Abstract
This study presents analytical models for simulating the thermal properties of linear triatomic systems, using the modified Rosen–Morse oscillator and harmonic oscillator potential to represent vibrational modes. The models employ existing partition functions to derive the thermodynamic functions for the symmetric, asymmetric, and 2-fold degenerate bending modes. These thermodynamic functions are applied to gaseous triatomic molecules such as BO2, HCN, N3, and Si2N. The results demonstrate high accuracy, with mean percentage absolute deviations (MPAD) of less than 0.17% for molar entropy and Gibbs free energy. For enthalpy and heat capacity, MPAD values are below 2% compared to National Institute of Standards and Technology (NIST) data. The findings are in strong agreement with the existing literature on gaseous triatomic molecules, confirming the reliability of the proposed models.
1. Introduction
Thermodynamics is a branch of physical science focused on the study of heat, work, energy, and how these quantities interact with matter. It provides insights into energy conversion processes and the transformation of energy between systems. Thermodynamic processes are governed by key state variables, such as temperature, pressure, volume, and energy, and are described by the following four fundamental laws: the Zeroth, First (energy conservation), Second (entropy), and Third (Nernst heat theorem) laws [1].
A deep understanding of thermodynamics is essential for advancing technologies such as fuel cells, enhanced oil recovery techniques, carbon capture and storage systems, and battery management systems. It also plays a critical role in manufacturing heat exchangers, which are widely used in industries like nuclear power, dyeing and printing, and steel production [2,3,4,5]. Furthermore, thermodynamics aids in determining equilibrium constants in chemical reactions [6,7,8], with applications in hydrogen production, carbon emission control, water treatment, material selection, and corrosion prevention [9].
Quantum mechanical methods, particularly non-fitting models, are increasingly embraced for analyzing the thermodynamic properties of molecular systems. These models offer a computationally efficient alternative for studying diatomic and polyatomic molecules. Fitted models, while supported by experimental data to improve accuracy, rely on data availability and are system-specific. They also require new data for different systems or conditions and are sensitive to temperature and pressure, limiting their broader use. In contrast, non-fitting models, based on fundamental physical principles, do not rely on experimental data and are valuable when such data are unavailable, offering a simplified approach to predicting thermal properties.
In polyatomic molecule analysis, both the number of atoms (atomicity) and the molecular shape are fundamental to identifying the vibrational modes. Each of these modes is associated with an oscillator similar to that of a diatomic molecule. The energy levels and partition function can then be calculated to explore different thermodynamic models. For a linear polyatomic molecule with N atoms, the number of vibrational modes is determined by the formula 3N–5.
A diatomic molecule (N = 2) has only one vibrational mode—the symmetric stretching mode. Different oscillator models, such as exponential-type and hyperbolic-type models, have been used to represent these vibrational modes [10,11,12,13,14,15]. Thermodynamic models derived from these oscillators, such as Helmholtz free energy, Gibbs free energy, mean thermal energy, heat capacities, enthalpy, and entropy, have been applied to various diatomic molecules [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31].
Despite these successes, the thermodynamic properties of triatomic molecules remain underexplored, particularly when analyzed using non-fitting computational models. Analytical expressions for Gibbs free energy, entropy, heat capacity, and enthalpy have been derived by using appropriate representations of the vibrational, rotational, and translational partition functions, and by being applied to molecules such as MgCl2, Si2N, BeF2, CNC, BO2, and N3 [32]. Non-fitting models have also been employed to study the thermodynamic properties of nonlinear triatomic molecules, such as NO2, BF2, and AlCl2 [33]. However, the exploration of thermodynamic properties using these non-fitting models remains limited. Further studies on polyatomic molecules using non-fitting models are listed in [34,35,36,37,38,39,40].
Unlike diatomic molecules, which have a single vibrational mode, triatomic molecules are more complex. A linear triatomic molecule has the following four vibrational modes: symmetric, asymmetric, and two bending modes (degenerate). Thus, accurately formulating the thermodynamic functions for these molecules requires incorporating all four vibrational modes. Previous models for both linear and nonlinear polyatomic systems have used the improved Tietz (IMTZ) oscillator [10] to approximate the internal stretching vibrations. These models are based on three molecular constants (equilibrium dissociation energy, chemical bond length, and harmonic frequency of vibration) and two oscillator parameters (screening parameter α and dimensionless parameter q). A key limitation of the IMTZ model is the need to adjust the dimensionless parameter for each molecular system.
To address this issue, this study employs the modified Rosen–Morse (MRM) oscillator [11], which has proven effective in modeling the internuclear potential energy curve of diatomic molecules and handling anharmonicity in molecular systems. The MRM oscillator is widely used in fields such as solid-state physics, chemical physics, computational chemistry, and material science. Despite its versatility, the MRM oscillator has yet to be applied to study the thermodynamic properties of linear triatomic molecules. This study is the first to employ the MRM oscillator to derive computational models for the thermodynamic properties of these molecules, providing novel insights into the thermodynamics of triatomic systems.
The paper is organized as follows: Section 2 derives the thermodynamic functions; Section 3 uses these functions to generate numerical data for linear triatomic molecules such as BO2 (boron oxide), HCN (hydrogen cyanide), N3 (azide), and Si2N (silicon nitride); and Section 4 provides a brief conclusion.
2. Computational Method
This section outlines the computational models that do not rely on fitting parameters for determining the thermodynamic properties of linear triatomic molecules. The non-fitting computational method uses quantum mechanics to develop model equations that analyze the physical properties of substances. Unlike the fitting functional approach, which is restricted to systems or conditions similar to those used for parameter calibration, the non-fitting method is more adaptable and can be applied to a variety of chemical and physical systems. In this approach, the thermal properties of a linear triatomic molecule are predicted through analytical formulations that start with the canonical partition function, given by , where , , and correspond to the vibrational, rotational, and translational partition functions, respectively [30].
2.1. Formulating the Vibrational Partition Function
In a linear triatomic molecule of the form XZY, where X, Y, and Z represent atoms, the system exhibits the following three distinct vibrational modes: symmetric stretching, asymmetric stretching, and two degenerate bending modes. The vibrational partition function is expressed as , where , , and represent the partition functions for the symmetric, asymmetric, and bending vibrations, respectively. The square in accounts for the two-fold degeneracy of the bending modes. If the molecule consists of more than three atoms while remaining linear, additional vibrational modes emerge, requiring an adjustment to the vibrational partition function to include these modes.
In a linear triatomic molecule XZY, where atoms X, Y, and Z are bonded in a straight line, symmetric stretching occurs when both the X-Z and Z-Y bonds stretch and compress in unison. If atoms X and Y are identical, the central atom Z shifts along the molecular axis, and both X and Y move symmetrically toward or away from Z. Since X and Y are the same, their movements are identical, resulting in equal changes in bond lengths and preserving the molecule’s symmetry.
