Abstract
Alzheimer’s disease is driven by multiple molecular drivers, including the pathological behavior of two intrinsically disordered proteins, amyloid-β (Aβ) and tau, whose aggregation is regulated by sequence-encoded ensembles and liquid–liquid phase separation (LLPS). This review integrates recent advances in biophysics, structural biology, and computational modeling to provide a multiscale perspective on how sequence determinants, post-translational modifications, and protein dynamics regulate the conformational landscapes of Aβ and tau. We discuss sequence-to-ensemble principles, from charge patterning and aromatic binders to familial mutations that reprogram structural ensembles and modulate LLPS. Structural studies, including NMR, SAXS, cryo-EM, and cryo-electron tomography, trace transitions from disordered monomers to fibrils and tissue-level structures. We highlight experimental challenges in LLPS assays, emerging standards for reproducibility, e.g., LLPSDB, PhaSePro, and FUS benchmarks, and computational strategies to refine and condensate modeling. Finally, we explore the therapeutic implications, including condensate-aware medicinal chemistry, ensemble-driven docking, and novel insights from clinical trials of anti-Aβ antibodies. Together, these perspectives underscore a paradigm shift toward environment- and ensemble-aware therapeutic design for Alzheimer’s and related protein condensation disorders.
1. Introduction
Brains with Alzheimer’s disease show the accumulation of β-amyloid (Aβ) and tau. In aqueous solution, neither proteins adopt a stable fold; instead, they populate shifting ensembles of conformations, which is the defining behavior of intrinsically disordered proteins (IDPs) [1,2]. Many cellular proteins can form liquid-like condensates through liquid–liquid phase separation (LLPS). This process forms membrane-less droplets that fuse and flow, and under certain conditions, can evolve into less dynamic, more solid assemblies relevant to pathology [3]. Tau undergoes LLPS in vitro and in cells, and phase-separated tau droplets can promote aggregation, which connects condensates to filament formation [4]. Structural methods now anchor these concepts across length scales. Recent cryo-electron microscopy and tomography initiatives solved patient-derived tau filaments at nearly atomic resolution, and protofilament folds disease-specific, and have actually viewed the three-dimensional structure of Aβ plaques and tau inclusions in human brain tissue [5]. In aqueous solution, the conformational behavior of Aβ and tau is largely governed by primary-sequence features. Global descriptors such as net charge per residue influence overall chain expansion or compaction, and the linear patterning of oppositely charged residues controls intrachain electrostatics and the extent of structural flexibility. Studies have shown that shuffling positive and negative residues, even without changing composition, measurably shifts ensemble dimensions and intrachain contacts. These sequence–ensemble rules provide a practical basis for anticipating how mutations or post-translational modifications will alter conformational states in Aβ or tau [6,7]. Many proteins that include disordered regions can also separate into two liquid phases under physiologic-like conditions. The dense phase behaves like a fluid, i.e., droplets fuse, flow, and exchange components with the surrounding solution. Because crowding agents, ionic strength, protein concentration, and surface effects can produce misleading readouts, the field has issued best-practice guidance. Recommended steps include titrating protein and salt to map phase diagrams, testing reversibility, combining microscopy with orthogonal probes of material properties, and interpreting Fluorescence Recovery After Photobleaching (FRAP) with care to avoid mistaking gels or aggregates for liquids [8,9,10]. For tau, LLPS has been demonstrated in vitro and in cells. Under crowding or salt conditions compatible with the cytoplasm, tau forms liquid-like droplets that can age to less dynamic states and seed filament assembly. These observations place LLPS on the mechanistic path to tau aggregation [4,11,12,13]. Evidence is also developing for Aβ, where soluble oligomers have been observed to form liquid-like droplets that modulate subsequent amyloid formation under defined conditions [14]. The current developments in structural methods have linked atomic structure to pathological outcome, i.e., cryo-EM structures of patient-derived tau filaments up to nearly atomic scale have characterized disease-specific protofilament folds, and cryo-electron tomography has given a direct view of the three-dimensional organization of Aβ plaques as well as tau inclusions in human brain tissue [5,15,16]. More recently, cryo-electron tomography has visualized the architecture of Aβ plaques and tau inclusions directly in the post-mortem human brain. These data reveal mixtures of fibrils, including branched forms, together with protofilaments arranged in parallel arrays and lattice-like assemblies in situ [5]. Clinical practice and trials now use a biological framework to stage disease. The National Institute on Aging—Alzheimer’s Association’s AT(N) research framework categorizes biomarkers into amyloid-β deposition (A), pathological tau (T), and neurodegeneration or neuronal injury (N), and views cognitive impairment as a clinical manifestation [17]. Blood-based tests have advanced rapidly- in multicohort studies, a plasma p-tau217 immunoassay identified abnormal Aβ and tau with accuracy comparable to cerebrospinal fluid measures and showed longitudinal changes in individuals with biomarker-positive Alzheimer’s disease [18]. On the therapeutic side, lecanemab met its primary endpoint in the phase-3 Clarity AD trial, significantly reducing brain amyloid as measured by PET imaging and leading to a moderately slower rate of cognitive decline over 18 months in early Alzheimer’s disease [19]. Similarly, Donanemab slowed clinical decline in the phase 3 TRAILBLAZER-ALZ 2 trial, which evaluated individuals with early symptomatic Alzheimer’s disease [20]. Together, these developments underscore the importance of connecting ensemble behavior, phase separation, and structured aggregate formation to measurable biomarkers and to therapeutic interventions now advancing in clinical practice (Figure 1).
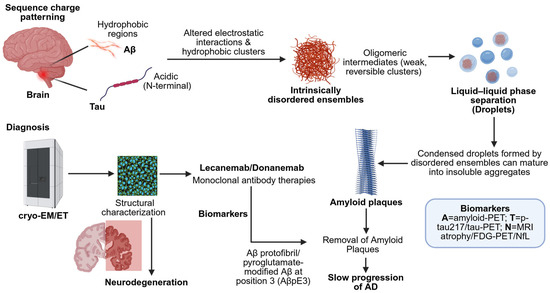
Figure 1.
Diagnostic scheme of the Aβ and Tau sequence-encoded charge/hydrophobic pattern stimulation of the conformational changes in Alzheimer’s disease. Distorted electrostatic interactions result in inherently disordered aggregates and LLPS, which advance to amyloid aggregates. Cryo-EM/ET offers structural information, whereas biomarkers allow for in vivo diagnosis. Lecanemab/Donanemab is a monoclonal antibody that encourages the breakdown of amyloid plaques, reducing the development of the disease (created using BioRender.com/v0zfo7e (accessed on 9 September 2025)).
2. Sequence-to-Ensemble Principles That Govern Aβ and Tau
2.1. Sequence Features and the Shape of the Ensemble
In a dilute buffer, the average size and the conformational heterogeneity of IDP chains are primarily governed by simple sequence-derived descriptors. One such parameter, net charge per residue (NCPR), modulates the coil–globule equilibrium, which is a higher absolute NCPR typically promotes chain expansion, as electrostatic repulsion between like-charged residues disfavors intrachain compaction. This trend was experimentally validated by Mao et al., who used single-molecule fluorescence and small-angle X-ray scattering (SAXS) to demonstrate how varying NCPR alters the conformational dimensions of synthetic IDPs [21].
A second key sequence descriptor is the linear patterning of oppositely charged residues, quantified by the κ (kappa) parameter. Although NCPR captures overall charge content, κ describes how charges are distributed along the sequence. Importantly, holding composition constant but rearranging positive and negative residues can significantly alter ensemble dimensions and intrachain electrostatic interactions. Sequences with more segregated or “blocky” charge distributions (higher κ) tend to compact, whereas well-mixed patterns (lower κ) favor more expanded conformations. This concept was introduced and validated by Das and Pappu, who used both computational modeling and synthetic IDP sequences to demonstrate κ’s influence on conformational ensembles [6]. These electrostatic rules are supported by independent work showing that charge interactions dominate chain dimensions for many IDRs under controlled solution conditions [22].
