Abstract
Epidemiological studies have recognized that daily consumption of dietary fiber-containing foods reduces the incidence of developing many chronic diseases, for example, by interacting with nutritionally relevant compounds. The low affinity nature that some of these interactions can have make the development of an analytical detection system for their study particularly difficult. Therefore, the mechanism of action of binding compounds, by which a dietary fiber exerts its potential health benefits, remains largely unknown. Here, a novel method based on glyco-nanotechnology is proposed for studying the interaction between galactomannan and target molecules. Starting from a bottom-up approach, gold nanoparticles and thiolated galactomannans of two different sizes were synthesized separately, and then mixed for auto-assembly of the two glyconanoparticle materials. In addition, a preliminary interaction study between the prepared glyconanoparticles and Concanavalin A was carried out using transmission electron microscopy (TEM) from which it could be deduced that the molecular weight and ligand density on the gold core play an important role in the interaction. Therefore, dietary fiber-tethered gold nanoparticles are a valuable tool to elucidate key parameters underlying dietary fiber interactions.
1. Introduction
In recent years, many studies have been conducted to establish a correlation between food-enriched dietary fibers and human well-being. Specifically, epidemiological studies demonstrated that daily intake of dietary fibers had an inverse relationship with the incidence chronical diseases [1,2]. Therefore, the willingness of societies to opt for a high-fiber diet approach increased significantly, and at the same time, consumers demanded the market introduction of dietary fiber supplements and functional foods enriched with these polysaccharides [3,4].
Based on the current definition (FAO/WHO, 2009 revised in 2010), dietary fibers are classified as “edible carbohydrate polymers naturally occurring in foods as well as isolated, modified, and synthetic polymers with proven physiological effects of benefit to health” [5]. The fibers’ solubility in water is one of the discriminating factors for their classification. Pectin, mixed-linkage β-glucans, galactomannan, fructans, hemicelluloses, gums, and mucilage are indigestible, (at least partially) water-soluble dietary fibers (SDF). Conversely, cellulose, lignin, and resistant starch are classified as insoluble dietary fibers (IDF) [6]. Although the latter also exert beneficial physiological effects such as increasing fecal bulk or decreasing intestinal transit, the growing attention has been focused on the soluble fraction. Solubility, water-holding capacity, viscosity, and gelling properties as well as binding ability and fermentability are the key factors that make SDFs the favored option for food processing operations and incorporation in food products [7].
In the upper intestine, the absorption of minerals, bile acids, drugs, sugars, and toxins can be affected by the ability of SDFs to entertain water molecules and form gels or viscous solutions that can reversibly retain these compounds as they pass through the gastrointestinal tract. It became clear among researchers that this physical entrapment mechanism based on the viscosity of the fiber solution as well as the formation of a fiber-based barrier in the small intestine and reversible molecular interactions between fiber and nutritionally relevant compounds were the main processes behind how soluble dietary fibers could exert their health-supporting activity by altering the bioavailability of certain undesirable molecules [8,9]. However, it is challenging to find studies in the literature that demonstrate the specificity of action of soluble dietary fibers toward a specific molecule, and the supramolecular mechanism by which SDFs may exert their protective effects. Thus, this unknown mechanism of interaction between dietary fibers and components present in the upper intestine might also cause undesirable effects. For example, it is important to consider the possibility of decreased mineral bioavailability in some specific population group. Findings from in vitro studies have shown that semi-purified insoluble (cellulose, hemicelluloses, and lignin) as well as soluble fibers (gums and pectin) have mineral binding properties closely related to the type of fiber and the concentration in solution [5]. Moreover, Douwina Bosscher and co-workers investigated the effect of increasing amounts of alginic acid, locust-bean gum, and guar gum in casein and whey-based infant formulas, discovering that SDFs inhibited iron, calcium, and zinc availability more in casein than in whey-based formulas [10]. In addition, the therapeutic effect of some orally administered drugs might be reduced when these pharmaceutical molecules are simultaneously taken with fibers. For example, Reppas and co-workers showed that in canine models, both the rate and extent of absorption of paracetamol and hydrochlorothiazide were significantly decreased when administered together with guar gum [11]. Currently, more and more research is supporting the hypothesis that weak chemical interactions between fiber and small molecule are additionally impacting the beneficial effect of fibers, which is strictly correlated to the physicochemical properties of the fiber and the nature of the ligand. For example, Espinal-Ruiz and Parada-Alfonso [12] used isothermal titration calorimetry (ITC) to show that pectin associates with bile acid and calcium through hydrophobic and electrostatic interactions, respectively. Elucidation of the binding behavior between λ-carrageenan and Fe3+/Al3+ was done by Cao and colleagues [13]. They discovered that the interaction was driven by an exothermic ion-binding behavior with a relatively large binding constant. In another study, response surface methodology (RSM) was used to explore the influence of different binding factors such as pH, temperature, and salt concentration on the binding nature of polyphenols and neutral cellulose. It was found that pH was the main factor affecting the nature of the interaction, which in turn was controlled by hydrophobic forces and hydrogen bonding [14].
Although some interaction studies between charged polysaccharides and nutritionally relevant ligands are available in the literature, the chemical mechanism governing the weak interaction between neutral soluble dietary fibers and nutritionally relevant small molecules remains almost entirely unexplored. This low affinity association makes the design of an adequate high-sensitivity detection system a particularly difficult challenge. Here, we propose a unique approach exploiting the latest innovations in the field of surface engineered glyco-nanotechnology to study the association between galactomannan and concanavalin A. This interaction experiment can be considered as a positive control, which is always an important gold standard in any new method development. Con A is a lectin that has a high binding affinity to the terminal α-D-mannopyranoside residues of a branched trimannoside unit (Man)3 [15]. More recent studies have shown that Con A interacts with galactomannan (GM), even though the glycosidic linkages of the mannose residues exists in a β-anomeric configuration. For example, the experiments suggested by Vilaro’ and coworkers have shown that the galactose side chains and the reducing end of the GM mannose interacted weakly with the active sites of lectins, and that these interactions were magnified by hydrophobic effects between non-ionic GM and lectins [16]. Zahang and colleagues used Quart Crystall Microbalance (QCM) technology to prove the biospecific interaction between galactomannan and Con A through a competitive binding study with Me-α-man, which is known to have a strong affinity to Con A [17]. From these studies, it looks like not only the terminal group of a polysaccharide chain but also the other monomer units present in the polymer can take part in the interaction with con A, and that the affinity between Con A and galactomannan is susceptible to a number of factors such as flexibility/stiffness of the polysaccharide chain, length and density of the polymer linker on the nanoparticle surface, and distance between them. Here, we want to emphasize the influence of the size and surface density of the polysaccharide in the interaction with con A using TEM. Specifically, we used a well-known procedure involving thiol-mediated functionalization of gold nanoparticles to create a powerful tool that can assess, with the help of advanced analytical methods, the nature of these weak interactions (Figure 1).
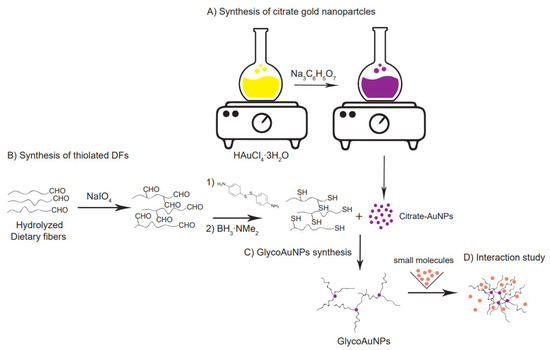
Figure 1.
Multi-step synthetic strategy for glyconanoparticle formation. (A) Synthesis of citrate-capped gold nanoparticles starting from HAuCl4 solution. (B) Thiolation by reductive amination of the partially hydrolyzed, by NaIO4 to a low degree oxidized neutral soluble dietary fibers. (C) Thiolated fiber conjugation by self-assembly onto the gold nanoparticle surface. (D) Molecules triggered aggregation of GlycoAuNPs indicating an interaction.
Using a bottom-up approach, sphere-like gold nanoparticles (AuNPs) were synthesized starting from a tetrachloroauric acid solution (HAuCl4∙3H2O) as a gold precursor [18]. Neutral, thiolated galactomannans with two different molecular weights (Mw) and dispersity (Đ) were used to functionalize the gold nanoparticle surface through citrate-ligand exchange. Finally, after exposing the glyco-nanoparticles (GlycoAuNPs) to a target compound (Concanavalin A, (Con A)) as model system, potential interaction was revealed by monitoring the spatial arrangement of the GlycoAuNPs before and after the addition of the target molecules.
The synthetic strategy used in this study leads to a satisfactory covalent functionalization of the gold particles with dietary fibers of relatively high molecular weight. Therefore, these glyco-nanoparticles can be considered a versatile platform for the detection of specific interactions between fibers and molecules, which can have positive or negative consequences on human health.