In an asymmetric linear triatomic molecule, where X, Y, and Z are all different, the central atom Z moves along the molecular axis, and atoms X and Y move in phase. However, due to differences in atomic size, mass, and electronegativity, the displacements of X and Y are unequal. This leads to one bond (X-Z or Z-Y) stretching more than the other, causing an asymmetric vibration despite the motion occurring in phase.
However, anharmonicity causes deviations from perfect harmonic motion, particularly at higher energy levels, leading to the uneven spacing of vibrational energy levels. While previous studies modeled anharmonicity using the IMTZ potential, the present work employs the simpler MRM oscillator, which avoids the need for an adjustable dimensionless parameter that varies across molecules. The energy–distance relationship for a system modeled by the MRM oscillator is given by [11]
where
is the potential screening parameter; is the equilibrium dissociation energy; is the equilibrium force constant for symmetric vibrations; is the reduced mass of atoms X and Y; c is the speed of light; is the symmetric harmonic vibrational frequency; and the parameters and are defined as and , where is the equilibrium bond length between atoms Z and X, is the equilibrium bond length between atoms Z and Y, W is the Lambert W function, and r is the internuclear separation between atoms X and Y.
Deng and Jia previously derived the vibrational partition function for a diatomic molecule using the MRM oscillator. Their expression for the symmetric stretching vibrations is adopted here, given by [41]
In Equation (3), , where β−1 = kBT (with T representing the temperature and kB the Boltzmann constant); Erfi(z) denotes the imaginary error function evaluated at z. refers to the number of excited bonded molecular states, which can be deduced by applying , where ν = 0, 1, 2, … is the vibrational quantum number, Eν represents the vibrational energy eigenvalues, ℏ is the reduced Planck constant, and λ is an optimization parameter. As noted in Ref. [42], is negligibly small for the molecular system. By neglecting the terms involving this factor, the partition function simplifies to
In the asymmetric stretching vibration, the bonds (X-Z and Z-Y) stretch and compress in opposite directions, causing unequal changes in bond lengths. During this vibration, atom Z moves along the molecular axis, while atoms X and Y move in opposite directions. This mode is termed “asymmetric” because the two sides of Z behave differently. The bending vibration, on the other hand, changes the angle between the bonds (X-Z and Z-Y), rather than the bond lengths. In this mode, atoms X and Y move perpendicular to the molecular axis, causing the bond angle to increase or decrease. There are two degenerate bending modes with the same frequency but different atomic motion directions, typically referred to as in-plane and out-of-plane bending. These bending vibrations occur at lower frequencies than the stretching modes.
Anharmonicity plays an important role in selecting oscillator models to represent the internal motion of molecular systems. It refers to how molecular vibrations deviate from the idealized harmonic oscillator behavior. In simpler systems, deviations are typically less pronounced in asymmetric and bending modes than in symmetric stretching modes. At typical temperatures, these deviations are smaller in asymmetric and bending vibrations compared to symmetric stretching vibrations, likely due to the lower vibrational frequencies of the atoms involved. Therefore, at standard temperatures, these vibrations can often be accurately modeled using harmonic oscillator potentials. As a result, the partition functions for the asymmetric and bending modes are given in compact form as [43]
where and are the equilibrium frequencies for the asymmetric and bending vibrations, respectively.
2.2. Formulating the Rotational and Translational Partition Functions
Using the rigid rotor approximation for molecular systems and neglecting molecular interactions, the rotational and translational partition functions can be expressed as [43]
In these expressions, , m denotes the molar mass, and p represents the pressure exerted by the system in a container of volume V.
2.3. Formulating the Thermodynamic Functions
In this work, analytical equations for assessing the thermal properties of a linear triatomic molecule are derived from the following expressions [43]
In these equations, , , and the Avogadro number, , with R denotes the universal gas constant. Additionally, S represents the molar entropy, H the molar enthalpy, G the molar Gibbs free energy, and is the constant-pressure (or isobaric) molar heat capacity. The thermodynamic functions are computed by substituting and into Equations (8)–(11). The first- and second-order derivatives required for these evaluations are derived from Equations (4)–(7), with the corresponding expressions for the first and second derivatives provided in (12) and (13), respectively.
This work introduces the application of these equations specifically to the thermal properties of linear triatomic molecules, providing new insights into the calculation of thermodynamic properties. The use of the MRM oscillator, a tool previously underexplored in this context, represents a significant advancement in computational thermodynamics.
3. Results and Discussion
The accuracy of the new model equations for analyzing the thermal properties of linear triatomic molecules is assessed using data on equilibrium dissociation energy (), harmonic vibrational frequencies (, , ), and bond lengths (, ). The molecules considered, including boron oxide (BO2), hydrogen cyanide (HCN), azide (N3), and silicon nitride (Si2N), are selected for their relevance to computational and theoretical chemistry, atomic and molecular physics, materials science, and chemical physics. The equilibrium dissociation energy represents the energy required to break a molecule XZY into X and ZY. For these molecules, the dissociation reactions are as follows: BO2 dissociates into O and BO [44], HCN dissociates into H and CN [45], N3 dissociates into N and N2 [46], and Si2N dissociates into Si and NSi [47]. Model parameters, along with experimental values for , , , , , and , are provided in Table 1, with data drawn from Refs. [32,34,44,45,46,47].

Table 1.
Equilibrium dissociation energy, bond lengths, vibrational frequencies, and screening and optimization parameters for linear triatomic molecules in this study.
3.1. Significance of the Optimization Parameter
Before exploring the applicability of the model equations, it is important to consider the role of the optimization parameter, λ, introduced into the partition function. By expressing the symmetric partition function as , where F(β) is solely a function of temperature, the combined partition function , where , can also be expressed as
The first derivative of this equation is given by
Equation (14) clearly depends on λ, which in turn affects the molar entropy and Gibbs free energy (Equations (8) and (10)). However, Equation (15) is independent of λ, meaning the molar enthalpy (Equation (9)) and heat capacity (Equation (11)) are unaffected by changes in the optimization parameter. Setting λ = 0 effectively disables the optimization parameter, which, in turn, recovers the original model equations. This recovery ensures that the system returns to the unoptimized form, where the entropy and Gibbs free energy models are no longer adjusted by λ, but the enthalpy and heat capacity remain unchanged. The optimization parameter, λ, is varied manually until optimized data for molar entropy and reduced Gibbs free energy are achieved.
3.2. Validating the Thermodynamic Models
This section explores the use of model equations to evaluate the thermodynamic properties of linear triatomic molecules, as listed in Table 1. The model equations generate numerical data, which are then compared to available experimental data. The accuracy of the model is assessed based on Lippincott’s specifications, which dictate that the mean percentage absolute deviation (MPAD) between observed and experimental values should not exceed 1%.