Beyond global charge, aromatic residues and their linear patterning serve as key determinants of cohesive interactions in intrinsically disordered regions. In many prion-like domains, aromatic side chains, especially tyrosine and phenylalanine, act as “stickers” that mediate π–π and π–cation interactions, influencing both single-chain compaction and phase-separation propensity. The valence (number) and spacing of these aromatic residues critically modulate this behavior. Martin and colleagues combined NMR spectroscopy, SAXS, mutational scanning, and coarse-grained simulations to demonstrate that higher aromatic valence and specific sequence patterns enhance intra- and intermolecular cohesion, thereby promoting LLPS [23]. These observations connect naturally to the stickers-and-spacers framework for condensates, where cohesive residues (stickers) are separated by more solvated linkers (spacers) [24] (Table 1).
2.2. Charge Patterning Connects Chain Compaction and LLPS in Tau
Residue-level simulations and experimental data converge on a central principle: the same sequence features that promote single-chain compaction, such as charge patterning and cohesive stickers, also enhance LLPS when the protein concentration exceeds a critical threshold. This mechanistic overlap reflects a continuity between intramolecular and intermolecular interactions. Coarse-grained models, calibrated using experimental data on radii of gyration for disordered proteins, successfully recapitulate phase diagrams and help pinpoint the specific interaction motifs, such as π–π, electrostatic, or cation–π interactions, which drive LLPS in intrinsically disordered regions [25]. In tau, electrostatics between negatively charged N-terminal regions and positively charged microtubule-binding repeats make an important contribution to LLPS. Boyko and colleagues showed that changing salt or charge balance strongly modulates droplet formation [26] (Table 1).
2.3. Disease Mutations as Sequence Edits
Mutations linked to familial Alzheimer’s disease (FAD) function as precise sequence-level edits that reshape the aggregation behavior of the Aβ peptide. Several of these mutations cluster near the central and C-terminal hydrophobic segments, particularly around residues 22–23, which are critical for modulating oligomerization and fibrillization. Classic examples include E22Q, E22G, E22K, and D23N, all of which enhance aggregation kinetics in vitro and shift the balance toward protofibril or fibril formation. These substitutions increase the structural cohesion of Aβ assemblies and are often associated with early-onset cerebrovascular or parenchymal amyloidosis. In contrast, N-terminal variants such as A2V and A2T exhibit divergent behavior; A2V can either promote or inhibit aggregation, while A2T has been reported to confer protective effects in certain genetic backgrounds. Together, these naturally occurring mutations illustrate how single-residue changes can reprogram the conformational ensemble of Aβ and influence its pathogenic trajectory [27,28,29] (Table 1).
2.4. Post-Translational Modifications (PTMs) That Shift Ensembles and Condensates
Tau undergoes numerous post-translational modifications in the human brain. Modifications that introduce negative charge, such as phosphorylation at AT8 epitopes, can disrupt long-range intramolecular contacts and promote LLPS under cytoplasmic conditions. Early AT8 immunoreactivity is identified in the onset stage of the disease before the development of obvious filaments, and thus is widely applied in neuropathological staging (Braak stages I–IV). This phosphorylation process depends largely on the activity of GSK3β and CDK5 among other kinases, and therefore, attributes AT8 positivity to tau kinase signaling dysregulation in the pathology of Alzheimer’s. The resulting droplets can then age into less dynamic assemblies that seed filament formation. Wegmann and colleagues were the first to place tau LLPS on the path to aggregation, while Kanaan and co-workers demonstrated that LLPS can induce disease-like conformations [4,11].
Acetylation modifies lysines in microtubule-binding repeats and other regions. Acetylated tau shows impaired microtubule binding and can form pathological aggregates in cellular and mouse models. Cohen and colleagues identified major acetylation sites, including K280, and demonstrated that acetylation is associated with loss of function and aggregation. Acetylation also reduces binding capacity by reducing electrostatic interactions between the negatively charged tubulin surface and the positively charged lysine residues in the inner portion of the microtubule-binding motif. At the same time, acetylation at KXGS motifs can block phosphorylation at these motifs and, in some systems, reduce aggregation, underscoring the context-dependent nature of this process [30].
O-GlcNAcylation on serine/threonine residues generally stabilizes tau against aggregation, and has been associated with slower neurodegeneration in mouse models when O-GlcNAc levels are pharmacologically increased. Yuzwa and co-workers reported that inhibition of O-GlcNAcase (OGA), the enzyme that removes O-GlcNAc modifications, elevates tau O-GlcNAcylation, which hinders tau aggregation and reduces neuronal cell loss in tau transgenic mice. Subsequent studies confirmed that O-GlcNAc modification directly inhibits tau fibrillization in vitro. This post-translational modification maintains tau in a soluble, non-toxic form, reducing pathological aggregation, and makes OGA inhibition a promising therapeutic approach for Alzheimer’s and other tauopathies. Several OGA inhibitors are in preclinical and early clinical development based on these findings [31,32,33].
Proteolytic truncations of tau are frequent in disease tissues, and contribute to increased aggregation by exposing amyloid-prone segments or altering intramolecular contacts. Early work demonstrated that caspase cleavage near the microtubule-binding region, especially at residue D421, promotes tau aggregation. Caspase-3 and other caspases cleave tau at this site, facilitating filament assembly. More recent studies have found that several N- and C-terminal tau truncations display enhanced oligomerization and seeding activity. N-terminal truncations toward the microtubule-binding repeat region modulate phosphorylation and aggregation propensity, and some truncations significantly increase tau’s ability to seed aggregation templated by Alzheimer’s disease tau. Specific fragments, such as Tau_151-391, are highly prone to aggregate and seed pathological tau assemblies. Truncations have been detected in oligomeric fractions, paired helical filaments, and tau neurofibrillary tangles from Alzheimer’s disease brains and related mouse models. These truncated tau fragments expose the microtubule-binding repeats, facilitating aggregation and potentially initiating tau pathology [34,35,36] (Table 1).
2.5. Implications for Aβ
Although Aβ is a short peptide, the same sequence-structure logic applies. It features a polar N-terminus, a central hydrophobic region around residues 17–21 that favors β-structure, and a hydrophobic C-terminus. Mutations or chemical modifications that alter charge at positions 2, 22, or 23, or that disrupt hydrophobic interactions in the C-terminal region, can shift monomer conformation distribution and affect both nucleation rates and fibril growth kinetics. Further mutational development could result in the dimer or aggregation formation, resulting in neurodegeneration. FAD variants in these regions illustrate how even subtle sequence changes can rewire aggregation pathways and alter the balance between oligomeric and fibrillar states [27,28].

Table 1.
Sequence and biophysical determinants of Aβ and tau ensembles.
Table 1.
Sequence and biophysical determinants of Aβ and tau ensembles.