2. Materials and Methods
2.1. Materials
Tetrachloroauric (III) acid trihydrate (HAuCl4∙3H2O ≥ 99%), distilled water (DI), nitric acid (HNO3 70.0%), concentrated hydrochloric acid (HCl, 37%), tri-sodium citrate dihydrate (Na3C6H5O7·2H2O ≥99.0%), sodium phosphate dibasic (Na2HPO4, 99.95%), isopropanol (i-PrOH, ≥99.7%), sulfuric acid (H2SO4, ACS reagent, 95.0–98.0%), anthrone (C14H10O, ACS reagent, 97.0%), bicinchoninic acid disodium salt hydrate (BCA, ≥98.0%), borane dimethylamine complex (DMAB, ≥97.0%), copper(II) sulfate pentahydrate (CuSO2·5H2O, ≥98.0%), sodium carbonate (Na2CO3, ReagentPlus®, ≥99.5%), sodium bicarbonate (NaHCO3, ACS reagent, ≥99.7%), sodium (meta)periodate (NaIO4, ≥99.0%), ethylene glycol ((CH2OH)2, ReagentPlus®, ≥99.0%), 4-aminophenyl disulfide (APDS, ≥99.0%), acetic acid (CH3COOH, glacial, ReagentPlus ®, ≥99%), sodium acetate (CH3COONa, ACS reagent, ≥99.0%), and Concanavalin A (Con A) from Canavalia ensiformis (Jack bean) were purchased from Sigma-Aldrich (St. Louis, MI, USA). Deuterium oxide (D2O, 99.9%) was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). Endo-1,4-β-Mannanase (Aspergillus niger) supplied at 600 U/mL was purchased from Megazyme (Bray, Ireland). One unit of mannanase activity is defined as the amount of enzyme required to release one µmole of mannose reducing-sugar equivalents per minute from carob galactomannan (10 mg/mL) in sodium acetate buffer (100 mM), pH 4.0 at 40 °C. Guar galactomannan (GM) high viscosity (Gal depleted, viscosity > 10 dL/g, Mw = 350 kDa, sugar ratio Gal:Man = 21:79 (Lot#10502 A), and (Lot#10502 B)) was purchased from Megazyme (Bray, Ireland). Tris-(hydroxymerhyl) aminomethane (Tris base, ≥99.8%) was purchased from Bio-Rad (Richmond, CA, USA). All aqueous solutions were prepared using Milli-Q water (Milli-Q® IQ 7000 Ultrapure Water System). Dialysis membranes made from regenerated cellulose with MWCO 12,000–14,000 Da (25 Å; 29 mm) were supplied by SERVA (Heidelberg, Germany). All reagents were used as received without further purification.
2.2. Synthesis and Characterization of Thiolated Galactomannan
2.2.1. Synthesis of Low Mw Galactomannans
Low molecular weight galactomannan was obtained by both acid and enzymatic treatment of the fiber. For the acid hydrolysis, galactomannan (GM (Lot A)) was hydrated in water at a concentration of 0.6% (w/v) for 1 h at 80 °C. To ensure a complete dissolution of the polysaccharide, the solution was kept at room temperature overnight under stirring. Concentrated HCl was added to reach a final concentration of 0.1 M HCl. Galactomannan was hydrolyzed for 24 h at 50 °C under stirring (800 rpm). The reaction was stopped by the addition of an equal volume of 0.2 M Na2HPO4. The product was purified by dialysis against water for 72 h at room temperature and freeze-dried for 48 h. The purified galactomannan sample was solubilized and separated to different molecular weight fractions with i-PrOH according to a previous published procedure with some modifications [19]. A 0.6% (w/v) solution of the purified galactomannan was dissolved for 1 h at 80 °C, and stirred at room temperature overnight. This polysaccharide solution (300 mL) was mixed with 60 mL of i-PrOH and kept on ice for 2 h. The precipitate (GMHCl) was collected by centrifugation at 12,000× g for 20 min at 5 °C. The precipitate was freeze-dried for 48 h and stored in a desiccator at room temperature.
For the enzymatic treatment, 2 g of galactomannan (GM (Lot B)) was dissolved in 400 mL of water at 0.5% (w/v) final concentration and hydrolyzed via a procedure reported by Cheng et al. using endo-1,4-β-mannanase [20]. Briefly, 432 µL of enzyme (supplied at 600 U/mL) was diluted into 2 mL of a 0.1 M sodium acetate/acetic acid pH = 6.0 buffer solution (2.432 mL final volume). Subsequently, 40 µL (4.3 U) of the enzyme solution was added to GM (0.01 U/mL enzyme concentration) and the mixture was reacted under stirring for 2.5 h at room temperature. After that, the enzyme was denatured by heating the polysaccharide solution for 20 min at 100 °C. Thereafter, the mixture was centrifuged at 4000 rpm for 20 min. The product was precipitated using three volumes of i-PrOH, and freeze-dried for 48 h. The resulting sample GMEnz was stored in a desiccator at room temperature.
2.2.2. Controlled Oxidation of Hydrolyzed Galactomannans
To facilitate the functionalization of galactomannan polymer chains with a thiol group via reductive amination, additional carbonyl groups (in addition to the one reducing end group per chain) were introduced into the polysaccharide. Dialdehyde galactomannans were prepared according to the method described by da Silvia et al. with some modifications (Figure 2) [21]. Briefly, 0.6% (w/v) of each GMHCl and GMEnz were hydrated in water under agitation for 24 h. After complete dissolution, 2 equiv. (with respect to the polysaccharide chains according to their Mn) of a 1% (w/v) NaIO4 solution (=47 mM) were added to each polysaccharide solution, and the mixtures were reacted for 6 h in the dark at room temperature. The reactions were quenched by addition of 2 equiv. ethylene glycol. Oxidized galactomannan solutions were purified by dialysis against water for 12 h. The products were precipitated with twice the volume of i-PrOH l and freeze-dried for 48 h. Solid oxidized galactomannans GMHCl/ox and GMEnz/ox were stored in a desiccator at room temperature until further analysis.
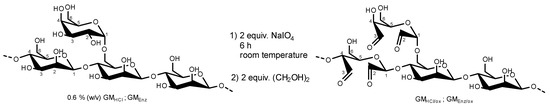
Figure 2.
Oxidation reaction of acid and enzymatically hydrolyzed galactomannans by 2 equiv. sodium (meta)periodate (stochiometry on the basis of polymer chains). The structure of the fibers is only partially represented and only very few oxidations per polymer chain occurred.
2.2.3. Conjugation of Oxidized Galactomannans with Thiol Linker
Oxidized galactomannans GMHCl/ox and GMEnz/ox were reacted with 4-aminophenyl disulfide (APDS), leading to the formation of thiol derivatives through reductive amination (see Figure 3). The synthesis was based on the procedure described by Seo and coworkers [22]. A 1% (w/v) aqueous solution of GMHCl/ox and GMEnz/ox was prepared in an amber glass tube by overnight dissolution. Separately, a 50 mM solution of APDS was prepared in water:acetic acid = 1:1. A total of 10 mL of the polysaccharides were mixed with 10 mL of the thiol-linker solution for 1 h at 30 °C. Dimethylamine borane was added into each of the reaction chambers at 100 mM final concentration and the reaction was stirred unsealed for 1 h at room temperature. Subsequently, the tubes were resealed and heated again for 1 h at 50 °C. The products were purified by dialysis for 48 h and freeze-dried for two nights. Thiolated galactomannans GMHCl/ox/ATP and GMEnz/ox/ATP were stored at −20 °C in the dark.
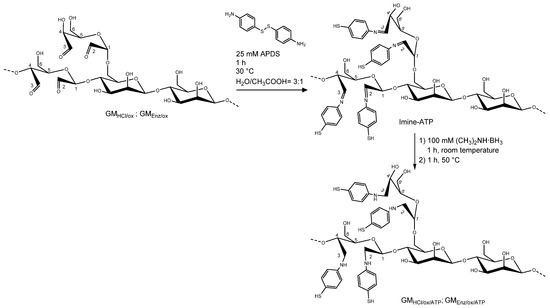
Figure 3.
Schematic representation of the reductive amination on an oxidized galactomannan chain. The first reaction consists of the conversion of carbonyl group to the intermediate imine. Secondly, the introduction of a reducing agent reduces the imine to the respective amine. The structure of the fibers is only partially represented and only a small percentage of repeating units per polymer chain are actually labelled.
2.2.4. Molecular Weight (Mw and Mn) and Dispersity (Đ) Analysis
Weight-average Mw and dispersity (Đ) of the hydrolyzed, oxidized, and thiolated galactomannans were measured using high-performance size exclusion chromatography (HPSEC) (OMNISEC, Malvern Panalytical Ltd., Malvern, UK) following the procedure described by Demuth et al. with slight modifications [23]. Briefly, two A’6000M columns in series (8.0 × 300 mm, exclusion limit of 20,000,000 g/mol, Viscotek, parent organization: Malvern Panalytical Ltd., Malvern, UK) were used for the separation. Low and right-angle laser light scattering detector (LALS/RALS), the refractive index (RI), and a viscometer detector were employed for the analysis. OMNISEC software version v.10.30 was used for data acquisition, analysis, and reporting. A solution of 0.1 M NaNO3 with 0.02% NaN3 was used as the mobile phase. The temperature of both columns was kept at 30 °C and the flow rate was 0.7 mL/min with an injection volume of 100 µL. The total run time was 60 min. Samples were dissolved in the mobile phase at a concentration of 0.1% (w/v) and filtered through a 0.45 µm nylon filter prior to injection. For the absolute molecular weight determination, a calibration was performed using the narrow molecular weight distribution polyethylene oxide (PEO-24K) standard. The refractive index increment (dn/dc) value of the galactomannan samples was set at 0.144 mL/g as determined in the previous publication [19]. Samples were measured in triplicate.