To calculate the MPAD, the following equation is applied: , where Np denotes the total number of experimental data points and represents the percentage absolute deviation [25]. The terms Aj and Bj refer to the predicted and experimental values at data point j, respectively. One source of experimental data for comparison is the NIST database [48]. NIST employs a fitting functional approach, using various fitting parameters to model the thermal properties of a substance, though its applicability is limited to certain temperature and pressure ranges.
The approach presented here offers a more efficient alternative by reducing the need for extensive experimental setups and the large number of fitting parameters used in the NIST method. Instead, only a few key molecular constants—such as equilibrium dissociation energy, vibrational frequencies, and bond lengths—are required to predict the thermal properties of linear triatomic systems. Since NIST results for molar enthalpy and Gibbs free energy are standardized to molar enthalpy at T = 298.15 K and p = 0.1 MPa, this study introduces theoretical adjustments to calibrate enthalpy and Gibbs free energy using the following equations
Here, H0 represents the enthalpy calculated from Equation (9) at T = 298.15 K and p = 0.1 MPa. The thermodynamic functions from Equations (8), (11), (16) and (17) are applied to the data in Table 1, where pressure is held constant at 0.1 MPa and temperature varies from 300 to 6000 K. MATLAB (version 2016a) is utilized for numerical computations and to generate graphical plots. The program includes molecular constants and other necessary parameters, such as those defining the canonical partition function.