| Determinant | Typical Experimental Readouts | Expected Qualitative Effect on Ensemble and LLPS | Representative References |
|---|---|---|---|
| Net charge per residue (NCPR) | SAXS Rg, smFRET, single-molecule methods | Larger absolute NCPR expands chains and generally disfavors LLPS at fixed ionic strength. | Mao et al., 2010; Müller-Späth et al., 2010 [21,22]. |
| Charge patterning (κ) | SAXS, smFRET, coarse-grained simulation with sequence variants | Blockier patterns compact chains and can favor condensate formation relative to well-mixed patterns. | Das & Pappu, 2013; Sherry et al., 2017 [6,7]. |
| Aromatic valence and spacing | NMR, SAXS, mutational scans, phase diagrams | More aromatics and certain patterns increase cohesion, compact single chains, and raise LLPS propensity. | Martin et al., 2020 [23]. |
| Stickers and spacers | FRAP, rheology, microscopy, simulations | Sticker enrichment strengthens cohesion; spacer changes the material properties of condensates. | Choi et al., 2020; Dignon et al., 2018 [24,25]. |
| Tau phosphorylation (e.g., AT8) | Phospho-specific immunoassays, LLPS assays, and microscopy | Adds a negative charge, shifts long-range contacts, and can increase LLPS, promoting ageing toward aggregates. | Wegmann et al., 2018; Kanaan et al., 2020 [4,11]. |
| Tau acetylation (e.g., K280, KXGS motifs) | MS mapping, MT-binding assays, aggregation assays | Impairs microtubule binding; it can promote aggregation in cells and mice. KXGS acetylation can also block phosphorylation and reduce aggregation in some systems. | Cohen et al., 2011; Cook et al., 2014 [30,37]. |
| Tau O-GlcNAcylation | O-GlcNAc proteomics, aggregation assays, in vivo models | Stabilizes tau and suppresses aggregation; slows neurodegeneration in mice when increased. | Yuzwa et al., 2012; Yuzwa et al., 2014 [38,39]. |
| Tau truncations | MS-based proteomics, seeding and aggregation assays | Many truncations increase oligomerization and seeding; caspase cleavage promotes aggregation. | Gu et al., 2020; Gamblin et al., 2003; Chu et al., 2023; Gao et al., 2018 [35,40,41,42]. |
| Aβ familial mutations (E22Q/G/K, D23N; A2V/T) | Kinetics, EM, solid-state NMR, cytotoxicity | Alter nucleation and growth rates and the balance of oligomer and fibril states. | Kim et al., 2008; Rezaei-Ghaleh et al., 2023; Park et al., 2021; Benilova et al., 2014 [27,28,29,43] |
3. Structural Transitions Across Biological Scales: From Monomer to Oligomer, Fibril, and Tissue
3.1. Solution Ensembles and Small Oligomers
Studies on intrinsically disordered proteins have shown that solution ensembles can be defined by integrating data from NMR, SAXS, and single-molecule FRET (smFRET), with each technique constraining different aspects of chain dimensions and conformational heterogeneity. Recent methodological advances describe how to combine these readouts within a unified modeling framework, an essential step for accurately capturing the highly dynamic, interconverting ensembles populated by disordered proteins like Aβ and tau [44,45,46]. These experiments can be extended to low-order oligomers, although short lifetimes and low abundance remain challenges (Figure 2).
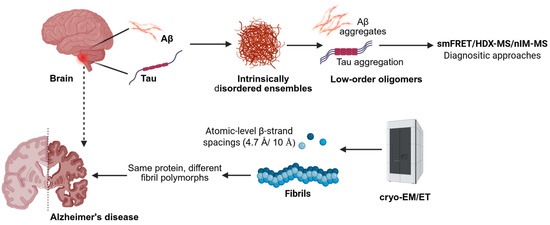
Figure 2.
Mass spectrometry and structural techniques map the progression from disordered monomers to mature fibrils and tissue-level assemblies in Alzheimer’s disease (created using BioRender.com/32702fs (accessed on 9 September 2025)).
3.2. Ex Vivo Fibrils at Near-Atomic Resolution
The field changed when tau filaments from Alzheimer’s disease brains were solved using cryo-EM at 3.4–3.5 Å resolution. The cores of paired helical and straight filaments were shown to comprise two identical protofilaments built from residues 306–378 of tau, establishing disease-defining folds and a structural basis for tau polymorphism [5]. Comparable progress has been made for Aβ. Cryo-EM structures of Aβ1–42 fibrils at ~4.0 Å revealed two intertwined protofilaments and provided atomic models that match solid-state NMR restraints [47]. More recently, cryo-EM structures of patient-derived Aβ1–42 fibril polymorphs grown by seeded growth have been reported, highlighting the structural diversity of Aβ aggregates and underscoring the importance of considering multiple fibril states in therapeutic development [48] (Figure 2).
3.3. In-Tissue Architecture by Cryo-Electron Tomography
Recent methodological advances that preserve the native ultrastructure have enabled the visualization of protein aggregates directly within the human brain. A 2024 study applied targeted cryo-fluorescence microscopy, focused-ion-beam milling, and cryo-electron tomography to post-mortem Alzheimer’s tissue to map the spatial organization of amyloid plaques and tau inclusions. The resulting tomograms revealed parallel fibril arrays, branched morphologies, and lattice-like architectures in situ. These findings provide a structural link between purified fibrils and their native arrangements within diseased brain tissue, offering new insight into the microenvironment of pathogenic aggregates [5] (Figure 2).
3.4. Mass Spectrometry Toolkits for Dynamic Assemblies
Hydrogen–deuterium exchange mass spectrometry (HDX-MS) offers insights into solvent accessibility and conformational dynamics in solution, and is increasingly used to monitor structural transitions during assembly processes and to map epitopes on large protein complexes. Recent reviews have outlined the strengths, limitations, and best practices for HDX-MS, and advances in millisecond-scale labeling have significantly improved temporal resolution [49,50]. Native ion-mobility mass spectrometry (nIM-MS) enables oligomeric stoichiometries detection and can assess the stability of complexes. In the context of Aβ–antibody interactions, nIM-MS has identified preferential binding to monomers, dimers, and higher-order oligomers, and characterized unfolding behaviors upon antibody binding; In the nIM-MS, it has been demonstrated that various antibodies exhibit specific binding preferences when using Aβ ensembles, with some preferentially attaching to monomers or small oligomers, whilst others to higher-order aggregates, indicating that there is conformation-selective recognition and unfolding behavior on binding [51]. Together with cross-linking mass spectrometry, these tools complement solution-state and cryo-EM/ET approaches by capturing transient, intermediate states that are often inaccessible to traditional structural methods (Figure 2).
4. LLPS of Tau and Aβ: Evidence, Mechanisms, and Experimental Challenges
4.1. Tau LLPS and Links to Aggregation
Tau protein undergoes LLPS under physiological conditions with the cytoplasm, forming dynamic liquid droplets that exhibit fusion and flow. Initial studies demonstrated that prolonged incubation of these tau droplets forms less dynamic, more solid-like material capable of seeding tau aggregation, positioning LLPS as a mechanistic intermediate along the path to tau assembly and aggregation [4]. Subsequent investigations revealed that extended phase separation drives tau into disease-relevant, non-filamentous conformations and promotes toxic oligomeric species formation, with mutants such as P301L exhibiting an enhanced propensity for LLPS and oligomerization. Notably, filament formation is not a prerequisite for acquiring pathogenic tau conformations [11,52]. The regulation of tau LLPS is profoundly influenced by electrostatic interactions, ionic strength, molecular crowding, and charge-altering post-translational modifications, including phosphorylation and acetylation, as well as disease-linked mutations, which together shift the phase boundary and modulate phase behavior [26,53]. Phosphorylation at the AT8 epitope introduces negative charges that disrupt tau’s long-range intramolecular contacts, increasing its LLPS propensity and accelerating droplet aging toward more static, aggregation-prone states [11]. Collectively, these studies provide compelling mechanistic insights into how tau LLPS functions as a dynamic precursor to pathological aggregation, bridging the soluble protein state and neurotoxic aggregate formation. Understanding the biophysical determinants and modulators of tau phase behavior elucidates fundamental processes underlying tauopathy progression and offers potential avenues for therapeutic intervention targeting early aggregation intermediates (Figure 3).
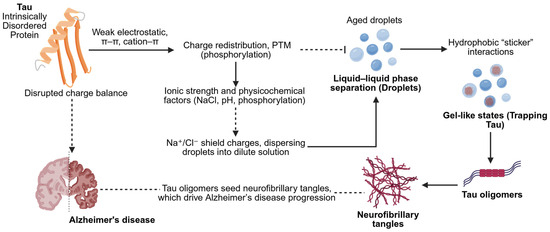
Figure 3.
Mechanistic description of IDP Tau-mediated LLPS. Intrinsically disordered Tau is charge-redistributed and physiochemically modulated (constituent ionic strength, pH, phosphorylation) to promote the formation of droplets of LLPS. These droplets have the potential to develop into oligomers and fibrils, which cause neurodegeneration. Various other factors besides NaCl have a role to play in condensate behavior and aggregation in Alzheimer’s disease (created using BioRender.com/1v6060u (accessed on 9 September 2025)).