2.2.5. Monosaccharide Composition Analysis
The monosaccharide ratio (Gal/Man) of acid-treated (GMHCl) and enzymatically treated (GMEnz) galactomannan was measured by high-performance anion-exchange chromatography-pulsed amperometric detection (HPAEC-PAD) after complete hydrolysis with HCl, as done previously [19].
An amount of 50 mg of the dried samples were completely hydrolyzed in 10 mL 2 M HCl solution at 100 °C for 45 min. After cooling to room temperature, the reaction mixture was neutralized with 4 M NaOH and centrifuged for 15 min at 4000 rpm to separate debris from the sample solution. The analysis was taken on an aliquot of the supernatant, which was diluted with water to reach a concentration of 10 mg/L and filtered through a 0.45 µm PTFE filter.
The analysis was performed with the Dionex ICS-5000+ System (Thermo Scientific, Sunnyvale, CA, USA). A disposable gold electrode, an Ag/AgCl reference electrode, and a CarboPAC PA1 (4 × 250 mm) column were used for the analysis. The temperature of the column was kept at 26 °C, and the flow rate was set at 1.0 mL/min. The mobile phase consisted of two eluents: (A) 200 mM NaOH (prepared from 50% (w/w) NaOH solution) and (B) water. An isocratic method was applied for the sugar separation, namely 8% (A) and 92% (B) for the first 22.5 min, followed by 100% (A) for 8.5 min, and back to the initial conditions for 8 min. The total run time was 39 min. For the determination of the absolute monosaccharide amount, an external standard calibration was performed using a mixture of standard sugars containing galactose and mannose with a concentration ranging between 1.25–30.0 mg/L. D-Sorbitol was used as internal standard and added at a constant concentration of 10 mg/L to each sample and calibrant solution. The monosaccharide concentration was quantified relative to the internal standard signal. Data processing was carried out on Chromeleon 7 (Thermo Fischer Scientific AG, Basel, Switzerland). All samples were measured in triplicate.
2.2.6. Determination of Carbonyl Groups in the Oxidized Samples
An estimation of the total carbonyl content of the oxidized samples GMHCl/ox and GMEnz/ox was performed by the 2,2′-bicinchoninate (BCA) assay usually applied for the determination of reducing end group concentrations [REG] [24,25].
Briefly, assay solution A (pH = 9.7) was prepared by dissolving 5.43 g of Na2CO3 and 2.42 g of NaHCO3 in 80 mL of water. A total of 0.5 mmol of BCA was added to the solution and the volume was adjusted to 100 mL with water. For assay solution B, 0.5 mmol CuSO4∙5H2O and 1.2 mmol L-serine were dissolved in 100 mL of water. The working solution was freshly prepared by mixing equal volumes of solution A and solution B. GMHCl/ox and GMEnz/ox were dissolved in water at a 0.1% (w/v) concentration. Afterward, equal volumes of working solution and oxidized galactomannan were mixed and incubated at 75 °C in a water bath for 30 min. Samples were cooled down at room temperature for 20 min and their absorbance was measured at 560 nm against a blank containing no carbohydrate using disposable polystyrene cuvettes of 1 cm path length. The instrument was zeroed with water. The calibration was accomplished on the basis of mannose reducing end groups (REG) with a mannose standard curve obtained with concentrations ranging from 1 to 50 µM, and all the standard solutions were treated with the BCA assay following the same procedure described above. All samples were measured in triplicate.
2.2.7. Raman and FTIR Spectroscopy
Fourier transform infrared absorption (FTIR) and Raman scattering spectroscopy are molecular spectroscopy techniques sensitive to different types of vibration and therefore they provide complementary vibrational spectra. Thiolated polysaccharides were analyzed by Raman and FTIR spectroscopy to detect the presence of the aromatic thiol linker in the polysaccharide structure.
FTIR measurements were recorded using a Varian 640 (Palo Alto, CA, USA). A mean of 64 scans were registered in the range of 4000 to 400 cm−1 at a 4 cm−1 resolution. Samples were prepared as KBr pellets with a sample to KBr ratio of 1:100.
Raman scattering spectra were recorded in a solid state, in the range of 100–3199 cm−1 using a Horiba LabRAM HR Evol spectrometer (HORIBA France SAS, Longjumeau, France) supplied with a Nd:Yag single frequency laser source with an excitation wavelength of 532 nm.
2.2.8. Proton Nuclear Magnetic Resonance (1H-NMR)
Thiolated galactomannans were characterized using nuclear magnetic resonance (1H-NMR) to obtain the number of 4-amino thiophenol linkers (ATP) per polymer chain. About 20 mg of GMHCl/ox/ATP and GMEnz/ox/ATP were solubilized overnight with 1.2 mL of D2O, and 700 µL of this solution were transferred into NMR tubes and analyzed by a Bruker AVANCE III-400 spectrometer (Bruker, Ettlingen, Germany) operating at room temperature at 400 MHz with 16 repetitive scans and an acquisition time of 4 s. Data processing was carried out on MestReNova 14 (Mestrelab Research SL, Santiago de Compostela, Spain). To determine the number of ATP molecules per polymer chain (n°(ATP)), the following equation was used
where Mn (PS-SH) is the number average molecular weight of the thiolated fibers, which was measured by SEC (Mn (GMHCl/ox/ATP) = 70.80 kDa, Mn (GM21Enz/ox/ATP) = 26.90 kDa). n°(Gal) and n°(Man) are the number of molecules of galactose and mannose in the polysaccharide chain. n°(ATP) is the number of molecules of thiol linker, and its molecular weight is Mw (ATP) = 107.18 g/mol. Mw (Gal) and Mw (Man) are the molecular weights of the monomeric unit Mw (Gal) = Mw (Man) = 162.14 g/mol, considering that each hexopyranose unit lost a water molecule in the polymerization process. Knowing the ratio n°(Gal)/n°(Man) from HPAEC analysis, and the relative peak area of the 2 aromatic ring protons, it was possible to solve Equation (1) as a function of n°(ATP) considering the following steps
- (1)
- from HPAEC analysis
- (2)
- from 1H-NMR data analysiswhere A(1HGal) is the relative peak area of the anomeric galactosyl proton and is the relative peak area of the aromatic group protons. These two values were determined by setting the total integral of the repeating unit [RU] protons equal to 6 [26]. By substituting Equation (5) into Equation (3), the number of mannose units in the polysaccharide chain can be given as
Substitution of Equations (5) and (6) into Equation (1) gives the number of thiol linkers per polymer chain (n°(ATP))
2.3. Synthesis and Characterization of Citrate-Gold Nanoparticles (Citrate-AuNPs) and Glyco-Gold Nanoparticles (Glyco-AuNPs)
2.3.1. Citrate-Functionalized Gold Nanoparticle Synthesis
Citrate-capped gold nanoparticles (citrate-AuNPs) were synthesized following the Turkevich method [27]. Briefly, 43 mg of HAuCl4∙H2O were dissolved in 100 mL water. The solution was heated to 100 °C under stirring (800 rpm). As soon as the solution boiled, 10 mL of a freshly prepared sodium citrate (38.8 mM) solution was quickly added (1.0 mM HAuCl4∙H2O final concentration). The mixture was heated for a further 15 min at 90 °C under stirring (800 rpm). Within 10 min, a change in the solution’s color from colorless to purple was observed. The particle solution was maintained under stirring for an additional 20 min, cooled to room temperature, and transferred into an amber bottle where the colloidal solution was stored at room temperature until further analysis. Assuming that the reduction from Au+3 to Au0 was 100% complete, that the density of AuNPs is equal to that of gold [ρ = 19.3 g/cm3], and that the particles are spherical in shape with an average diameter of d = 18.3 nm (see Section 3.2.1), the number of nanoparticles in 110 mL of solution is
where m(Au0) is the mass of gold atoms calculated from the mg of HAuCl4∙H2O used, and V is the volume of a sphere and from which the concentration of the particles can be derived as
where V is the volume of the particle solution and NA is Avogadro’s number (6.022 × 1023 mol−1).
2.3.2. Synthesis of Glyconanoparticles
Glyconanoparticles (glyco-AuNPs) were prepared based on the procedure described by Hone and colleagues [28]. A total of 50 mL of citrate-capped gold nanoparticle solution was prepared as described in Section 2.3.1, and containing approximately 2.4 nM Au particles were mixed with 50 mg or 30 mg of thiolated polysaccharides GM21HCl/ox/ATP or GM21Enz/ox/ATP, respectively. The solutions were stirred for 48 h at room temperature in the dark where the self-assembly mechanism took place. To ensure a complete removal of unbound thiolated polysaccharides, a washing step of the glyconanoparticle solution was performed. Briefly, the particles were centrifuged at 23,710× g for 25 min, after which the supernatant solution was discarded and replaced with 10 mL of 10 mM Tris buffer (pH = 7.6). After being strongly mixed, the glyco-AuNPs solution was centrifuged again, and the procedure described above was repeated two more times. Modified nanoparticles (GM21HCl/ox/ATP-AuNPs and GM21Enz/ox/ATP-AuNPs) were resuspended in 10 mM Tris buffer (pH = 7.6) solution to give a concentration of about 6–10 nM of Au particles.