The computational procedure is illustrated using data for the HCN molecule. When T = 298.15 K, p = 0.1 MPa, and λ is set to zero, the dimensionless parameters γ1 and γ2 are calculated as −20.3332 and 10.2903, respectively. These values are substituted into Equation (9), yielding H0 = 51.48 kJmol−1. This value of H0 is required for the calculation of HSTA and GSTA at other temperatures.
When T = 300 K, p = 0.1 MPa, and λ set to 2.3, the dimensionless parameters are γ1 = −20.2361 and γ2 = 8.9915. These values are inserted into Equations (8), (11), (16) and (17), resulting in S = 200.095 Jmol−1K−1, Cp = 38.146 Jmol−1K−1, HSTA = 0.070 kJmol−1, and GSTA = 199.860 Jmol−1K−1. The same procedure is repeated for the other temperature values.
The program algorithm also calculates the MPAD values, enabling λ to be adjusted gradually to optimize S and GSTA. The numerical results for BO2, HCN, N3, and Si2N, along with the NIST data and MPAD values, are summarized in Table 2, Table 3, Table 4 and Table 5. A MATLAB script for computing the thermodynamic properties of nonlinear triatomic molecules is available upon request.
Table 2.
Comparison of thermodynamic properties of the BO2 molecule at 0.1 MPa: NIST data vs. MRM model predictions.
Table 3.