4.2. Aβ Condensation as a Context-Dependent Intermediate in Amyloid Assembly
Recent evidence increasingly supports the ability of Aβ to access LLPS. In vitro studies have demonstrated that soluble Aβ oligomers can form droplets exhibiting classic liquid behaviors such as fusion and dynamic molecular rearrangements. These condensates modulate the subsequent formation of amyloid fibrils, suggesting that LLPS plays a regulatory role in Aβ aggregation [14]. Furthermore, microfluidic experiments that precisely control protein concentration and incubation timescales reveal that Aβ40 can form a transient liquid-like intermediate state that feeds into the fibril formation pathway. This highlights the kinetics and concentration dependence of Aβ phase behavior, providing new insights into amyloid aggregation mechanisms [54]. Complementary studies show that Aβ undergoes phase separation on lipid membranes both in vitro and in cells, linking Aβ condensation directly to membrane environments relevant to Alzheimer’s pathology. Presence of lipid surfaces appears to facilitate and modulate Aβ phase separation and aggregation, emphasizing the critical role of interfaces in modulating phase behavior and amyloid assembly [55]. Taken together, these findings support a condensate-mediated pathway for Aβ assembly and amyloid fibrillogenesis, dependent on solution chemistry, peptide concentration, timescale, and interfacial contexts, thus enhancing our mechanistic understanding of Aβ aggregation and its pathological implications.
4.3. Conserved Biophysical Mechanisms Underlying LLPS Across Systems
Common mechanistic principles have emerged from LLPS studies of tau and Aβ. First, sequence features that promote chain compaction and increase intermolecular “stickiness” also tend to lower the concentration threshold required for phase separation. Second, ionic conditions strongly modulate LLPS, i.e., monovalent salts typically suppress electrostatically driven condensation by screening long-range interactions, whereas polyvalent ions can either promote or inhibit phase separation, depending on the charge distribution and valence. Third, condensates often “age” over time, with their material properties transitioning from liquid to gel-like or solid-like states under multiple conditions, e.g., high protein concentration or enhanced cohesive interactions. These principles align with quantitative models for biomolecular condensates and underscore the importance of explicitly assessing time dependence and reversibility in LLPS experiments [52,56,57,58].
4.4. Experimental Challenges and Reporting Standards
Because multiple variables can mimic droplet formation or conceal gelation, LLPS experiments require rigorous controls and standardized reporting. It is recommended for (i) comprehensive mapping of phase diagrams by systematically varying protein concentration, salt, pH, and additives to define the boundaries and conditions driving phase separation; (ii) verification of phase reversibility, typically by dilution or salt-dependent dissolution assays, to distinguish true LLPS from irreversible aggregation; (iii) integration of microscopy with orthogonal methods that probe material properties, such as passive microrheology or droplet fusion-time analysis, for quantitative characterization of condensate dynamics; and (iv) use of FRAP, recognizing partial fluorescence recovery may reflect molecular exchange with surrounding or permeable gel states rather than true liquid-like behaviour. Surface adhesion and high concentrations of crowding agents (e.g., PEG) are known sources of artifacts and should be minimized or explicitly controlled. Emerging microfluidic platforms enhance experimental reproducibility by enabling rapid, high-throughput control over concentration, mixing time, and droplet geometry, thereby facilitating quantitative analysis of phase behavior and kinetic properties [8,59].
5. Multiscale Computational Modeling of IDPs
5.1. All-Atom Force Fields for IDPs: Advances Toward Physically Realistic Ensemble Simulations
The development of all-atom force fields tailored for IDPs is pivotal in achieving accurate multiscale computational models to reflect experimental observations. Traditional force fields often misrepresented the balance required among backbone conformations, side-chain interactions, and protein–water dispersion, leading to either overly collapsed or overly expanded IDP ensembles that deviated from solution measurements. CHARMM36m (a forcefield) emerged as a significant advance by refining backbone torsional terms, which yielded more realistic coil and helix populations and reproduced the radii of gyration seen in SAXS and NMR analyses of IDPs, thus improving upon previous CHARMM generations [60]. In parallel, the a99SB-disp force field was optimized by simultaneously tuning protein and water dispersion interactions. This parameterization brought theoretical ensembles into quantitative agreement with SAXS and NMR benchmarks for both IDPs and folded proteins, without sacrificing accuracy for structured domains [61]. The choice of water model, particularly the use of TIP4P-D over TIP3P, also plays a crucial role, as TIP4P-D enhances water dispersion and prevents non-physical compaction of IDPs, further aligning simulated and experimental ensembles [62,63]. Collectively, consistency across biophysical techniques, driven by improved force fields and state-of-the-art sampling strategies such as replica-exchange simulations, whereby a system is simulated repeatedly at different temperatures and different exchange configurations to complement conformational sampling has enabled computational models to produce IDP ensembles that are highly congruent with SAXS, SANS, and NMR data, underscoring the importance of matching simulation physics with experimental multiscale reality (Figure 4).
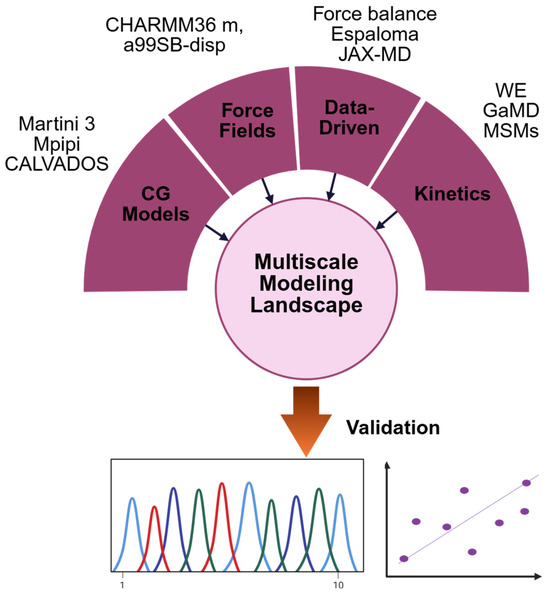
Figure 4.
Multiscale computational modeling of IDPs, highlighting force field development, data-driven refinement, coarse-grained models, ensemble validation, and kinetic approaches.
5.2. Emerging Data-Driven and Differentiable Approaches in Force Field Refinement
Recent advances in molecular modeling have increasingly leveraged automated and differentiable frameworks to refine force field parameters with greater accuracy and reproducibility. Tools such as ForceBalance offer a systematic platform for parameter optimization against combined experimental and quantum mechanical (ab initio) datasets. This methodology has been applied to the development of improved point-charge water models and polarizable force fields, enhancing the transferability and physical realism of molecular simulations [64,65,66]. In parallel, differentiable molecular mechanics frameworks, notably Espaloma and JAX-MD, have introduced the capability to compute gradients through entire physical simulations. These platforms integrate machine learning architectures with physical simulation engines, enabling the direct learning of force field parameters from structural, energetic, or dynamical observables [67,68,69]. Furthermore, differentiable coarse-grained training methods allow end-to-end optimization of simplified models while preserving key thermodynamic behaviors. Although still in early stages relative to conventional all-atom force fields, these approaches offer promising directions for the next generation of data-driven biomolecular models (Figure 4).
5.3. Coarse-Grained Modeling of Condensates and Long-Timescale Dynamics
Coarse-grained (CG) simulations make simulations accessible to larger spatial and time scales to allow for small mesoscale processes such as LLPS, condensate dynamics, and material properties to be studied, which would have been inaccessible to all-atom molecular dynamics. CG models do not have the atomic detail, but still have the necessary physicochemical interactions. The Martini 3 force field provides a general-purpose framework with refined bead types and interaction schemes for diverse biomolecular systems. However, several studies have reported that unmodified Martini 3 tends to over-compact IDPs and over-stabilize protein–protein interactions, thereby distorting single-chain dimensions and phase behavior unless interaction parameters are empirically rescaled [70,71,72]. Complementing these general-purpose models, sequence-specific CG models tailored to IDP properties and LLPS have shown improved predictive efficacy. The Mpipi model reproduces single-chain compaction and shifts in critical temperature with near-quantitative agreement for diverse IDRs that drive phase separation [73]. The CALVADOS 2 model further refines conformational ensembles and LLPS trends across varied sequences and has recently been extended to simulate macromolecular crowding environments (e.g., with polyethylene glycol, PEG) [74,75]. Additionally, IDP-tuned variants of Martini are being developed to correct for over-compaction and to better replicate SAXS- and NMR-derived IDP dimensions [72] (Figure 4).