2.3.3. UV–Vis Absorption Measurements
The absorbance values of citrate-AuNPs and glyco-AuNPs solutions were measured on a Cary 100 spectrophotometer (Agilent Technologies Inc., Santa Clara, CA, USA). A total of 500 µL of citrate-AuNPs were diluted with 500 µL water, vortex mixed, and placed in a 1 cm path length disposable polystyrene cuvette. UV–Vis spectra were acquired from 350 nm to 700 nm and the instrument was zeroed with water. For the derivatized nanoparticles, 500 µL of glyco-AuNPs were diluted with 500 µL 10 mM Tris buffer (pH = 7.6), vortexed, and placed into a 1 cm path length disposable cuvette. The wavelength was scanned from 350 nm to 700 nm and 10 mM Tris buffer (pH = 7.6) was used as a blank. All the measurements were performed in triplicate.
2.3.4. DLS (for dh) and Zeta Potential (ζ)
The hydrodynamic diameter (dh) and polydispersity index (PdI) of citrate-capped gold nanoparticles were measured by dynamic light scattering (DLS) using the Zetasizer Nano-ZS (Malvern Instrument Ltd., Malvern, UK). In addition, the net surface charge of the nanoparticle was determined by measuring the zeta potential (ζ) using the Zetasizer Nano-ZS. Before analysis, 1 mL of citrate-capped gold nanoparticle solution was filtered through a 0.45 µm PTFE filter. All the measurements were performed in triplicate. In addition, the dh and PdI of the glyco-AuNPs were measured. Before analysis, 1 mL of glyco-AuNPs solution was filtered through a 0.45 µm PTFE filter. All the measurements were performed in triplicate.
2.3.5. Determination of Morphology and Size Distribution
The citrate-AuNPs size (d) and morphology were analyzed by a transmission electron microscope (FEI TalosTM F200X) operating at 200 kV. Scanning transmission electron microscope acquisition (STEM) was used to map the sample in dark-field mode (HAADF). An aliquot of about 5 µL of the sample was dropped onto copper grids coated with carbon film (300 mesh) and placed on absorbent paper to absorb the excess solution. The STEM images were analyzed using ImageJ software. A total of 454 particles were counted, and their area was measured. Approximating the shape of each particle to that of a circle, the average diameter (d) of the analyzed particle samples were calculated by the following equation
where A is the area of a circle and r is the radius. Knowing r of the particle, the diameter (d) was calculated as
Glyco-AuNPs were also analyzed by TEM using the same procedure described above.
2.3.6. Ligand Surface Density
The anthrone/H2SO4 assay was used as a colorimetric method for the determination of the amount of carbohydrate ligands coupled to the surface of the AuNPs [29]. Sulfuric acid promotes the hydrolysis of the polysaccharide chains into monoscaccharide units of which the open-form reacts with an anthrone molecule, leading to a green colored complex. A calibration curve was created using different concentrations of monosaccharide mixtures equivalent to the Gal:Man molar ratio of the respective GM. The calibration ranged from 12 µg/mL to 192 µg/mL. A 0.2% (w/v) anthrone solution was prepared in concentrated H2SO4 in iced water. A total of 0.5 mL of each standard solution was transferred inside an amber bottle and stored in iced water under stirring. One mL of the freshly prepared anthrone solution was added into the standard solutions. The samples were heated to 100 °C for 10 min under stirring. After that, the standard solutions were cooled in an iced water bath and the absorbance was measured at λ = 625 nm. The blank sample consisted of 1 mL of anthrone solution in 0.5 mL water, and the instrument was zeroed with water. All standards were measured in triplicate.
Glyco-AuNP solutions were extensively mixed and 2 mL were centrifuged at 23,710× g for 25 min. The supernatant was discarded, and the particles were resuspended in 1.5 mL water. The particle solution was divided into three aliquots (0.5 mL each) and placed inside an amber bottle previously cooled in an iced water bath. The solutions were treated with anthrone reagent following the same procedure described for the standard solutions, with the exception that glyconanoparticles were reacted with the anthrone reagent for 20 min [30]. The blank sample consisted of 1 mL of anthrone solution in 0.5- mL water, and the instrument was zeroed with water. A control experiment was performed by measuring the absorbance of Glyco-AuNPs at λ = 625 nm in the absence of the anthrone reagent. All samples were measured in triplicate.
2.4. Interaction Study by TEM
Transmission electron microscopy (Thermo Fisher Talos F200X and Hitachi HD-2700) operating at 200 kV was used as a preliminary interaction study method between guar galactomannan and concanavalin A (con A). Scanning transmission electron microscope (STEM) acquisition was used to map the sample in a high angle annular dark field (HAADF) mode. For the experiment, 500 µL of GMHCl/ox/ATP-AuNPs (6.0 nM), GMEnz/ox/ATP-AuNPs (5.5 nM) were incubated with 88 µL of a 2.35 µM Con A solution in 10 mM Tris buffer pH = 7.6, containing 99 mM NaCl, 1 mM CaCl2 and 1 mM MnCl2. The solutions were left under shaking for 3 h at room temperature. Afterward, 5 µL of each sample solution was placed onto a 300 mesh copper grid and the excess liquid was absorbed by paper tissue. The control sample consisted of a 500 µL glyconanoparticle solution with 88 µL of 10 mM Tris buffer pH = 7.6 (including NaCl, CaCl2, and MnCl2). A control experiment was conducted by exposing the citrate-stabilized nanoparticles to the same conditions described above.
3. Results and Discussion
3.1. Characterization of Hydrolyzed, Oxidized, and Thiolated Galactomannan
3.1.1. Molecular Weight, Dispersity, and Monosaccharide Composition Analysis
Native galactomannan from two production batches, acid treated galactomannan, galactomannan treated with endo 1,4 β-mannanase, and additionally oxidized and thiolated galactomannans were analyzed for their molecular weights and dispersity by SEC (Table 1).

Table 1.
Gal/Man monosaccharide ratio of native and hydrolyzed GM. Mw, Mn, and Đ analysis of GM. Samples were analyzed in the native form, after enzymatic hydrolysis (Lot A) and acid hydrolysis (Lot B), after oxidation with sodium periodate, and after reductive amination.
Enzymatically treated galactomannan (GMEnz) showed a significantly lower Mw than GMNative (Lot A) and GMHCl after 2.5 h reaction time, with a decrease in dispersity. Sodium (meta)periodate (NaIO4) was used to chemically modify the polysaccharides with aldehyde groups to enhance the subsequent labelling. In both oxidized products, the molecular weight was significantly reduced while an increase in the dispersity of the samples was observed (Table 1). The reason behind this substantial reduction of the average degree of polymerization and a wider dispersion is most likely related to β-elimination processes. Indeed, the introduction of oxidized functionalities at C2 and/or C3, upon periodate oxidation, results in an acidification of the α protons in position C1 and C4, respectively, making the polysaccharide’s backbone prone to chain scission, leading to enones under elimination of ROH, where R is one side of the GM chain [31]. Reductive amination of these oxidized materials with an amino-thiol linker produced thiolated galactomannans with almost double (GMHCl/ox/ATP) or triple (GMEnz/ox/ATP) their Mw and Mn values. A dimerization process of the thiol group to disulfides is very common and can occur by thiol oxidation in aqueous solution [32].
Monosaccharide composition of native and enzymatically treated galactomannans was assessed by HPAEC after complete hydrolysis of the samples. The galactose (Gal) to mannose (Man) ratio was determined to be 10:90 for the enzymatically treated sample (GMEnz) and 11:89 for the untreated galactomannan (GMNative). This lack of significant change in Gal:Man ratio is in accordance with the enzyme’s specificity for (1,4)-β-d-mannosidic linkages, with no loss of Gal side chains. As reported in our previous study, the Gal/Man ratio of the acid treated polysaccharide (GMHCl) was 16:84, which was significantly different from the monosaccharide ratio of its native galactomannan (19:81) [19]. Note that the monosaccharide composition between the native galactomannans determined in this study (used for enzymatic treatment) and the previous study (HCl treatment) varied slightly between the different batches.
3.1.2. Determination of Aldehyde Groups of Oxidized Galactomannans
Preliminary labelling experiments with APDS were conducted on GMNative and GMHCl without performing the oxidation step with NaIO4. However, based on 1H-NMR analysis, labelling efficiency was very poor, regardless of the fiber’s Mw (data not shown). The newly introduced carbonyl groups are more reactive than the reducing end groups, and are therefore potentially more susceptible to being involved in the subsequent reductive amination reaction. The BCA assay was used to determine the extent of the oxidation process in terms of the number of carbonyl groups [C=O] per polysaccharide chain. In both samples, GMHCl/ox and GMEnz/ox, the concentration of aldhehyde groups increased after oxidation, indicating that the ring opening reaction caused by periodate in position C2 and C3 was successful (Table 2).
Table 2.
Theoretical [µmol C=O/g] reducing end group (from Mn) and experimental [µmol C=O/g] carbonyl group contents before and after periodate oxidation. n°[C=O]polymer chain is the number of aldehyde groups per polymer chain related to the conversion of hydroxyl groups to carbonyl groups exclusively due to the oxidation reaction. The ratio between carbonyl groups per polymer chain (n°[C=O]polymer chain) of oxidized and unoxidized polysaccharides is also presented.
The discrepancy between the theoretical and experimental [µmol C=O/g] values, whose ratio should not deviate from 1, of the non-oxidized galactomannan samples (GMHCl and GMEnz), may be either due to a different reactivity of the reducing end compared to the used monosaccharide calibrant in the BCA assay, and/or a slight overestimation of the processed Mn obtained from the analysis of the samples by SEC. Indeed, aggregates of higher molecular weight were visible in these samples (data not shown), which may affect the peak integration process, leading to an underestimation of theoretical reducing ends. This explains how the number of carbonyl groups per polymer chain (n°[C=O]polymer chain) of the unoxidized samples (GMHCl and GMEnz) deviated slightly from 1.