Comparison of thermodynamic properties of the HCN molecule at 0.1 MPa: NIST data vs. MRM model predictions.
Table 4.
Comparison of thermodynamic properties of the N3 molecule at 0.1 MPa: NIST data vs. MRM model predictions.
Table 5.
Comparison of thermodynamic properties of the Si2N molecule at 0.1 MPa: NIST data vs. MRM model predictions.
The results show that the MPAD values for all molecules fall within the acceptable range. However, the models for enthalpy and heat capacity slightly deviate from the NIST data for the HCN molecule, with the MPAD exceeding the Lippincott error threshold. These discrepancies may result from the exclusion of quantum correction terms in the partition function for the MRM oscillator [41]. Nevertheless, the models for molar enthalpy and heat capacity provide accurate predictions for most molecules.
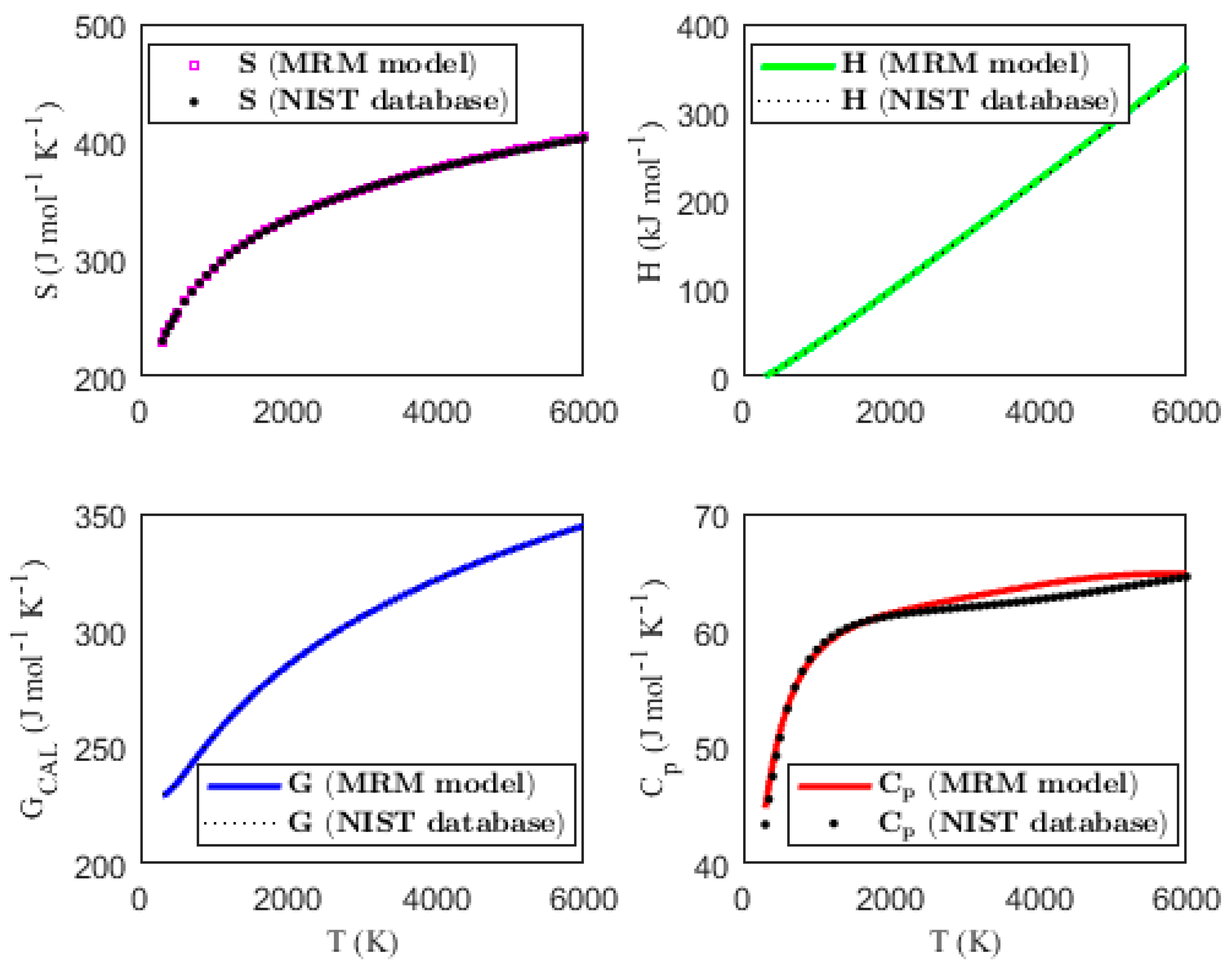
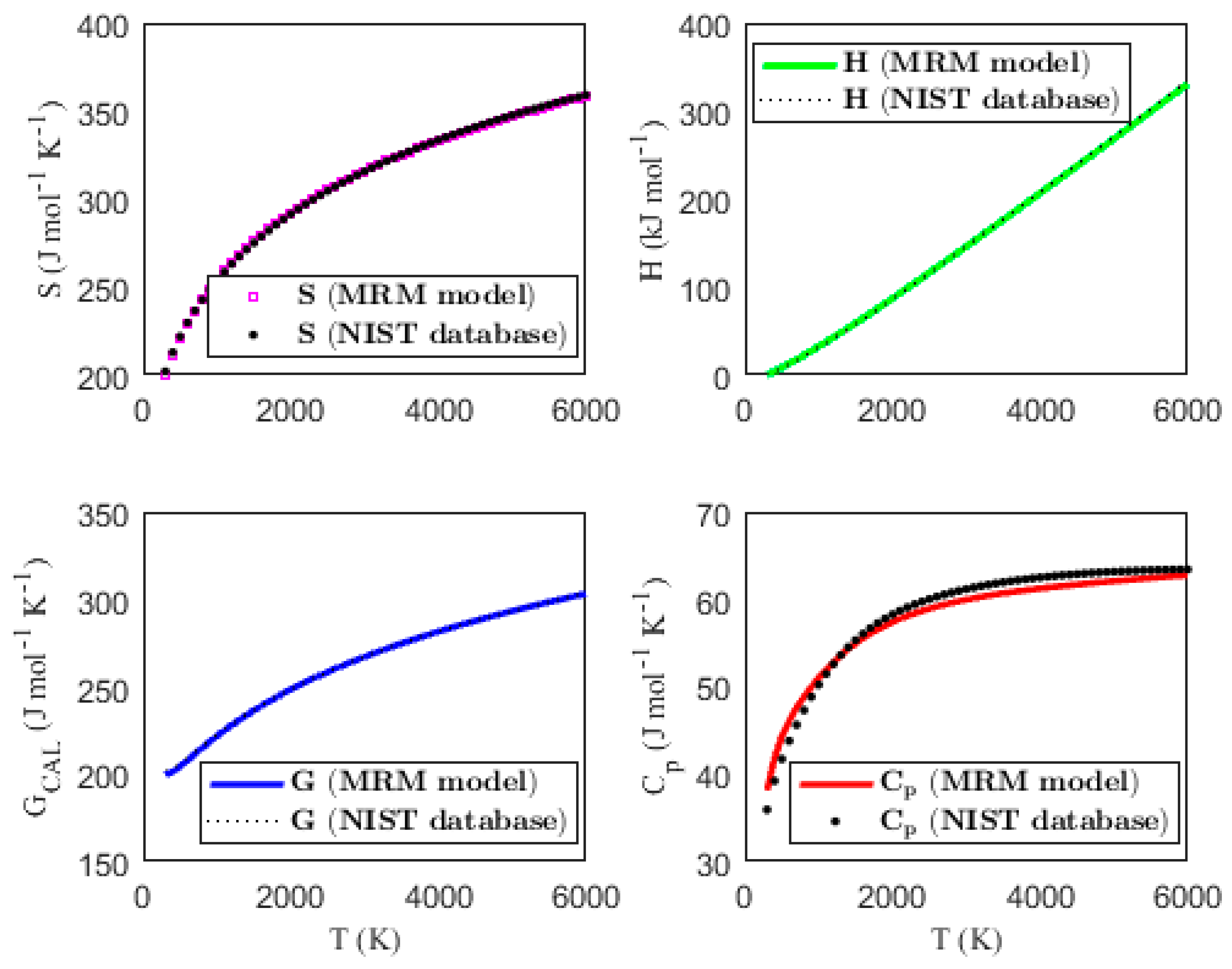
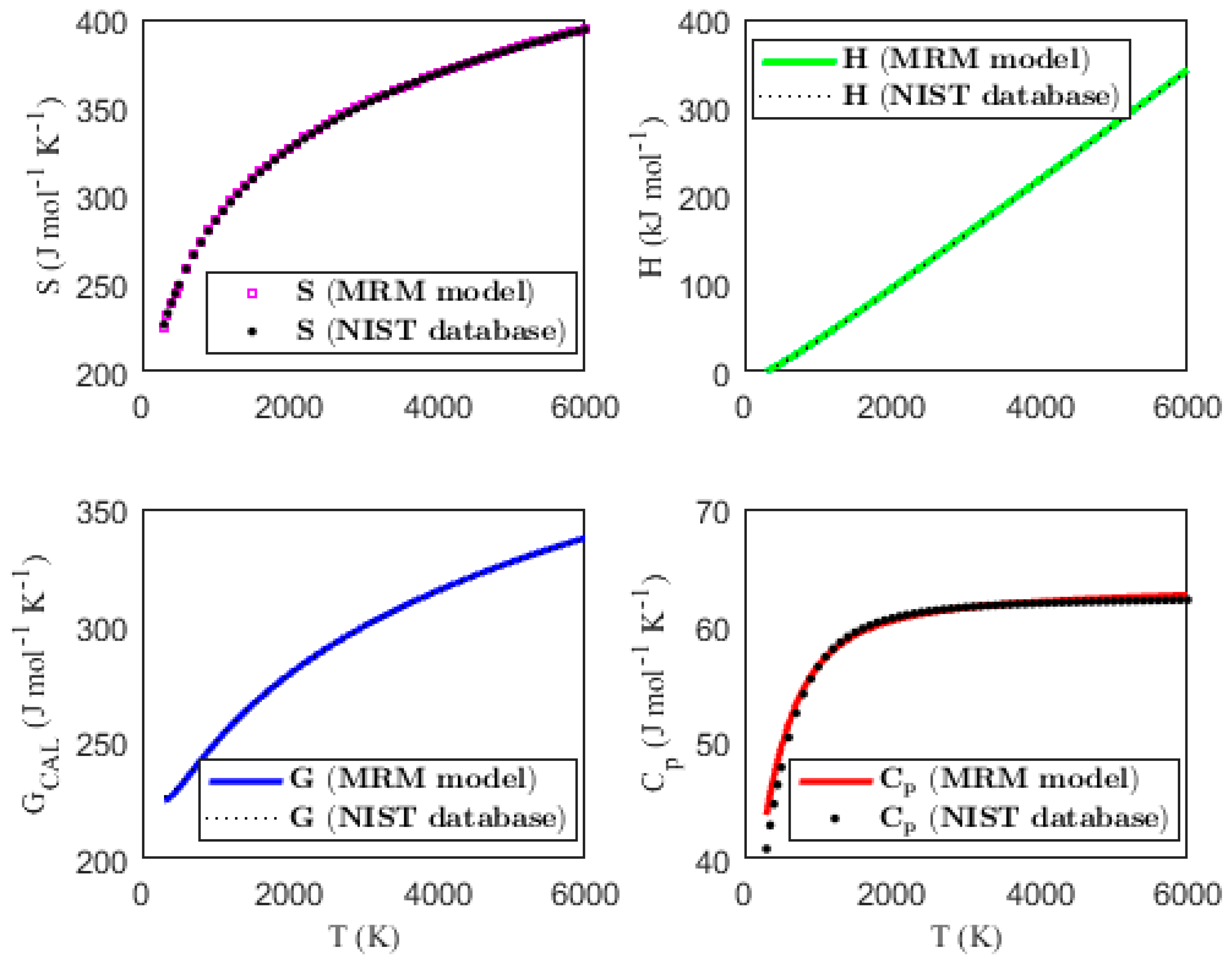
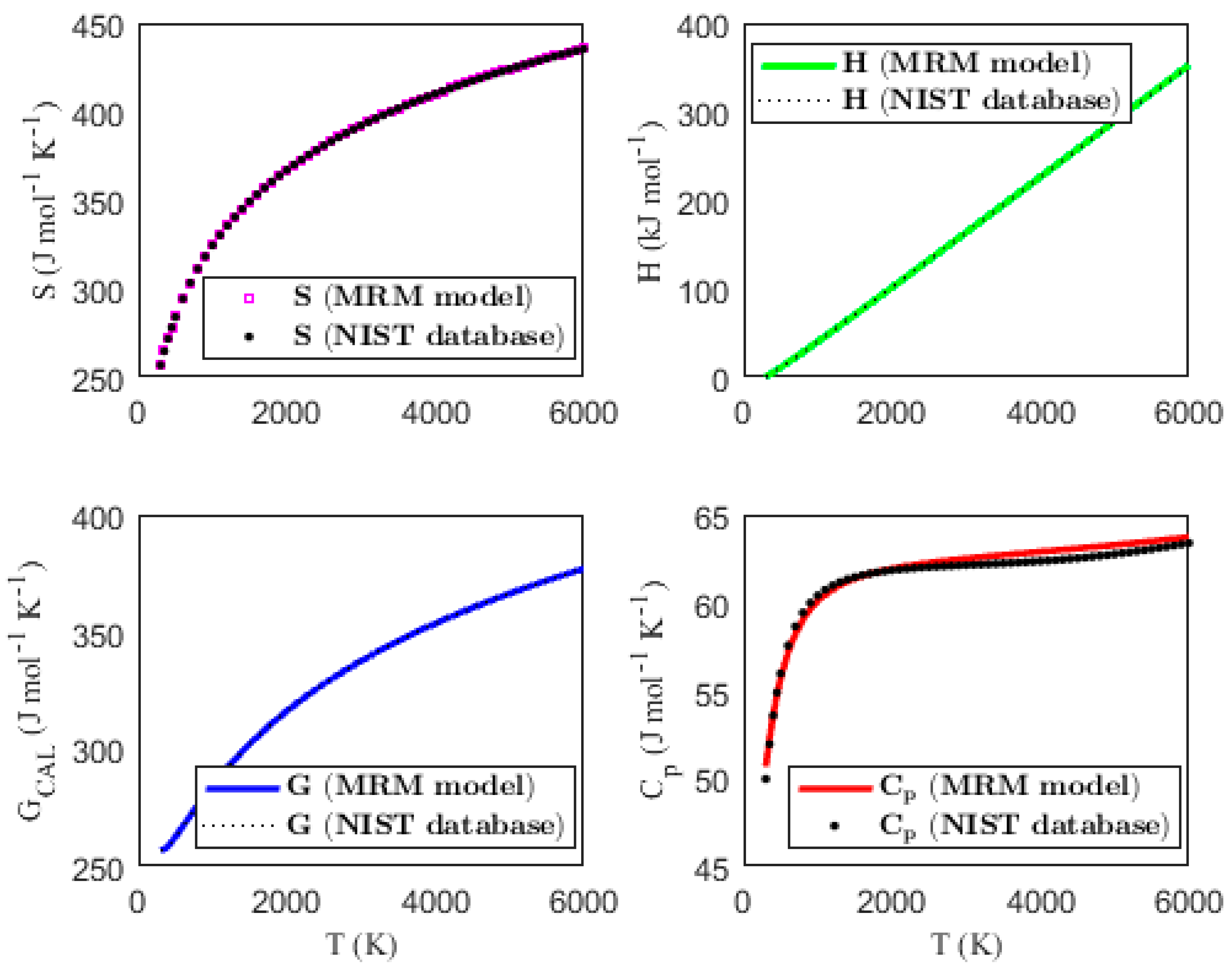
Figure 1, Figure 2, Figure 3 and Figure 4 present the temperature dependence of the thermal functions, with the corresponding NIST data shown for comparison. The alignment of the predicted thermal functions with the NIST data further supports the validity of the proposed thermodynamic models in predicting the thermal properties of the selected triatomic molecules.
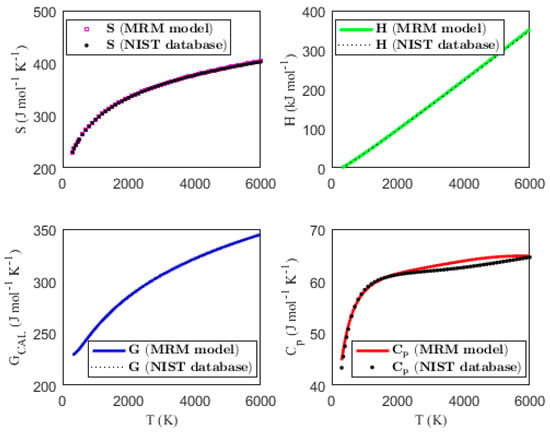
Figure 1.
Characterization of molar thermodynamic functions for the BO2 molecule, with comparison to NIST data.