5.4. Ensemble Validation and Optimization Guided by Experimental Data
Despite the use of advanced, well-parameterized force fields and enhanced sampling techniques, conformational ensembles generated using molecular simulations often show residual discrepancies from experimental data. This arises from limitations in sampling efficiency and inherent biases in force field parameters. To reconcile simulations with experimental data, ensemble refinement methods have become standard. These methods adjust the statistical weights of individual conformations in an ensemble to better reflect observables measured experimentally. Bayesian or Maximum Entropy (BME) frameworks are widely adopted for this purpose. BME systematically modifies the probability distribution over simulation frames so that ensemble-averaged observables, such as SAXS, NMR chemical shifts, paramagnetic relaxation enhancements, or smFRET distances-match experiments within experimental uncertainties. Crucially, these frameworks minimize deviations from the original simulated distribution, preserving as much of the unbiased simulation information as possible, thus providing statistically principled refinement consistent with uncertainties [76]. An alternative is metainference, which explicitly incorporates experimental noise and conformational heterogeneity by running multiple replicas and averaging observables across them during simulation. When combined with metadynamics, it can enhance sampling of rarely visited states while integrating data-driven correction [77,78] (Figure 4).
5.5. Kinetics and Mechanisms at Relevant Timescales
Many fundamental questions in Aβ and tau biology involve kinetics, such as ligand binding/unbinding, conformational exchange, and oligomerization steps, which occur over microsecond to second timescales. These timescales often exceed the reach of conventional molecular dynamics (MD), but modern enhanced-sampling and path-sampling techniques now allow for mechanistic insight and rate constant estimation under controlled approximations. The Weighted Ensemble (WE) approach orchestrates many short, unbiased MD trajectories to capture rare events efficiently, enabling the computation of rate constants and mechanistic pathways. Tools like WESTPA 2.0 support scalable implementations and have been successfully applied to protein folding, association, and ligand dissociation [79,80]. Milestoning divides configuration space into discrete interfaces and reconstructs kinetics from trajectories initiated at these milestones, allowing estimation of mean first-passage times and dissociation rates with good efficiency [81,82]. Replica-based methods enhance sampling by selectively raising the effective solute temperature, followed by maintaining realistic solvent conditions, generating equilibrated ensembles that can be used for kinetic modelling or reweighting [83]. Gaussian accelerated MD (GaMD) applies a recoverable bias to smooth the energy landscape, accelerating conformational transitions and binding events by orders of magnitude. Hybrid approaches like GaMD + WE combine GaMD’s exploration power with WE’s accurate rate predictions [84,85,86]. Finally, Markov State Models aggregate diverse trajectory data from unbiased or enhanced sampling into kinetic networks that predict long-timescale dynamics; recent reviews outline best practices for Markov State Models construction and their integration with methods like WE [87] (Figure 4).
6. Druggability of Dynamic and Condensed States
6.1. Condensate Microenvironments and Small-Molecule Enrichment
Biomolecular condensates create physicochemical niches that can concentrate or exclude small molecules by orders of magnitude. A large-scale screen of ~1700 metabolites and approved drugs across four unrelated condensates found partition coefficients spanning ~106, with strong correlations across condensate types. A simple model using computed physicochemical descriptors, dominated by hydrophobicity/solubility, predicted enrichment, arguing that broad solvent compatibility rather than high-affinity site binding controls partitioning for many compounds [88,89]. Some condensates exhibit unique solvating environments that influence small-molecule partitioning beyond conventional affinity-based interactions. Complementary studies show that these chemical microenvironments can be systematically characterized and predicted using molecular descriptors, showing a new direction in “condensate-aware” medicinal chemistry that considers both binding and differential enrichment within condensates [90,91]. Together, these findings highlight medicinal chemistry strategies for tuning small-molecule enrichment in condensates, i.e., increasing hydrophobic surface area, adjusting hydrogen-bond donor and acceptor counts, and balancing ionizable groups to optimize both solubility and compatibility with condensate interiors. However, due to the diverse chemical nature of different condensates, empirical profiling remains essential for accurate prediction and optimization [88,90].
6.2. Ensemble-Based Druggability Profiling
For dynamic targets like Aβ and tau, which exist as structural ensembles and participate in condensates, druggability assessments require methods that go beyond static structures. Pocket discovery on trajectories leverages MD to identify transient or cryptic binding pockets using tools like TRAPP or FTMap, which detect ligandable hot spots across frames [92,93]. Ensemble docking across experimentally guided or MD-generated conformers has become a common approach for targeting disordered regions, with pipelines increasingly tailored for IDPs [94,95]. Chemical-free energy methods offer rigorous ligand ranking when binding modes are known and sampling is sufficient. Despite advances in automation and best-practice protocols, their reliability is higher for well-defined pockets (such as those in folded tau segments or Aβ oligomers) than for diffuse or interface-driven condensate environments [96,97].
6.3. Insights from Clinical Trials of Anti-Aβ Antibodies
Clinical trials of monoclonal antibodies targeting Aβ have provided key insights into which Aβ assemblies are tractable in humans, thereby shaping future small-molecule strategies. Central or N-terminal epitopes of soluble monomeric Aβ recognized by first-generation antibodies such as Solanezumab and Bapineuzumab reduce peripheral Aβ levels and fail to confer benefit in cognitive phase III trials. Such antibodies had no affinity with oligomeric and fibrillar forms, which become the most toxic to synapses of neurodegeneration [98]. Lecanemab, which preferentially binds to soluble Aβ protofibrils, demonstrated a statistically significant, though modest, slowing of cognitive decline in the phase 3 CLARITY-AD trial [19,99]. Mechanistic studies and phase 2/3 analyses support protofibril selectivity as a defining feature of its therapeutic profile [100,101]. Donanemab, by contrast, targets pyroglutamate-modified Aβ at position 3 (pGlu3-Aβ), an epitope enriched in dense-core plaques, and showed clinical benefit in the TRAILBLAZER-ALZ2 trial, as measured by the iADRS and additional cognitive endpoints [20]. Meta-analyses across anti-Aβ monoclonal antibodies indicate class-wide efficacy signals alongside amyloid-related imaging abnormalities, highlighting trade-offs in safety profiles depending on the targeted Aβ species [102]. Contrarily, Lecanemab and Donanemab were intended to bind protofibrils and pyroglutamate-modified Aβ (pGlu3-Aβ), respectively. N-terminal truncation resulting in the production of pGlu3-Aβ, which is a cyclized glutamate residue at position 3, is more hydrophobic, aggregation-prone, and resistant to proteolysis as compared to canonical Aβ1-42. It is also a seed of nucleation, which enhances the increase and propagation of amyloid. Selective clearance of this species using Donanemab will result in clearance of the most pathogenic aggregates and sparing of physiological Aβ pools, which has already demonstrated potent plaque-reduction and a lesser, but significant cognitive improvement in recent phase III studies [103].
6.4. Design Implications for Small Molecules and Biologics
If local enrichment within tau or Aβ condensates increases drug concentration by 10–1000-fold, even modest intrinsic affinity can yield meaningful effective potency. This highlights two linked optimization strategies, i.e., tuning condensate partitioning, guided by experimental measurements or predictive models, and optimizing target engagement in the condensate environment. Several studies now offer practical frameworks to support both, combining partition-coefficient assays (e.g., mass spectrometry, fluorescence microscopy) with machine learning–based predictors and ensemble-informed docking or free-energy calculations [88,90]. For biologics, epitope selection is equally critical, i.e., protofibril-selective antibodies target dynamic aggregates and may act synergistically with small molecules that alter condensate material properties or modulate oligomer kinetics.