Normalizing the data to correct for potential reactivity differences and SEC peak integration effects was accomplished by taking the ratio between the number of carbonyls per polymeric chain of the oxidized and unoxidized samples (n°ox[C=O])/n°[C=O]), resulting in very similar values between 1.3–1.4 for the two classes of materials (Table 2), demonstrating that NaIO4 has similar effects on both acidic and enzymatically treated samples of differing Mn. Therefore, the BCA assay in combination with Mn data from SEC allowed us to verify whether the observed increase in the concentration of carbonyl groups was caused by the lowered Mn, or whether additional medium-chain carbonyls were introduced during treatment with NaIO4. Based on the ratio n°ox[C=O]/n°[C=O] (1.4 and 1.3), we can conclude that new carbonyl groups were introduced into the polysaccharide structure through the oxidation step with NaIO4. In particular, the sample GMHCl/ox had a 43% increase in the number of carbonyl groups per chain while GMEnz/ox increased the carbonyl group content by 27%.
3.1.3. Raman and FTIR Spectroscopy of Thiolated Galactomannan
Unmodified and thiolated galactomannans were characterized by FTIR and Raman scattering spectroscopy to confirm the presence of the aminothiophenol (ATP) linker in the polysaccharide structure (Figure 4).
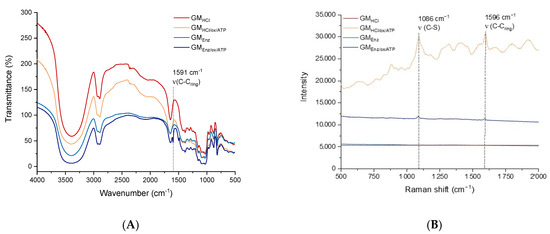
Figure 4.
(A) FTIR spectra, and (B) Raman spectra of galactomannans before and after oxidation + reductive amination to the thiolated product.
In the FTIR spectra (Figure 4A), all the samples presented a broad absorption band at 3400 cm−1, which can be attributed to the (OH) stretching mode vibration of hydroxyl groups of galactomannans. Peaks at 2923 cm−1 and 2885 cm−1 were assigned to (CH) stretching of alkanes. An additional peak was found in the thiolated polysaccharides. Specifically, an absorption band at 1591 cm−1, representing the stretching mode vibration (aromatic C-C) of the thiol-linker (ATP), was found in both the functionalized galactomannans [33]. Regarding the Raman spectra, two prominent peaks at 1086 cm−1 and 1596 cm−1 were present in both the thiolated galactomannans (Figure 4B), which can be assigned to the (C–S) and (C–Cring) stretching modes of ATP, respectively [34].
3.1.4. Nuclear Magnetic Resonance (1H-NMR) of Thiolated Galactomannan
Modification of galactomannans with ATP linkers was studied by 1D-NMR analysis. In both 1H-NMR spectra, typical signals of the aromatic ring were found (Figure 5). Specifically, signals related to the two protons directly attached to the arene ring were found at δ = 7.4 ppm and δ = 6.8 ppm, confirming the functionalization of the galactomannan with the aromatic thiol linker. The peak at δ = 5.03 ppm was assigned to the anomeric proton (H-1) of the α-d-galactopyranoside residue [26], and was used as a quantitative probe for the determination of the number of ATP molecules conjugated to a polysaccharide chain.
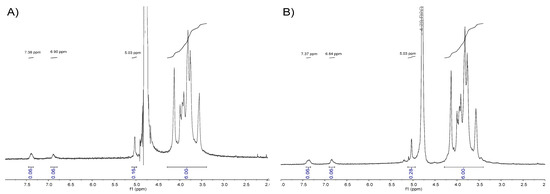
Figure 5.
(A) 1H-NMR of GMHCl/ox/ATP in D2O. (B) 1H-NMR of GMEnz/ox/ATP in D2O.
The region of the spectrum related to the repeating unit [RU] protons of galactomannan, ranging from approximately 3.3 ppm to 4.2 ppm, and containing a “fingerprint type” of information was used to determine the relative peak areas of the anomeric galactose signal (~5.03 ppm) and aromatic group protons (~7.4 and 6.8 ppm). Knowing the monosaccharide ratio of the polysaccharides, GMHCl (Gal:Man = 16:84) and GMEnz (Gal:Man = 10:90), the Mn of the hydrolyzed fibers, and the relative peak area of aromatic protons = 0.06 and = 0.07, it was possible to calculate the number of ATP molecules per polymer chain (n°(ATP)) based on Equation (1). Specifically, it was found that n°(ATP) GMHCl/ox/ATP = 2.1 and n°(ATP) GMEnz/ox/ATP = 1.7, which is the same general trend as the number of carbonyl groups per polymer chain as previously calculated for the oxidized substrates used for the labelling (Table 2).
3.2. Characterization of Citrate-AuNPs and Glyco-AuNPs
3.2.1. UV–Vis Absorption, DLS, and TEM Analysis of Citrate-AuNPs
Citrate gold nanoparticles were synthetized through reduction of Au3+ to Au0 by tri-sodium citrate. Individual replicates of the UV–Visible spectrum of the resultant nanoparticles are shown in Figure 6A, where in all the measured samples, the characteristic gold surface plasmon resonance absorption band (λ(SPR)) was observed at 524 nm, confirming the synthesis of nanoparticles.
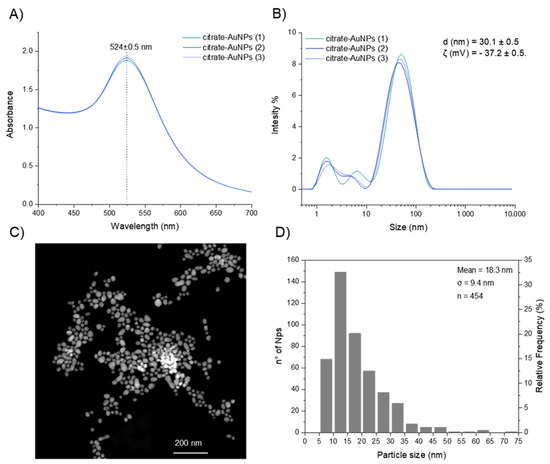
Figure 6.
(A) UV–Vis absorbance spectra of gold colloid (≈0.012 nM Au particles). (B) Intensity-based DLS analysis of citrate AuNPs. (C) TEM images (with a sample of ≈5.3 nM Au particles) and (D) size distribution of colloidal nanoparticles (by TEM).
Moreover, dynamic light scattering (DLS) and zeta potential measurements (ζ) were performed to investigate the hydrodynamic diameter (dh) and surface charge of the citrate-capped AuNPs, respectively. The mean hydrodynamic size of the colloid sample was dh = 30.1 ± 0.5 nm (Figure 6B) and the zeta potential was ζ = −37.2 ± 0.5 mV, suggesting an efficient stabilization of the particles with negative charges on the surface, in accordance with anionic citrate caps on the AuNP surface [35].
In addition, the average particle size (d) and morphology were determined by TEM (Figure 6C). Sphere-like gold nanoparticles were produced, however, the particles had a broad range of sizes in the colloidal solution with an average size of 18.3 ± 9.4 nm (Figure 6D). This broad particle size distribution can be a consequence of the initial concentration of sodium citrate added into the reaction mixture. Kumar and Meenan reported that the Au/citrate molar ratio was the parameter that had a significant effect on the dispersity of the particle solution [36]. In particular, a lower amount of citrate led to a wider distribution and larger average size. According to the authors and considering that the polydispersity index (0 ≤ PdI ≤ 1) measures the broadness of the size distribution where values close to 1 indicate that the distribution is highly polydisperse, an average dispersity value of PdI = 0.48 was obtained when the Au/citrate molar ratio increased from 0.09 (PdI = 0.25) to 1.73. In this study, the Au/citrate molar ratio was kept at 0.29 and the average dispersity value was 0.63 ± 0.03, a value that falls within the range of dispersity reported in literature.
3.2.2. UV–Vis Absorption, DLS, and TEM Analysis of Glyco-AuNPs
The success of the covalent coupling reaction between the thiolated linkers and gold nanoparticle surface was monitored by UV–Vis spectroscopy (Figure 7A,B). Gold particles have several polarizable conduction electrons that when interacting with an electromagnetic field, generate collective oscillations of free electrons, known as surface plasmon resonance (SPR). SPR induces a strong absorption of the incident light at ca. λ(SPR) = 520 nm. Upon displacement of citrate and the self-assembly of thiolated galactomannan, a change in the polarizable conduction electrons region may occur, leading to a small red-shift of the maximum absorption band [37]. The two glyco-AuNPs samples, GMHCl/ox/ATP-AuNPs and GMEnz/ox/ATP-AuNPs, had a λ(SPR) value that differed by 3 nm (λ(SPR) = 527 nm) and 7 nm (λ(SPR) = 532 nm) from the value obtained for the citrate-AuNPs sample (λ(SPR) = 524 nm) (see Section 3.2.1), confirming the functionalization of the metal surface.