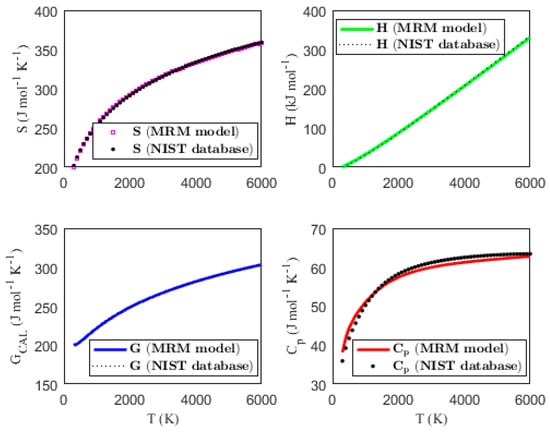
Figure 2.
Characterization of molar thermodynamic functions for the HCN molecule, with comparison to NIST data.
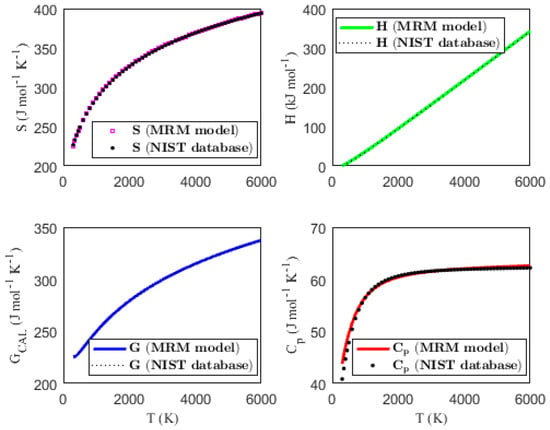
Figure 3.
Characterization of molar thermodynamic functions for the N3 molecule, with comparison to NIST data.
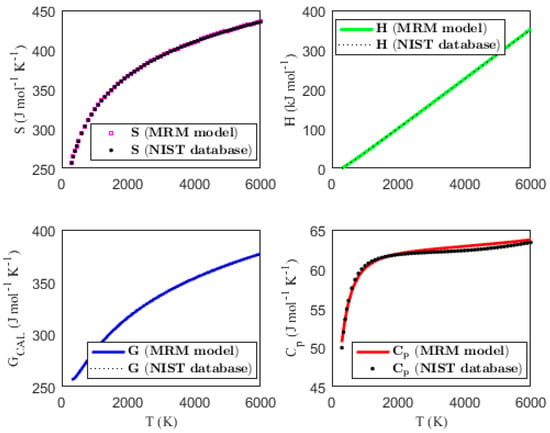
Figure 4.
Characterization of molar thermodynamic functions for the Si2N molecule, with comparison to NIST data.
Finally, Table 6 compares the performance of the MRM models (developed in the present work) with the existing IMTZ models from the literature. The MPAD values for the MRM models are smaller than those for the IMTZ models, indicating that the MRM models offer improved accuracy in predicting the thermal properties of BO2, HCN, N3, and Si2N. Both the MRM and IMTZ models incorporate optimization parameters for efficiency and rely on the same input data. However, the simplicity of the MRM models lies in their ability to predict thermal properties without requiring a dimensionless parameter.
Table 6.
Benchmarking APAD (%) data for MRM thermodynamic models against the literature-based IMTZ models.
4. Conclusions
In this study, computational models are developed to estimate the thermodynamic properties of linear triatomic molecules, including molar enthalpy, molar Gibbs free energy, constant pressure molar heat capacity, and molar entropy. These models are derived from the partition functions of the modified Rosen–Morse (MRM) potential, which simulates symmetric vibrational modes, and the harmonic oscillator, which represents asymmetric and 2-fold degenerate bending vibrational modes. The models incorporate key parameters such as equilibrium dissociation energy (De), vibrational frequencies (ωes, ωea, ωeb), and bond lengths (reZX, reZY). Additionally, an optimization parameter (λ) is introduced to refine the expressions for molar entropy and Gibbs free energy. The models are then applied to determine the thermodynamic properties of linear triatomic molecules, including BO2, HCN, N3, and Si2N. The determined values are compared to experimental data from the National Institute of Standards and Technology (NIST) database. When assessed using the mean percentage absolute deviation (MPAD), the models show strong accuracy, as follows: the MPAD for molar entropy and Gibbs free energy is no greater than 0.17%, while the MPAD for molar enthalpy and isobaric heat capacity does not exceed 2%. The determined thermodynamic properties agree with values found in the literature for triatomic molecules. These computational models provide an efficient and straightforward approach for estimating thermodynamic properties, making them highly relevant for applications in the chemical industry, environmental chemistry, materials science, and chemical engineering.
Author Contributions
E.S.E.: Conceptualization; Supervision; Data curation; Writing—Original draft; Writing—Review and editing; Methodology; Validation; Formal analysis; Resources; and Software. A.D.A.: Data curation; Writing—Original draft; Writing—Review and editing; Methodology; Formal analysis; Resources; and Software. C.A.O.: Writing—Original draft; Writing—Review and editing; Methodology; Data curation; and Formal analysis. E.O.: Writing—Original draft; Writing—Review and editing; Methodology; Data curation; Formal analysis; Investigation; Validation; and Visualization. E.P.I.: Data curation; Formal analysis; Investigation; Resources; Software; Visualization; Writing—Original draft; Writing—Review and editing; and Methodology. S.A.: Formal analysis; Investigation; Resources; Software; Visualization; Writing—Original draft; Writing—Review and editing; and Methodology. E.K.M.: Supervision; Writing—Original draft; Writing—Review and editing; Methodology; Project administration; Resources; and Validation. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
We have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
Glossary
| NIST | National Institute of Standards and Technology |
| N | Atomicity or number of atoms in a molecule |
| IMTZ | Improved Tietz |
| MRM | Modified Rosen–Morse |
| Q | Canonical partition function |
| Vibrational partition function | |
| Rotational partition function | |
| Translational partition function | |
| Partition function for symmetric vibration | |
| Partition function for asymmetric vibration | |
| Partition function for bending vibration | |
| XZY | Linear triatomic molecule with atoms X, Y, and Z |
| Equilibrium dissociation energy | |
| Equilibrium force constant for the symmetric vibration | |
| Reduced mass of atoms X and Y | |
| c | Speed of light |
| Symmetric harmonic vibrational frequency | |
| Asymmetric harmonic vibrational frequency | |
| Bending vibration harmonic frequency | |
| Equilibrium bond length between atoms Z and X | |
| Equilibrium bond length between atoms Z and Y | |
| W | Lambert W function |
| r | Internuclear separation between atoms X and Y |
| T | Temperature and kB the Boltzmann constant |
| Boltzmann constant | |
| Erfi(z) | Imaginary error function evaluated at z |
| Number of excited bonded molecular states | |
