021, 27, 264–280. [Google Scholar] [CrossRef]
- Bai, W.; Wang, P.; Yu, F. CAR-T cells shed light on the treatments of fatal liver diseases. Biotarget 2018, 2, 6. [Google Scholar] [CrossRef]
- Akdoğan, Ö.; Atak Yücel, A.; Gök Sargin, Z.; Sönmez, C.; Esendağli Yilmaz, G.; Özenirler, S. Evaluation of Plasma Urokinase-Type Plasminogen Activator Receptor (UPAR) in Patients With Chronic Hepatitis B, C and Non-Alcoholic Fatty Liver Disease (NAFLD) as Serological Fibrosis Marker. J. Clin. Exp. Hepatol. 2019, 9, 29–33. [Google Scholar] [CrossRef]
- Li, G.; Lin, J.; Peng, Y.; Qin, K.; Wen, L.; Zhao, T.; Feng, Q. Curcumol may reverse early and advanced liver fibrogenesis through downregulating the uPA/uPAR pathway. Phytother. Res. 2020, 34, 1421–1435. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.H.; Zhu, Q.; Li, H.H.; Ra, H.J.; Majumdar, S.; Gulick, D.L.; Jerome, J.A.; Madsen, D.H.; Christofidou-Solomidou, M.; Speicher, D.W.; et al. Fibroblast Activation Protein (FAP) Accelerates Collagen Degradation and Clearance from Lungs in Mice. J. Biol. Chem. 2016, 291, 8070–8089. [Google Scholar] [CrossRef] [PubMed]
- Lay, A.J.; Zhang, H.E.; McCaughan, G.W.; Gorrell, M.D. Fibroblast activation protein in liver fibrosis. Front. Biosci. (Landmark Ed) 2019, 24, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Monslow, J.; Klampatsa, A.; Leibowitz, M.; Sun, J.; Liousia, M.; Woodruff, P.; Moon, E.; Todd, L.; Puré, E.; et al. Loss of cells expressing fibroblast activation protein has variable effects in models of TGF-β and chronic bleomycin-induced fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 317, 271–282. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef]
- Pang, N.; Zhang, F.; Ma, X.; Zhu, Y.; Zhao, H.; Xin, Y.; Wang, S.; Chen, Z.; Wen, H.; Ding, J. TGF-β/Smad signaling pathway regulates Th17/Treg balance during Echinococcus multilocularis infection. Int. Immunopharmacol. 2014, 20, 248–257. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef]
- Chen, X.; Oppenheim, J.J. Resolving the identity myth: Key markers of functional CD4+FoxP3+ regulatory T cells. Int. Immunopharmacol. 2011, 11, 1489–1496. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).