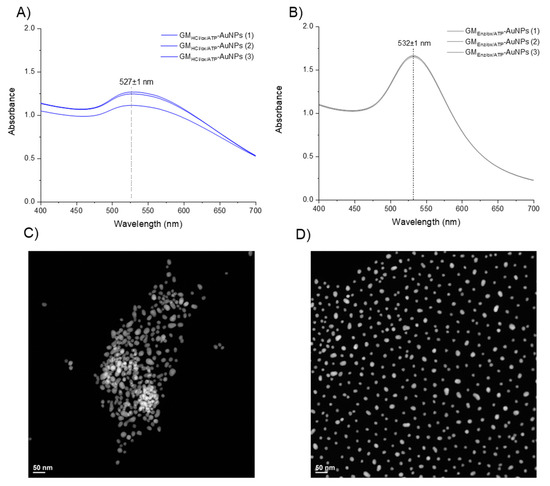
Figure 7.
(A,B) Surface plasmon resonance of gold particles upon functionalization with GMHCl/ox/ATP and GMEnz/ox/ATP, respectively (≈ 0.012 nM Au particles). (C,D) TEM images of gold particles upon functionalization with GMHCl/ox/ATP and GMEnz/ox/ATP, respectively.
A study of particle size upon functionalization with polysaccharide and a morphological analysis of the glyco-NP samples was done by TEM (Figure 7C,D). While the particle size of Au-cores underwent almost no change after the coupling reaction (dtem [GMHCl/ox/ATP-AuNPs] = 18.6 nm; dtem [GMEnz/ox/ATP-AuNPs] = 17.0 nm), a clear influence of the type of Mw and Đ of the ligand on the spatial arrangement of the particles in solution was observed. Indeed, the particles produced using the 5–6× larger low-dispersity galactomannan (GMHCl/ox l; 37 kDa) were more likely to agglomerate (Figure 7C) than those with short-chain higher dispersity galactomannan synthesized using the enzymatic procedure (GMEnz/ox; 6.4 kDa) (Figure 7D). In addition, polysaccharide-coated nanoparticles were characterized by dynamic light scattering (DLS) for their hydrodynamic radius and zeta potential (ζ) to measure the stability of the colloid particles under the measurement condition. As a result of the capping process, the average diameter of the glyco-AuNPs measured by DLS increased significantly compared to that measured for the citrate-capped AuNPs (dh = 30.1 ± 0.5 nm) (see Section 3.2.1). The new hydrodynamic diameters of the coated particles GMHCl/ox/ATP-AuNPs and GMEnz/ox/ATP-AuNPs were 250 ± 9 nm and 86.6 ± 0.5 nm, respectively. In addition, the zeta potential increased drastically in particles that underwent ligand exchange reaction, where anionic citrate molecules were replaced by neutral polysaccharide chains. Indeed, both the glyco-AuNPs presented a similar value of ca. ζ = −1.5 ± 0.3 mV. It is well-established that the magnitude of the zeta potential correlates with the stability of a colloidal system. In particular, a colloidal suspension is stable when the absolute value of the zeta potential of the particles in solution is closed to either +30 mV or −30 mV as the particles tend to repel each other without flocculating [38]. In our system, the surface charge of the particles after the self-assembly reaction with higher and lower molecular weight derivatized fibers increased significantly (from ζ = −37.2 ± 0.5 mV to ζ = −1.5 ± 0.3 mV), indicating possible reduced stability of the colloidal solution. However, no change in solution coloring or particle aggregates were visible after functionalization. After 15 days, the same samples were analyzed again by TEM and nanoclusters were observed in the sample prepared using galactomannan at lower molecular weight (GMEnz/ox/ATP-AuNPs), indicating that this functionalization affected the dispersion of particles in solution over time (see Figure 8A,B), while the sample prepared using the higher molecular weight polysaccharide maintained the same original distribution (GMHCl/ox/ATP-AuNPs).
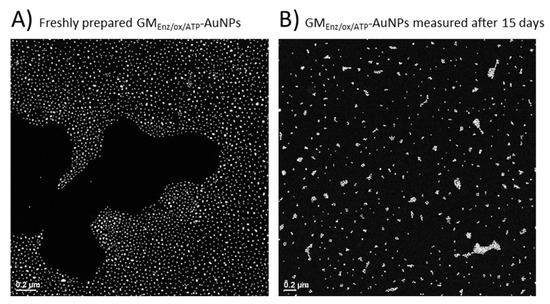
Figure 8.
TEM images of gold particles upon functionalization with GMEnz/ox/ATP. (A) The image was taken withthe freshly prepared sample. (B) The image was taken after 15 days.
3.2.3. Ligand Surface Density
From the data obtained by the anthrone/H2SO4 assay, we can conclude that only a small percentage of labelled substrate was successfully bound to the AuNP surface (Table 3). The unreacted excess ligand was washed away during the glyco-NP washing process. However, we can consider our coupling efficiency satisfactory. Indeed, even with what turned out to be a just 5- or 10-fold excess of polysaccharide, one can efficiently produce Glyco-AuNPs, which is much more economical than large excesses used in the literature for functionalization with monosaccharides. Hone and co-workers, who functionalized the gold surface with 2-mercaptoethyl α-d-mannopyranoside, used ~4400 nmol/ng AuNPs, and hence a 22–78-fold excess compared to the amounts employed in this study [28]. Moreover, the number of conjugated polymer chains per nanoparticles was within 5–203 as expected, well below the maximal number of D-mannose molecules that could theoretically occupy the surface of one AuNP with a diameter of about 20 nm, which was calculated by Wang et al. to be ~3500 [29]. In addition, the coupling efficiency decreased with the size of the polysaccharide, which is reasonable, as the added nmol chains/ng AuNPs were also 3.5-fold lower for GMHCl/ox/ATP compared to GMEnz/ox/ATP (56 vs. 195 nmol/ng AuNPs, respectively). In addition, the probability to adhere on the surface of the gold particle via the ligand-exchange mechanism is lower for the higher Mw thiolated polysaccharide due to higher steric hindrance and lower diffusion rates.
Table 3.
Amounts in nmol of thiolated polysaccharide (GMox/ATP) per ng of AuNPs added and of AuNPs conjugated. Coupling efficiency (%) of thiolated polysaccharides expressed as the ratio between GM(ox/ATP)conjugated and GM(ox/ATP)added. Number of repeat units [RU] per AuNPs. Number of conjugated polymer chains (GM(ox/ATP)conjugated) per AuNP.
All values shown in Table 3 were derived from calculations made using the Mn of the oxidized material (GMHCl/ox and GMEnz/ox), namely 37 kDa and 6.4 kDa, respectively, since it is related to the average number molecular weight value of a single polysaccharide chain, while the Mn of derivatized-ATP fibers resulted from a combination of two or more polysaccharide chains due to assumed dimerization via disulfide formation, which are broken up again during the reaction on the gold surface.
As already specified in Section 3.2.2, the type of Mw and Đ of the ligand significantly influences the spatial organization of the particles in solution. Specifically, short-chain higher dispersity ligands lead to a highly ordered spatial organization of the particles with particles equally spaced between each other (Figure 7C,D). A possible explanation for this phenomenon may lie in the different dispersity of the starting polysaccharide sample, which in turn is related to the molecular weight distribution of the thiolated polysaccharides. The sample fractionated with isopropanol (GMHCl) has a lower dispersity than the unfractionated sample (GMEnz) (Table 1). It is likely that the derivatization reaction of GMEnz with the thiol group was favored for lower molecular weight polymer chains. Non-derivatized galactomannan chains were removed during the washing step after the synthesis of the glyconanoparticles. As a result, small, monodispersed polymer chains covalently bonded on the surface of the particles, leading to a highly ordered spatial particle distribution. However, the dispersion of the polysaccharide solution only has an indirect effect as the main reason remains related to the small Mw (or Mn) used during the synthesis of the glycoAuNPs. Indeed, the same effect could be obtained with a monodisperse GM material of small size, which can completely saturate the surface of the Au particles, and thus keep the Au nuclei away from each other. As demonstrated by the loading efficiency (n° GM(ox/ATP) chains/n° AuNPs) calculated using the anthrone method (Table 3), the high dispersion of GMEnz/ox/ATP allows for the smaller chains to react preferentially, due to the faster diffusion and the higher ratio of label to monomer units due to the shorter chains.
3.3. Interaction Studies of GlycoNPs and Con A by TEM
An interaction experiment between galactomannan-coated gold nanoparticles and Con A was designed to be studied by TEM. After exposing the glyco-AuNPs to the Con A, potential interaction was revealed by monitoring the spatial arrangement of the particles before and after the addition of the protein. Under the experimental condition (pH = 7.6), the macromolecule con A has an overall negative charge (pI ~ 5) and exists as a tetramer with a molecular weight of about 25.5 kDa for each monomeric unit [39]. TEM images showed clear differences in the aggregation pattern between the two glyconanoparticle samples. When the lower Mw ligand (GMEnz/ox/ATP) was coupled to the gold surface, essentially no aggregation was visible in TEM, but a rather highly ordered spatial distribution of the particles was observed (Figure 9C). Once Con A was added, the gold nanoparticle density seemed to decrease, with a small increase in distance between the gold cores (Figure 9D). Hence, based on relative dimensions of the polysaccharide chains estimated to be 20 nm, the whole glycoNP, and Con A, one can propose that either interactions between GMEnz/ox/ATP and Con A occurred, but the surface density of the particles was so high (203 per Au core) that Con A with its four pockets went fully saturated by the same glyco-AuNP as the chains were so close to each other that the four pockets could simply be filled with adjacent chains on the same Au surface, not allowing Con A to act as a bridge between gold cores to bring them closer together, therefore leading to no aggregation, or no interaction visible by TEM happened simply because the polysaccharide chains were too small (interactions below a certain chain length threshold) [40,41].