| ν | Vibrational quantum number |
| Vibrational energy eigenvalues | |
| ℏ | Reduced Planck constant |
| λ | Optimization parameter |
| m | Molar mass of molecule |
| p | Pressure exerted by gaseous molecules |
| V | Volume of gaseous molecules |
| Avogadro number | |
| R | Universal gas constant |
| S | Molar entropy |
| H | Molar enthalpy |
| Calibrated molar enthalpy | |
| G | Molar Gibbs free energy |
| Calibrated molar Gibbs free energy | |
| Constant-pressure (or isobaric) molar heat capacity | |
| PAD | Percentage absolute deviation |
| MPAD | Mean percentage absolute deviation |
References
- Bera, M.N.; Riera, A.; Lewenstein, M.; Winter, A. Generalized laws of thermodynamics in the presence of correlations. Nat. Commun. 2017, 8, 2180. [Google Scholar] [CrossRef]
- Xie, C.Y.; Tao, H.Z.; Li, W.; Wang, H.J. Numerical simulation and experimental investigation of heat pipe heat exchanger applied in residual heat removal system. Ann. Nucl. Energy 2019, 144, 568. [Google Scholar] [CrossRef]
- Ma, H.; Du, N.; Zhang, Z.; Lyu, F.; Deng, N.; Li, C.; Yu, S. Assessment of the optimum operation conditions on a heat pipe heat exchanger for waste heat recovery in steel industry. Renew. Sustain. Energy Rev. 2017, 79, 50. [Google Scholar] [CrossRef]
- Ma, H.; Yin, L.; Shen, X.; Lu, W.; Sun, Y.; Zhang, Y.; Deng, N. Experimental study on heat pipe assisted heat exchanger used for industrial waste heat recovery. Appl. Energy 2016, 169, 177. [Google Scholar] [CrossRef]
- Ding, Y.; Guo, Q.; Guo, W.; Chu, W.; Wang, Q. Review of Recent Applications of Heat Pipe Heat Exchanger Use for Waste Heat Recovery. Energies 2024, 17, 2504. [Google Scholar] [CrossRef]
- Wang, C.W.; Peng, X.L.; Liu, J.Y.; Jiang, R.; Li, X.P.; Liu, Y.S.; Liu, S.Y.; Wei, L.S.; Zhang, L.H.; Jia, C.S. A novel formulation representation of the equilibrium constant for water gas shift reaction. Int. J. Hydrogen Energy 2022, 47, 27821. [Google Scholar] [CrossRef]
- Wang, C.W.; Li, J.; Zhang, L.H.; Ding, Q.C.; Liu, G.H.; Li, G.; Jiang, R.; Peng, X.L.; Wei, L.S.; Tang, H.M.; et al. Non-fitting functional representation for the equilibrium constant subject to reaction between H2S and CO2. Fuel 2024, 362, 130916. [Google Scholar] [CrossRef]
- Wang, J.F.; Zhang, H.; Liang, L.X.; Peng, X.L.; Wang, C.W.; Deng, P.; Ding, Q.C.; Jia, C.S. A novel formulation representation regarding the equilibrium constant subject to reactions between N2 and O2. Comput. Theor. Chem. 2024, 1239, 114758. [Google Scholar] [CrossRef]
- Eyube, E.S.; Makasson, C.R.; Omugbe, E.; Onate, C.A.; Inyang, E.P.; Tahir, A.M.; Ojar, J.U.; Najoji, S.D. Improved energy equations and thermal functions for diatomic molecules: A generalized fractional derivative approach. J. Mol. Model. 2024, 30, 419. [Google Scholar] [CrossRef]
- Jia, C.S.; Diao, Y.F.; Liu, X.J.; Wang, P.Q.; Liu, J.Y.; Zhang, G.D. Equivalence of the Wei potential model and Tietz potential model for diatomic molecules. J. Chem. Phys. 2012, 137, 014101. [Google Scholar] [CrossRef]
- Zhang, G.D.; Liu, J.Y.; Zhang, L.H.; Zhou, W.; Jia, C.S. Modified Rosen-Morse potential-energy model for diatomic molecules. Phys. Rev. A 2012, 86, 062510. [Google Scholar] [CrossRef]
- Yanar, H.; Taş, A.; Salti, M.; Aydoğdu, O. Ro-vibrational energies of CO molecule via improved generalized Pöschl–Teller potential and Pekeris-type approximation. Eur. Phys. J. Plus 2020, 135, 292. [Google Scholar] [CrossRef]
- Eyube, E.S.; Notani, P.P.; Izam, M.M. Potential parameters and eigen spectra of improved Scarf II potential energy function for diatomic molecules. Mol. Phys. 2022, 120, e1979265. [Google Scholar] [CrossRef]
- Eyube, E.S.; Nyam, G.G.; Notani, P.P. Improved q-deformed Scarf II oscillator. Phys. Scr. 2021, 96, 125017. [Google Scholar] [CrossRef]
- Eyube, E.S.; Notani, P.P.; Dikko, A.B. Modeling of diatomic molecules with modified hyperbolical-type potential. Eur. Phys. J. Plus 2022, 137, 329. [Google Scholar] [CrossRef]
- Roshanzamir, M. Thermal responses and the energy spectral of diatomic molecules using Nikiforov-Uvarov methodology. Mathematics 2023, 11, 3338. [Google Scholar] [CrossRef]
- Khordad, R. Rashba effect in Frost-Musulin quantum dots: Analytical study. Opt. Quantum Electron. 2024, 56, 963. [Google Scholar] [CrossRef]
- Ghanbari, A.; Khordad, R. Thermodynamic properties of several substances using Tietz-Hua potential. Indian J. Phys. 2022, 96, 1413. [Google Scholar] [CrossRef]
- Onate, C.A.; Okon, I.B.; Eyube, E.S.; Omugbe, E.; Emeje, K.O.; Onyeaju, M.C.; Ajani, O.O.; Akinpelu, J.A. Fisher Information for a System Composed of a Combination of Similar Potential Models. Quantum Rep. 2024, 6, 184. [Google Scholar] [CrossRef]
- Khordad, R.; Avazpour, A.; Ghanbari, A. Exact analytical calculations of thermodynamic functions of gaseous substances. Chem. Phys. 2019, 517, 30. [Google Scholar] [CrossRef]
- Peng, X.L.; Jiang, R.; Jia, C.S.; Zhang, L.H.; Zhao, Y.L. Gibbs free energy of gaseous phosphorus dimer. Chem. Eng. Sci. 2018, 190, 122. [Google Scholar] [CrossRef]
- Eyube, E.S. Entropy and Gibbs free energy equations for the specialized Pöschl-Teller potential. Eur. Phys. J. Plus 2022, 137, 760. [Google Scholar] [CrossRef]