7. Conclusions
Alzheimer’s disease involves two intrinsically disordered proteins, Aβ and tau, whose aggregation behavior is shaped by their sequence-encoded ensembles and context-dependent condensation. Three key insights emerge from the current review. First, sequence features dictate phase separation, conformational diversity, and the emergence of ligandable pockets. Second, structural studies across multiple scales are increasingly anchoring these dynamic states in experimental reality. Third, multiscale simulations using IDP-refined force fields, coarse-grained models, and data-driven reweighting quantitatively link molecular interactions to mesoscale behaviors like LLPS and oligomer formation. These advances support a shift toward context-dependent therapeutic design, where biophysical properties of the local environment are explicitly integrated into drug discovery strategies. A practical workflow involves assessing compound enrichment in condensates, probing cryptic pockets across ensembles, validating mechanisms with orthogonal data, and using enhanced sampling methods to resolve kinetic steps. This mirrors clinical lessons from anti-Aβ antibodies, where agents targeting protofibrils and plaque cores show distinct efficacy and safety profiles, and offers direction for tau-targeted strategies. Progress will depend on standardizing LLPS assays, cross-validating simulations, linking biophysical features to biomarkers, and enabling data sharing. With these tools, we can move beyond single-structure models and design therapies that reshape structural ensembles, modulate partitioning, and redirect aggregation kinetics, offering new hope for disease-modifying interventions for Alzheimer’s and other protein condensation disorders.
Author Contributions
Conceptualisation, K.B. and P.K.; Literature Review and Analysis, K.B., P.K., J.C., N.R.C. and D.D.; resources, K.B., P.K., A.B. and J.C.; data curation, A.B., D.D., N.R.C. and J.C.; writing—original draft preparation, K.B., P.K., J.C., N.R.C., A.B. and D.D.; writing—review and editing, K.B., P.K., J.C. and N.R.C.; supervision, K.B., P.K.; project administration, K.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work did not receive any external funding from any national or international agencies to declare.
Data Availability Statement
No new data were created or analysed in this study. Data sharing does not apply to this article.
Conflicts of Interest
The authors declare that they have no conflicts of interest, whether financial or non-financial.
References
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2014, 16, 18–29. [Google Scholar] [CrossRef]
- Heller, G.T.; Aprile, F.A.; Michaels, T.C.T.; Limbocker, R.; Perni, M.; Ruggeri, F.S.; Mannini, B.; Löhr, T.; Bonomi, M.; Camilloni, C.; et al. Small-molecule sequestration of amyloid-β as a drug discovery strategy for Alzheimer’s disease. Sci. Adv. 2020, 6, eabb5924. [Google Scholar] [CrossRef]
- Peng, H.; Hsu, W.; Wu, J. Liquid-liquid phase separation (LLPS) in cellular physiology and tumor biology. Am. J. Cancer Res. 2021, 11, 3766. [Google Scholar] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.A.G.; Fatima, N.; Jenkins, J.; O’sUllivan, T.J.; Schertel, A.; Halfon, Y.; Wilkinson, M.; Morrema, T.H.J.; Geibel, M.; Read, R.J.; et al. CryoET of β-amyloid and tau within postmortem Alzheimer’s disease brain. Nature 2024, 631, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef]
- Sherry, K.P.; Das, R.K.; Pappu, R.V.; Barrick, D. Control of transcriptional activity by design of charge patterning in the intrinsically disordered RAM region of the Notch receptor. Proc. Natl. Acad. Sci. USA 2017, 114, E9243–E9252. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid–Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus–Host Interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef]
- Meca, E.; Fritsch, A.W.; Iglesias-Artola, J.M.; Reber, S.; Wagner, B. Predicting disordered regions driving phase separation of proteins under variable salt concentration. Front. Phys. 2023, 11, 1213304. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef]
- Boyko, S.; Surewicz, W.K. Study of Tau Liquid-Liquid Phase Separation In Vitro. Methods Mol. Biol. 2023, 2551, 245–252. [Google Scholar] [CrossRef]
- Boyko, S.; Surewicz, K.; Surewicz, W.K. Regulatory mechanisms of tau protein fibrillation under the conditions of liquid–liquid phase separation. Proc. Natl. Acad. Sci. USA 2020, 117, 31882–31890. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Feng, S.; Li, Z.; Li, Y.; Reif, B.; Shi, B.; Niu, Z. Liquid–liquid phase separation of amyloid-β oligomers modulates amyloid fibrils formation. J. Biol. Chem. 2023, 299, 102926. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Murzin, A.G.; Falcon, B.; Epstein, A.; Machin, J.; Tempest, P.; Newell, K.L.; Vidal, R.; Garringer, H.J.; Sahara, N.; et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol. 2021, 141, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Lövestam, S.; Li, D.; Wagstaff, J.L.; Kotecha, A.; Kimanius, D.; McLaughlin, S.H.; Murzin, A.G.; Freund, S.M.V.; Goedert, M.; Scheres, S.H.W. Disease-specific tau filaments assemble via polymorphic intermediates. Nature 2023, 625, 119–125. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024, 81, 255–263. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Nery, E.S.M.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Gomes, G.-N.W.; Krzeminski, M.; Namini, A.; Martin, E.W.; Mittag, T.; Head-Gordon, T.; Forman-Kay, J.D.; Gradinaru, C.C. Conformational Ensembles of an Intrinsically Disordered Protein Consistent with NMR, SAXS, and Single-Molecule FRET. J. Am. Chem. Soc. 2020, 142, 15697–15710. [Google Scholar] [CrossRef]
- Müller-Späth, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Rüegger, S.; Reymond, L.; Nettels, D.; Schuler, B. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Choi, J.-M.; Holehouse, A.S.; Pappu, R.V. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys. 2020, 49, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Dignon, G.L.; Zheng, W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLoS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef] [PubMed]
- Boyko, S.; Qi, X.; Chen, T.-H.; Surewicz, K.; Surewicz, W.K. Liquid–liquid phase separation of tau protein: The crucial role of electrostatic interactions. J. Biol. Chem. 2019, 294, 11054–11059. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Hecht, M.H. Mutations Enhance the Aggregation Propensity of the Alzheimer’s Aβ Peptide. J. Mol. Biol. 2008, 377, 565–574. [Google Scholar] [CrossRef]
- Rezaei-Ghaleh, N.; Amininasab, M.; Giller, K.; Becker, S. Familial Alzheimer’s Disease-Related Mutations Differentially Alter Stability of Amyloid-Beta Aggregates. J. Phys. Chem. Lett. 2023, 14, 1427–1435. [Google Scholar] [CrossRef]
- Park, K.-W.; Wood, C.A.; Li, J.; Taylor, B.C.; Oh, S.; Young, N.L.; Jankowsky, J.L. Gene therapy using Aβ variants for amyloid reduction. Mol. Ther. 2021, 29, 2294–2307. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef]