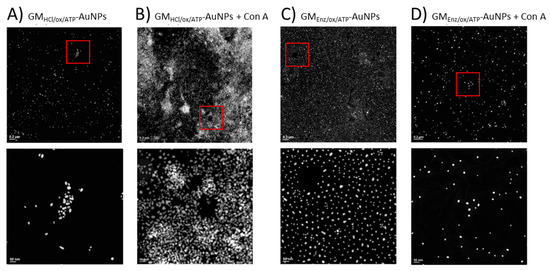
Figure 9.
TEM images of GMHCl/ox/ATP glyconanoparticles (6.0 nM) (A) before and (B) after the addition of 0.352 µM Con A. TEM images of GMEnz/ox/ATP glyconanoparticles (5.5 nM) (C) before and (D) after the addition of 0.352 µM Con A. The overall concentration of nanoparticles in the samples remained constant (≈4.7–4.8 nM). Scale bars are 0.2 µm (top row) and 50 nm (bottom row).
Conversely, with the higher Mw ligand (GMHCl/ox/ATP), a strong aggregation of the glyconanoparticles after Con A addition was visible by TEM (Figure 9A,B). The strong aggregation in the case of GMHCl/ox/ATPAuNP is likely due to multiple binding sites of Con A for mannose and galactose residues. Indeed, upon addition of the protein, particles are brought together, forming large nanoclusters (see Figure 9B). From this evidence, it is possible to assume that the size of the ligand can affect the number of lectins that can simultaneously interact with one or more polymer chains. Con A binding site consists of a rather deep pocket of approximately 6 Å × 7.5 Å × 18 Å [41], where the polysaccharide may fold into a conformation suitable for filling the binding site. However, as the size of the molecule increases, like in the case of GMHCl/ox/ATP, the flexible terminal binding portion of the polymer chain is more exposed to the active site of the protein and may align in a different way with respect to the protein, leading to favorable interactions.
Although the ligand density of GMEnz/ox/ATP-AuNPs expressed as [n°GM21(ox-ATP) conjugated/n°AuNPs] is more than 40× as high as with GMHCl/ox/ATP (Table 3), a visual inspection of the TEM images makes it clear that a higher surface density with 6× shorter chains of slightly lower Gal/Man ratio negatively affects the interaction with the protein to be visible by TEM.
Spectroscopic studies in the literature conducted on a large number of mono- and oligosaccharides (i.e., d-glucose, D-mannose, d-sorbitol, maltose, sucrose, and lactose) proposed that unmodified hydroxyl groups at the C3, C4, and C6 positions of the d-mannopyranose (or d-glucopyranose) ring were crucial for the interaction with the binding site of con A, while the hydroxyl group in position C2 appeared to have less impact. In addition, the α-linked glucobioses bound more strongly than the β-forms. Further evidence confirmed that concanavalin preferentially combined with the terminal, nonreducing portion of these molecules [42]. Based on this information and considering that galactomannan is a polysaccharide consisting of a linear backbone of β-(1→4)-linked D-mannose randomly substituted with α-(1→6)-linked galactopyranosyl units, we can propose that the unsubstituted mannose unit at the non-reducing end of each chain is the best candidate for the interaction, followed by galactose units, due to their α-configuration, and free hydroxyl groups (with the exception of C1). The reducing end and unsubstituted mid-chain mannose units are less likely to interact with the protein because the hydroxyl group at position C4 is involved in the β-(1→4) bond. Conversely, the mid-chain Gal-substituted Man units might not be directly involved in the recognition of con A, but only as mentioned potentially through their α-Gal C6 substituents.
A control experiment was carried out by exposing the citrate-capped gold particles to Con A under the same experimental conditions as for the glyconanoparticles. After 3 h of incubation, an unexpected change in color from violet to transparent was observed due to the collapse of the nanoparticles after the addition of the protein. The sample was also analyzed by TEM, revealing large, compact aggregates consisting of sintered gold particles. The lack of nanoparticle collapse of the synthesized GlycoAuNPs after exposure to the protein is an indirect evidence of the functionalization of the nanoparticles with galactomannan.
By comparing the size of the conjugated polysaccharides obtained by the SEC analysis, the value of the surface particle density calculated by the anthrone assay, and TEM images from particle screening before and after the addition of con A, we can conclude that the length and density of the galactomannan on the gold surface play an important role in determining the extent of the biomolecule–ligand interaction.
The creation of an analytical method that is sufficiently sensitive to such weak interactions is often difficult. This study shows that the combination of nanotechnology with advanced microscopy techniques such as TEM is certainly a valid alternative for determining whether two molecules interact.
4. Conclusions
The emphasis of this study was placed on the development of an analytical tool sensitive enough to recognize the interaction between neutral soluble fibers and target molecules. Based on the obtained data, we were successful in synthesizing glyco-NP-probes and demonstrated their use in an interaction study. A surface-bound carbohydrate ligand was used as a valuable approach for interaction studies. Specifically, shorter and longer thiolated guar galactomannans were successfully immobilized on gold nanoparticles. During the synthesis of glyconanoparticles, particular attention was given to optimize the functionalization of galactomannan with the thiol groups, possible by a prior controlled oxidation of the fiber with NaIO4, which led to an increase in reactive aldehyde groups on the polymer chain without significantly compromising the structure and properties of the original fiber. In addition, an estimation of the carbohydrate density was performed by the anthrone/H2SO4 assay where it was found that 6× smaller polysaccharide chains provide a higher coupling efficiency on the basis of number of chains. An interaction study was conducted between the particles glycated with the two galactomannans and concanavalin A (Con A). The study was based on inspection of the samples using TEM, where it was possible to observe a different spatial arrangement of the particles before and after exposure to Con A. In particular, it appeared that interactions visible as aggregates due to supramolecular interactions were favored for a lower surface density, but higher molecular weight of the anchored ligand at the particle surface. In conclusion, this approach can be considered a valid and flexible procedure for the creation of an analytical tool that can estimate and classify the type of process underlying the interaction between fibers and target molecules, leading to a better understanding of the mechanism that promotes the beneficial effect of dietary fibers. The analytical sensitivity and performance of this tool can be tuned by modifying the type and the length of the polysaccharide chain linked to the particle surface as well as the density of the carbohydrate ligands on the metal nanoparticles, leading to the creation of a library of glyconanoparticles with different compositions and chemical-physical properties that can be used for interaction studies with different target molecules.
Author Contributions
Conceptualization, C.L., S.B. and L.N.; Data curation, C.L. and C.D.V.; Formal analysis, C.L., C.D.V. and F.G.; Funding acquisition, S.B. and L.N.; Investigation, C.L., C.D.V. and F.G.; Methodology, C.L., S.B. and L.N.; Project administration, C.L., S.B. and L.N.; Resources, L.N.; Supervision, S.B. and L.N.; Visualization, C.L., C.D.V. and F.G.; Writing—original draft, C.L.; Writing—review & editing, C.D.V., F.G., S.B. and L.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by European Research Council ERC, under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 679037).
Acknowledgments
The authors are very thankful to Pflug Nicholas Craig and Xiaowen Wu for their support with the NMR analysis; Lee Sung Sik for help with Raman spectroscopy analysis; Yao Yang for the introduction to the FTIR machine; and Victorelli Francesca and Cao Yiping for the DLS introduction and data exportation assistance.
Conflicts of Interest
The authors would like to declare no conflicts of interest in the publication of this research.
Sample Availability
Not available.