- Edet, C.O.; Osang, J.E.; Ali, N.; Agbo, E.P.; Aljunid, S.A.; Endut, R.; Ettah, E.B.; Khordad, R.; Ikot, A.N.; Asjad, M. Non-relativistic energy spectra of the modified Hylleraas potential and its thermodynamic properties in arbitrary dimensions. Quantum Rep. 2022, 4, 239. [Google Scholar] [CrossRef]
- Habibinejad, M.; Khordad, R.; Ghanbari, A. Specific heat at constant pressure, enthalpy and Gibbs free energy of boron nitride (BN) using q-deformed exponential-type potential. Phys. B 2021, 613, 412940. [Google Scholar] [CrossRef]
- Eyube, E.S.; Notani, P.P.; Nyam, G.G.; Jabil, Y.Y.; Izam, M.M. Pure vibrational state energies and statistical-mechanical models for the reparameterized Scarf oscillator. Front. Phys. 2023, 11, 978347. [Google Scholar] [CrossRef]
- Jia, C.S.; Zhang, L.H.; Peng, X.L.; Luo, J.X.; Zhao, Y.L.; Liu, J.Y.; Guo, J.J.; Tang, L.D. Prediction of entropy and Gibbs free energy for nitrogen. Chem. Eng. Sci. 2019, 202, 70. [Google Scholar] [CrossRef]
- Onyenegecha, C.P.; Njokua, I.J.; Opara, A.I.; Echendua, O.K.; Omoko, E.N.; Eze, F.C.; Okereke, C.J.; Onyeocha, E.; Nwaneho, F.U. Nonrelativistic solutions of Schrödinger equation and thermodynamic properties with the proposed modified Mobius square plus Eckart potential. Heliyon 2022, 8, e08952. [Google Scholar] [CrossRef]
- Eyube, E.S. Prediction of thermal properties of phosphorus dimer—The analytical approach Chem. Phys. Lett. 2022, 801, 139702. [Google Scholar] [CrossRef]
- Jia, C.S.; Wang, C.W.; Zhang, L.H.; Peng, X.L.; Tang, H.M. Enthalpy of gaseous phosphorus dimer. Chem. Eng. Sci. 2018, 183, 26. [Google Scholar] [CrossRef]
- Eyube, E.S.; Bitrus, B.M.; Samaila, H.; Notani, P.P. Model Entropy Equation for Gaseous Substances. Int. J. Thermophys. 2022, 43, 55. [Google Scholar] [CrossRef]
- Eyube, E.S.; Notani, P.P.; Samaila, H. Analytical prediction of enthalpy and Gibbs free energy of gaseous molecules. Chem. Thermodyn. Therm. Anal. 2022, 6, 100060. [Google Scholar] [CrossRef]
- Ding, Q.C.; Chen, J.Q.; Peng, X.L.; Wang, C.W.; Liu, G.H.; Jiang, R.; Yuan, H.; Jia, C.S. A general formulation of the Gibbs free energy regarding six symmetric triatomic molecules. Eur. Phys. J. Plus 2024, 139, 660. [Google Scholar] [CrossRef]
- Liu, G.H.; Ding, Q.C.; Wang, C.W.; Jia, C.S. Unified explicit formulations of thermodynamic properties for the gas NO2 and gaseous BF2 and AlCl2 radicals. Chem. Phys. Lett. 2023, 830, 140788. [Google Scholar] [CrossRef]
- Liu, G.H.; Ding, Q.C.; Wang, C.W.; Jia, C.S. Unified non-fitting explicit formulation of thermodynamic properties for five compounds. J. Mol. Struct. 2023, 1294, 136543. [Google Scholar] [CrossRef]
- Liang, D.C.; Zeng, R.; Wang, C.W.; Ding, Q.C.; Wei, L.S.; Peng, X.L.; Liu, J.Y.; Yu, J.; Jia, C.S. Prediction of thermodynamic properties for sulfur dioxide. J. Mol. Liq. 2022, 352, 118722. [Google Scholar] [CrossRef]
- Wang, C.W.; Wang, J.; Liu, Y.S.; Li, J.; Peng, X.L.; Jia, C.S.; Zhang, L.H.; Yi, L.Z.; Liu, J.Y.; Li, C.J.; et al. Prediction of ideal-gas thermodynamic properties for water. J. Mol. Liq. 2021, 321, 114912. [Google Scholar] [CrossRef]
- Jia, C.S.; Li, J.; Liu, Y.S.; Peng, X.L.; Jia, X.; Zhang, L.H.; Jiang, R.; Li, X.P.; Liu, J.Y.; Zhao, Y.L. Predictions of thermodynamic properties for hydrogen sulfide. J. Mol. Liq. 2020, 315, 113751. [Google Scholar] [CrossRef]
- Jia, C.S.; Wang, Y.T.; Wei, L.S.; Wang, C.W.; Peng, X.L. Predictions of entropy and Gibbs energy for carbonyl sulfide. ACS Omega 2019, 4, 20000–20004. [Google Scholar] [CrossRef]
- Wang, J.; Jia, C.S.; Li, C.J.; Peng, X.L.; Zhang, L.H.; Liu, J.Y. Thermodynamic properties of carbon dioxide. ACS Omega 2019, 4, 19193. [Google Scholar] [CrossRef]
- Chen, X.Y.; Li, J.; Jia, C.S. Thermodynamic properties of gaseous carbon disulfide. ACS Omega 2019, 4, 16121. [Google Scholar] [CrossRef]
- Deng, M.; Jia, C.S. Prediction of enthalpy of nitrogen gas. Eur. Phys. J. Plus 2018, 133, 258. [Google Scholar] [CrossRef]
- Eyube, E.S.; Notani, P.P.; Dlama, Y.; Omugbe, E.; Onate, C.A.; Okon, I.B.; Nyam, G.G.; Jabil, Y.Y.; Izam, M.M. Isobaric molar heat capacity model for the improved Tietz potential. Int. J. Quantum Chem. 2023, 123, e27040. [Google Scholar] [CrossRef]
- Schwabl, F. Statistical Mechanics, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Johns, J.W.C. The absorption spectrum of BO2. Can. J. Phys. 1961, 39, 1738. [Google Scholar] [CrossRef]
- Nobes, R.H.; Pople, J.A.; Radom, L.; Handy, N.C.; Knowles, P.J. Slow convergence of the møller-plesset perturbation series: The dissociation energy of hydrogen cyanide and the electron affinity of the cyano radical. Chem. Phys. Lett. 1987, 138, 481. [Google Scholar] [CrossRef]
- Martin, J.M.L.; François, J.P.; Gijbels, R. The dissociation energy of N3. J. Chem. Phys. 1990, 93, 4485. [Google Scholar] [CrossRef]
- Ornellas, F.R.; Iwata, S. Ab Initio studies of silicon and nitrogen clusters: Cyclic or linear Si2N. J. Phys. Chem. 1996, 100, 10919. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology (NIST). NIST Chemistry JANAF; NIST Standard Reference Database Number 69; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).