- Graham, D.L.; Gray, A.J.; Joyce, J.A.; Yu, D.; O’Moore, J.; Carlson, G.A.; Shearman, M.S.; Dellovade, T.L.; Hering, H. Increased O-GlcNAcylation reduces pathological tau without affecting its normal phosphorylation in a mouse model of tauopathy. Neuropharmacology 2014, 79, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Hastings, N.B.; Wang, X.; Song, L.; Butts, B.D.; Grotz, D.; Hargreaves, R.; Hess, J.F.; Hong, K.-L.K.; Huang, C.R.-R.; Hyde, L.; et al. Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol. Neurodegener. 2017, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Permanne, B.; Sand, A.; Ousson, S.; Nény, M.; Hantson, J.; Schubert, R.; Wiessner, C.; Quattropani, A.; Beher, D. O-GlcNAcase Inhibitor ASN90 is a Multimodal Drug Candidate for Tau and α-Synuclein Proteinopathies. ACS Chem. Neurosci. 2022, 13, 1296–1314. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Shea, T.B. Caspase-Mediated Truncation of Tau Potentiates Aggregation. Int. J. Alzheimer’s Dis. 2012, 2012, 731063. [Google Scholar] [CrossRef]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.-X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- Wang, Y.P.; Biernat, J.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E.-M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Cheung, A.H.; Okon, M.; McIntosh, L.P.; Vocadlo, D.J. O-GlcNAc Modification of tau Directly Inhibits Its Aggregation without Perturbing the Conformational Properties of tau Monomers. J. Mol. Biol. 2014, 426, 1736–1752. [Google Scholar] [CrossRef]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; Lapointe, N.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef]
- Chu, D.; Yang, X.; Wang, J.; Zhou, Y.; Gu, J.-H.; Miao, J.; Wu, F.; Liu, F. Tau truncation in the pathogenesis of Alzheimer’s disease: A narrative review. Neural Regen. Res. 2023, 19, 1221–1232. [Google Scholar] [CrossRef]
- Gao, Y.-L.; Wang, N.; Sun, F.-R.; Cao, X.-P.; Zhang, W.; Yu, J.-T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Gallardo, R.; Ungureanu, A.-A.; Cano, V.C.; Snellinx, A.; Ramakers, M.; Bartic, C.; Rousseau, F.; Schymkowitz, J.; De Strooper, B. The Alzheimer Disease Protective Mutation A2T Modulates Kinetic and Thermodynamic Properties of Amyloid-β (Aβ) Aggregation. J. Biol. Chem. 2014, 289, 30977–30989. [Google Scholar] [CrossRef] [PubMed]
- Naudi-Fabra, S.; Tengo, M.; Jensen, M.R.; Blackledge, M.; Milles, S. Quantitative Description of Intrinsically Disordered Proteins Using Single-Molecule FRET, NMR, and SAXS. J. Am. Chem. Soc. 2021, 143, 20109–20121. [Google Scholar] [CrossRef] [PubMed]
- Metskas, L.A.; Rhoades, E. Single-Molecule FRET of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem. 2020, 71, 391–414. [Google Scholar] [CrossRef]
- Sil, S.; Datta, I.; Basu, S. Use of AI-methods over MD simulations in the sampling of conformational ensembles in IDPs. Front. Mol. Biosci. 2025, 12, 1542267. [Google Scholar] [CrossRef]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef]
- Lee, M.; Yau, W.-M.; Louis, J.M.; Tycko, R. Structures of brain-derived 42-residue amyloid-β fibril polymorphs with unusual molecular conformations and intermolecular interactions. Proc. Natl. Acad. Sci. USA 2023, 120, e2218831120. [Google Scholar] [CrossRef]
- Ozohanics, O.; Ambrus, A. Hydrogen-Deuterium Exchange Mass Spectrometry: A Novel Structural Biology Approach to Structure, Dynamics and Interactions of Proteins and Their Complexes. Life 2020, 10, 286. [Google Scholar] [CrossRef]
- Seetaloo, N.; Zacharopoulou, M.; Stephens, A.D.; Schierle, G.S.K.; Phillips, J.J. Millisecond Hydrogen/Deuterium-Exchange Mass Spectrometry Approach to Correlate Local Structure and Aggregation in α-Synuclein. Anal. Chem. 2022, 94, 16711–16719. [Google Scholar] [CrossRef]
- Han, Y.; Desai, A.A.; Zupancic, J.M.; Smith, M.D.; Tessier, P.M.; Ruotolo, B.T. Native ion mobility-mass spectrometry reveals the binding mechanisms of anti-amyloid therapeutic antibodies. Protein Sci. 2024, 33, e5008. [Google Scholar] [CrossRef]
- Boyko, S.; Surewicz, W.K. Tau liquid–liquid phase separation in neurodegenerative diseases. Trends Cell Biol. 2022, 32, 611–623. [Google Scholar] [CrossRef]
- Ainani, H.; Bouchmaa, N.; Ben Mrid, R.; El Fatimy, R. Liquid-liquid phase separation of protein tau: An emerging process in Alzheimer’s disease pathogenesis. Neurobiol. Dis. 2023, 178, 106011. [Google Scholar] [CrossRef]
- Morris, O.M.; Toprakcioglu, Z.; Röntgen, A.; Cali, M.; Knowles, T.P.J.; Vendruscolo, M. Aggregation of the amyloid-β peptide (Aβ40) within condensates generated through liquid–liquid phase separation. Sci. Rep. 2024, 14, 22633. [Google Scholar] [CrossRef] [PubMed]
- Šneiderienė, G.; Díaz, A.G.; Das Adhikari, S.; Wei, J.; Michaels, T.; Šneideris, T.; Linse, S.; Vendruscolo, M.; Garai, K.; Knowles, T.P.J. Lipid-induced condensate formation from the Alzheimer’s Aβ peptide triggers amyloid aggregation. Proc. Natl. Acad. Sci. USA 2025, 122, e2401307122. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Sun, H.; Cai, Q.; Tai, H.-C. The Enigma of Tau Protein Aggregation: Mechanistic Insights and Future Challenges. Int. J. Mol. Sci. 2024, 25, 4969. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, A.R.; Collepardo-Guevara, R.; Ramírez, J.; Espinosa, J.R. Time-Dependent Material Properties of Aging Biomolecular Condensates from Different Viscoelasticity Measurements in Molecular Dynamics Simulations. J. Phys. Chem. B 2023, 127, 4441–4459. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Kim, T.H.; Das, S.; Pal, T.; Wessén, J.; Rangadurai, A.K.; Kay, L.E.; Forman-Kay, J.D.; Chan, H.S. Electrostatics of salt-dependent reentrant phase behaviors highlights diverse roles of ATP in biomolecular condensates. eLife 2025, 13, RP100284. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.; Ma, Y.; Zhong, D.; Hou, S. Study liquid–liquid phase separation with optical microscopy: A methodology review. APL Bioeng. 2023, 7, 021502. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Henriques, J.; Skepö, M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: On the Accuracy of the TIP4P-D Water Model and the Representativeness of Protein Disorder Models. J. Chem. Theory Comput. 2016, 12, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Hamid, M.K.; Månsson, L.K.; Meklesh, V.; Persson, P.; Skepö, M. Molecular dynamics simulations of the adsorption of an intrinsically disordered protein: Force field and water model evaluation in comparison with experiments. Front. Mol. Biosci. 2022, 9, 958175. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-P.; Chen, J.; Van Voorhis, T. Systematic Parametrization of Polarizable Force Fields from Quantum Chemistry Data. J. Chem. Theory Comput. 2013, 9, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-P.; Martinez, T.J.; Pande, V.S. Building Force Fields: An Automatic, Systematic, and Reproducible Approach. J. Phys. Chem. Lett. 2014, 5, 1885–1891. [Google Scholar] [CrossRef]
- Polêto, M.D.; Lemkul, J.A. Integration of experimental data and use of automated fitting methods in developing protein force fields. Commun. Chem. 2022, 5, 38. [Google Scholar] [CrossRef]
- Wang, Y.; Fass, J.; Kaminow, B.; Herr, J.E.; Rufa, D.; Zhang, I.; Pulido, I.; Henry, M.; Macdonald, H.E.B.; Takaba, K.; et al. End-to-end differentiable construction of molecular mechanics force fields. Chem. Sci. 2022, 13, 12016–12033. [Google Scholar] [CrossRef]