References
- Kim, Y.; Je, Y. Dietary fiber intake and total mortality: A meta-analysis of prospective cohort studies. Am. J. Epidemiol. 2014, 180, 565–573. [Google Scholar] [CrossRef]
- Veronese, N.; Solmi, M.; Caruso, M.G.; Giannelli, G.; Osella, A.R.; Evangelou, E.; Maggi, S.; Fontana, L.; Stubbs, B.; Tzoulaki, I. Dietary fiber and health outcomes: An umbrella review of systematic reviews and meta-analyses. Am. J. Clin. Nutr. 2018, 107, 436–444. [Google Scholar] [CrossRef]
- Redgwell, R.J.; Fischer, M. Dietary fiber as a versatile food component: An industrial perspective. Mol. Nutr. Food Res. 2005, 49, 521–535. [Google Scholar] [CrossRef]
- McGill, C.R.; Devareddy, L. Ten-year trends in fiber and whole grain intakes and food sources for the United States population: National Health and Nutrition Examination Survey 2001–2010. Nutrients 2015, 7, 1119–1130. [Google Scholar] [CrossRef]
- Baye, K.; Guyot, J.P.; Mouquet-Rivier, C. The unresolved role of dietary fibers on mineral absorption. Crit. Rev. Food Sci. Nutr. 2016, 57, 949–957. [Google Scholar] [CrossRef] [PubMed]
- González-Aguilar, G.A.; Blancas-Benítez, F.J.; Sáyago-Ayerdi, S.G. Polyphenols associated with dietary fibers in plant foods: Molecular interactions and bioaccessibility. Curr. Opin. Food Sci. 2017, 13, 84–88. [Google Scholar] [CrossRef]
- Mudgil, D.; Barak, S. Composition, properties and health benefits of indigestible carbohydrate polymers as dietary fiber: A review. Int. J. Biol. Macromol. 2013, 61, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gunness, P.; Flanagan, B.M.; Shelat, K.; Gilbert, R.; Gidley, M.J. Kinetic analysis of bile salt passage across a dialysis membrane in the presence of cereal soluble dietary fibre polymers. Food Chem. 2012, 134, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Gunness, P.; Gidley, M.J. Mechanisms underlying the cholesterol-lowering properties of soluble dietary fibre polysaccharides. Food Funct. 2010, 1, 149–155. [Google Scholar] [CrossRef]
- Bosscher, D.; Van Caillie-Bertrand, M.; Deelstra, H. Effect of thickening agents, based on soluble dietary fiber, on the availability of calcium, iron, and zinc from infant formulas. Nutrition 2001, 17, 614–618. [Google Scholar] [CrossRef]
- Reppas, C.; Eleftheriou, G.; Macheras, P.; Symillides, M.; Dressman, J. Effect of elevated viscosity in the upper gastrointestinal tract on drug absorption in dogs. Eur. J. Pharm. Sci. 1998, 6, 131–139. [Google Scholar] [CrossRef]
- Espinal-Ruiz, M.; Parada-Alfonso, F.; Restrepo-Sánchez, L.P.; Narváez-Cuenca, C.E.; McClements, D.J. Interaction of a dietary fiber (pectin) with gastrointestinal components (bile salts, calcium, and lipase): A calorimetry, electrophoresis, and turbidity study. J. Agric. Food Chem. 2014, 62, 12620–12630. [Google Scholar] [CrossRef]
- Cao, Y.; Li, S.; Fang, Y.; Nishinari, K.; Phillips, G.O.; Lerbret, A.; Assifaoui, A. Specific binding of trivalent metal ions to λ-carrageenan. Int. J. Biol. Macromol. 2018, 109, 350–356. [Google Scholar] [CrossRef]
- Phan, A.D.T.; D’Arcy, B.R.; Gidley, M.J. Polyphenol–cellulose interactions: Effects of pH, temperature and salt. Int. J. Food Sci. Technol. 2016, 51, 203–211. [Google Scholar] [CrossRef]
- Naismith, J.H.; Field, R.A. Structural basis of trimannoside recognition by concanavalin A. J. Biol. Chem. 1996, 271, 972–976. [Google Scholar] [CrossRef]
- Vilaró, P.; Sampl, C.; Teichert, G.; Schlemmer, W.; Hobisch, M.; Weissl, M.; Panizzolo, L.; Ferreira, F.; Spirk, S. Interactions and Dissociation Constants of Galactomannan Rendered Cellulose Films with Concavalin A by SPR Spectroscopy. Polymers 2020, 12, 3040. [Google Scholar] [CrossRef]
- Zhang, C.; Qu, X.; Li, J.; Hong, H.; Li, J.; Ren, J.; Payne, G.F.; Liu, C. Biofabricated Nanoparticle Coating for Liver-Cell Targeting. Adv. Healthc. Mater. 2015, 4, 1972–1981. [Google Scholar] [CrossRef]
- Alex, S.; Tiwari, A. Functionalized gold nanoparticles: Synthesis, properties and applications: A review. J. Nanosci. Nanotechnol. 2015, 15, 1869–1894. [Google Scholar] [CrossRef]
- Lupo, C.; Boulos, S.; Nyström, L. Influence of Partial Acid Hydrolysis on Size, Dispersity, Monosaccharide Composition, and Conformation of Linearly Branched Water-Soluble Polysaccharides. Molecules 2020, 25, 2982. [Google Scholar] [CrossRef]
- Cheng, Y.; Prud’homme, R.K. Enzymatic degradation of guar and substituted guar galactomannans. Biomacromolecules 2000, 1, 782–788. [Google Scholar] [CrossRef]
- Da Silva, L.M.; Araújo, L.F.S.; Alvez, R.C.; Ono, L.; Sá, D.A.T.; da Cunha, P.L.; de Paula, R.C.M.; Maciel, J.S. Promising alternative gum: Extraction, characterization, and oxidation of the galactomannan of Cassia fistula. Int. J. Biol. Macromol. 2020, 165, 436–444. [Google Scholar] [CrossRef]
- Seo, J.H.; Adachi, K.; Lee, B.K.; Kang, D.G.; Kim, Y.K.; Kim, K.R.; Lee, H.Y.; Kawai, T.; Cha, H.J. Facile and Rapid Direct Gold Surface Immobilization with Controlled Orientation for Carbohydrates. Bioconjugate Chem. 2007, 18, 2197–2201. [Google Scholar] [CrossRef]
- Demuth, T.; Betschart, J.; Nyström, L. Structural modifications to water-soluble wheat bran arabinoxylan through milling and extrusion. Carbohydr. Polym. 2020, 240, 116328. [Google Scholar] [CrossRef]
- Doner, L.W.; Irwin, P.L. Assay of reducing end-groups in oligosaccharide homologues with 2, 2′-bicinchoninate. Anal. Biochem. 1992, 202, 50–53. [Google Scholar] [CrossRef]
- Waffenschmidt, S.; Jaenicke, L. Assay of reducing sugars in the nanomole range with 2, 2′-bicinchoninate. Anal. Biochem. 1987, 165, 337–340. [Google Scholar] [CrossRef]
- Nobre, K.A.; Soares, C.E.; Vieira, Í.G.; de Almeida, R.R.; de Moreira, R.A.; de Araújo, T.G.; Ribeiro, M.E.; Ricardo, N.M. Adenanthera pavonina galactomannan for controlled delivery of rutin: A preliminary study. Quím. Nova 2018, 41, 607–612. [Google Scholar] [CrossRef]
- Frens, G. Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Hone, D.C.; Haines, A.H.; Russell, D.A. Rapid, quantitative colorimetric detection of a lectin using mannose-stabilized gold nanoparticles. Langmuir 2003, 19, 7141–7144. [Google Scholar] [CrossRef]
- Wang, X.; Ramström, O.; Yan, M. A photochemically initiated chemistry for coupling underivatized carbohydrates to gold nanoparticles. J. Mater. Chem. 2009, 19, 8944–8949. [Google Scholar] [CrossRef] [PubMed]
- Turula, V.E., Jr.; Gore, T.; Singh, S.; Arumugham, R.G. Automation of the anthrone assay for carbohydrate concentration determinations. Anal. Chem. 2010, 82, 1786–1792. [Google Scholar] [CrossRef]
- Ahn, K.; Hennniges, U.; Banik, G.; Potthast, A. Is cellulose degradation due to β-elimination processes a threat in mass deacidification of library books? Cellulose 2012, 19, 1149–1159. [Google Scholar] [CrossRef]
- Houk, J.; Whitesides, G.M. ChemInform Abstract: Structure-Reactivity Relations for Thiol-Disulfide Interchange. ChemInform 1988, 109, 6825–6836. [Google Scholar] [CrossRef]
- Esmaeilzadeh, P.; Köwitsch, A.; Heyroth, F.; Schmidt, G.; Fischer, S.; Richter, K.; Groth, T. Synthesis of thiolated polysaccharides for formation of polyelectrolyte multilayers with improved cellular adhesion. Carbohydr. Polym. 2017, 157, 1205–1214. [Google Scholar] [CrossRef]
- Uetsuki, K.; Verma, P.; Yano, T.A.; Saito, Y.; Ichimura, T.; Kawata, S. Experimental Identification of Chemical Effects in Surface Enhanced Raman Scattering of 4-Aminothiophenol. J. Phys. Chem. C 2010, 114, 7515–7520. [Google Scholar] [CrossRef]
- Sujitha, M.V.; Kannan, S. Green synthesis of gold nanoparticles using Citrus fruits (Citrus limon, Citrus reticulata and Citrus sinensis) aqueous extract and its characterization. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 102, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Meenan, B.J.; Mutreja, I.; D’Sa, R.; Dixon, D. Controlling the size and size distribution of gold nanoparticles: A design of experiment study. Int. J. Nanosci. 2012, 11, 1250023. [Google Scholar] [CrossRef]
- Marín, M.J.; Schofield, C.L.; Field, R.; Russell, D.A. Glyconanoparticles for colorimetric bioassays. Analyst 2015, 140, 59–70. [Google Scholar] [CrossRef]
- Bhattacharjee, S. DLS and zeta potential—What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef]
- Locke, A.K.; Cummins, B.M.; Abraham, A.A.; Coté, G.L. PEGylation of Concanavalin A to Improve Its Stability for an In Vivo Glucose Sensing Assay. Anal. Chem. 2014, 86, 9091–9097. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.W.; Reeke, G.N., Jr.; Edelman, G.M. Location of the saccharide binding site of concanavalin A. J. Biol. Chem. 1971, 246, 6123–6125. [Google Scholar] [CrossRef]
- Edelman, G.M.; Cunningham, B.A.; Reeke, G.N.; Becker, J.W.; Waxdal, M.J.; Wang, J.L. The Covalent and Three-Dimensional Structure of Concanavalin A. Proc. Natl. Acad. Sci. USA 1972, 69, 2580–2584. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.J.; Hollerman, C.E.; Smith, E.E. Protein-Carbohydrate Interaction. II. Inhibition Studies on the Interaction of Concanavalin A with Polysaccharides*. Biochemistry 1965, 4, 876–883. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).