- Schoenholz, S.S.; Cubuk, E.D. JAX, MD A framework for differentiable physics*. J. Stat. Mech. Theory Exp. 2021, 2021, 124016. [Google Scholar] [CrossRef]
- Takaba, K.; Friedman, A.J.; Cavender, C.E.; Behara, P.K.; Pulido, I.; Henry, M.M.; MacDermott-Opeskin, H.; Iacovella, C.R.; Nagle, A.M.; Payne, A.M.; et al. Machine-learned molecular mechanics force fields from large-scale quantum chemical data. Chem. Sci. 2024, 15, 12861–12878. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- Thomasen, F.E.; Skaalum, T.; Kumar, A.; Srinivasan, S.; Vanni, S.; Lindorff-Larsen, K. Rescaling protein-protein interactions improves Martini 3 for flexible proteins in solution. Nat. Commun. 2024, 15, 6645. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brasnett, C.; Borges-Araújo, L.; Souza, P.C.T.; Marrink, S.J. Martini3-IDP: Improved Martini 3 force field for disordered proteins. Nat. Commun. 2025, 16, 2874. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.A.; Reinhardt, A.; Aguirre, A.; Chew, P.Y.; Russell, K.O.; Espinosa, J.R.; Garaizar, A.; Collepardo-Guevara, R. Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy. Nat. Comput. Sci. 2021, 1, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Tesei, G.; Lindorff-Larsen, K. Improved predictions of phase behaviour of intrinsically disordered proteins by tuning the interaction range. Open Res. Eur. 2023, 2, 94. [Google Scholar] [CrossRef]
- Rauh, A.S.; Tesei, G.; Lindorff-Larsen, K. A coarse-grained model for disordered proteins under crowded conditions. Protein Sci. 2025, 34, e70232. [Google Scholar] [CrossRef]
- Bottaro, S.; Bengtsen, T.; Lindorff-Larsen, K. Integrating Molecular Simulation and Experimental Data: A Bayesian/Maximum Entropy Reweighting Approach. Methods Mol. Biol. 2020, 2112, 219–240. [Google Scholar] [CrossRef]
- Bonomi, M.; Camilloni, C.; Cavalli, A.; Vendruscolo, M. Metainference: A Bayesian inference method for heterogeneous systems. Sci. Adv. 2016, 2, e1501177. [Google Scholar] [CrossRef]
- Bonomi, M.; Camilloni, C.; Vendruscolo, M. Metadynamic metainference: Enhanced sampling of the metainference ensemble using metadynamics. Sci. Rep. 2016, 6, 31232. [Google Scholar] [CrossRef]
- Zuckerman, D.M.; Chong, L.T. Weighted Ensemble Simulation: Review of Methodology, Applications, and Software. Annu. Rev. Biophys. 2017, 46, 43–57. [Google Scholar] [CrossRef]
- Russo, J.D.; Zhang, S.; Leung, J.M.G.; Bogetti, A.T.; Thompson, J.P.; DeGrave, A.J.; Torrillo, P.A.; Pratt, A.J.; Wong, K.F.; Xia, J.; et al. WESTPA 2.0: High-Performance Upgrades for Weighted Ensemble Simulations and Analysis of Longer-Timescale Applications. J. Chem. Theory Comput. 2022, 18, 638–649. [Google Scholar] [CrossRef]
- Elber, R. Milestoning: An Efficient Approach for Atomically Detailed Simulations of Kinetics in Biophysics. Annu. Rev. Biophys. 2020, 49, 69–85. [Google Scholar] [CrossRef]
- Rathnayake, S.; Narayan, B.; Elber, R.; Wong, C.F. Milestoning simulation of ligand dissociation from the glycogen synthase kinase 3β. Proteins Struct. Funct. Bioinform. 2023, 91, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Arantes, P.R.; Bhattarai, A.; Hsu, R.V.; Pawnikar, S.; Huang, Y.M.; Palermo, G.; Miao, Y. Gaussian accelerated molecular dynamics (GaMD): Principles and applications. Wiley interdisciplinary reviews. Comput. Mol. Sci. 2021, 11, e1521. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef]
- Ahn, S.-H.; Ojha, A.A.; Amaro, R.E.; McCammon, J.A. Gaussian-Accelerated Molecular Dynamics with the Weighted Ensemble Method: A Hybrid Method Improves Thermodynamic and Kinetic Sampling. J. Chem. Theory Comput. 2021, 17, 7938–7951. [Google Scholar] [CrossRef]
- Konovalov, K.A.; Unarta, I.C.; Cao, S.; Goonetilleke, E.C.; Huang, X. Markov State Models to Study the Functional Dynamics of Proteins in the Wake of Machine Learning. JACS Au 2021, 1, 1330–1341. [Google Scholar] [CrossRef]
- Thody, S.A.; Clements, H.D.; Baniasadi, H.; Lyon, A.S.; Sigman, M.S.; Rosen, M.K. Small-molecule properties define partitioning into biomolecular condensates. Nat. Chem. 2024, 16, 1794–1802. [Google Scholar] [CrossRef]
- Johnson, R. Predicting small-molecule partitioning. Nat. Chem. Biol. 2024, 20, 1388. [Google Scholar] [CrossRef]
- Kilgore, H.R.; Mikhael, P.G.; Overholt, K.J.; Boija, A.; Hannett, N.M.; Van Dongen, C.; Lee, T.I.; Chang, Y.-T.; Barzilay, R.; Young, R.A. Distinct chemical environments in biomolecular condensates. Nat. Chem. Biol. 2024, 20, 291–301. [Google Scholar] [CrossRef]
- Kilgore, H.R.; Young, R.A. Learning the chemical grammar of biomolecular condensates. Nat. Chem. Biol. 2022, 18, 1298–1306. [Google Scholar] [CrossRef]
- Stank, A.; Kokh, D.B.; Horn, M.; Sizikova, E.; Neil, R.; Panecka, J.; Richter, S.; Wade, R.C. TRAPP webserver: Predicting protein binding site flexibility and detecting transient binding pockets. Nucleic Acids Res. 2017, 45, W325–W330. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Bowman, G.R.; Juarez-Jimenez, J.; Michel, J.; Gervasio, F.L. Investigating Cryptic Binding Sites by Molecular Dynamics Simulations. Accounts Chem. Res. 2020, 53, 654–661. [Google Scholar] [CrossRef]
- Chen, Q.-H.; Krishnan, V.V. Identification of ligand binding sites in intrinsically disordered proteins with a differential binding score. Sci. Rep. 2021, 11, 22583. [Google Scholar] [CrossRef]
- Dhar, A.; Sisk, T.R.; Robustelli, P. Ensemble Docking for Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2025, 65, 6847–6860. [Google Scholar] [CrossRef] [PubMed]
- Mey, A.S.J.S.; Allen, B.K.; Macdonald, H.E.B.; Chodera, J.D.; Hahn, D.F.; Kuhn, M.; Michel, J.; Mobley, D.L.; Naden, L.N.; Prasad, S.; et al. Best Practices for Alchemical Free Energy Calculations [Article v1.0]. Living J. Comput. Mol. Sci. 2020, 2, 18378. [Google Scholar] [CrossRef] [PubMed]
- Gapsys, V.; Yildirim, A.; Aldeghi, M.; Khalak, Y.; van der Spoel, D.; de Groot, B.L. Accurate absolute free energies for ligand–protein binding based on non-equilibrium approaches. Commun. Chem. 2021, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Nussinov, R.; Ma, B. Mechanisms of recognition of amyloid-β (Aβ) monomer, oligomer, and fibril by homologous antibodies. J. Biol. Chem. 2017, 292, 18325–18343. [Google Scholar] [CrossRef]
- Cohen, S.; van Dyck, C.H.; Gee, M.; Doherty, T.; Kanekiyo, M.; Dhadda, S.; Li, D.; Hersch, S.; Irizarry, M.; Kramer, L.D. Lecanemab Clarity AD: Quality-of-Life Results from a Randomized, Double-Blind Phase 3 Trial in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2023, 10, 771–777. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Jeremic, D.; Navarro-López, J.D.; Jiménez-Díaz, L. Efficacy and safety of anti-amyloid-β monoclonal antibodies in current Alzheimer’s disease phase III clinical trials: A systematic review and interactive web app-based meta-analysis. Ageing Res. Rev. 2023, 90, 102012. [Google Scholar] [CrossRef]
- Osaka, H.; Nishida, K.; Kanazawa, T. Beyond lecanemab: Examining Phase III potential in Alzheimer’s therapeutics. Psychiatry Clin. Neurosci. Rep. 2024, 3, e185. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).