
New Derivatives of Oleanolic Acid: Semi-Synthesis and Evaluation of Their Anti-15-LOX, Anti-α-Glucosidase and Anticancer Activities and Molecular Docking Studies



Abstract

1. Introduction
2. Materials and Methods
2.1. General Experimental Methods
2.2. Extraction and Isolation OA (1)
2.3. Synthesis
2.3.1. Synthesis of Compounds 2, 2a, 3 and 3a
2.3.2. Synthesis of Compounds 4a–n and 5a–f
2.4. Anti-Inflammatory Activity (Anti-15-LOX)
2.5. Anti-Diabetic Activity (Anti-α-Glucosidase)
2.6. Cytotoxic Activity
2.7. Statistical Analysis
2.8. The Process of Molecular Docking
3. Results and Discussion
3.1. Chemistry
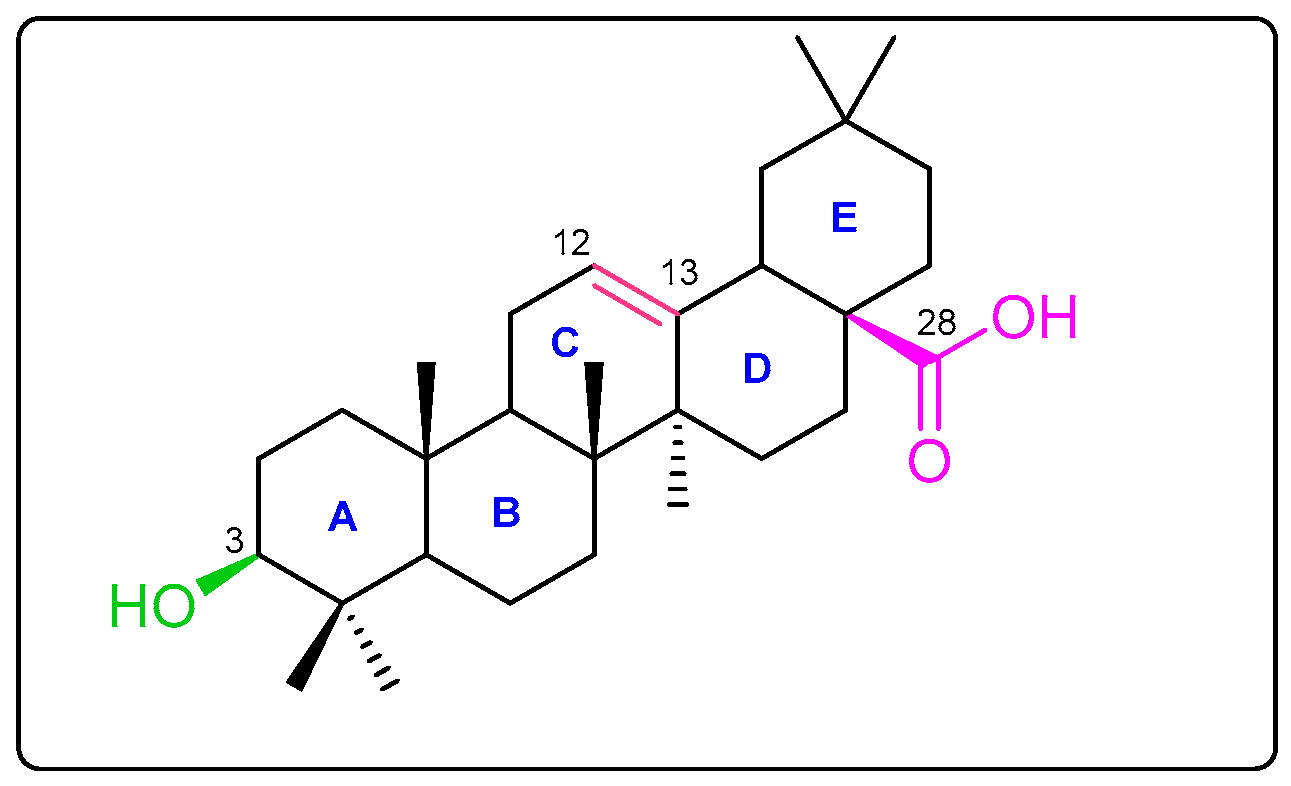
3.1.1. Extraction of OA (1)
3.1.2. Synthesis of New Derivatives
3.2. Biological Activity
3.2.1. Anti-15-Lipoxygenase Activity
3.2.2. Anti-α-Glucosidase Activity
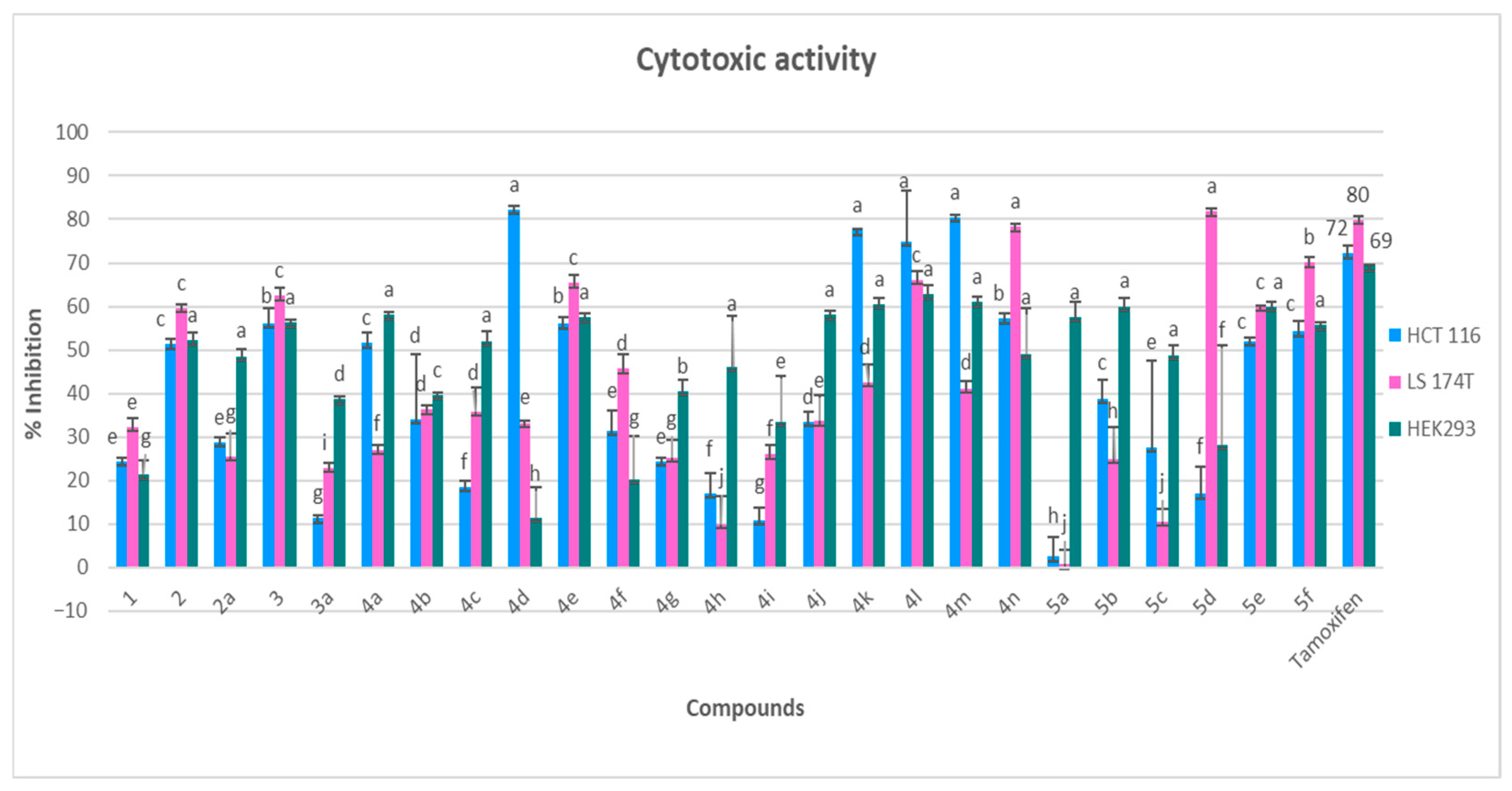
3.2.3. Cytotoxicity Evaluation
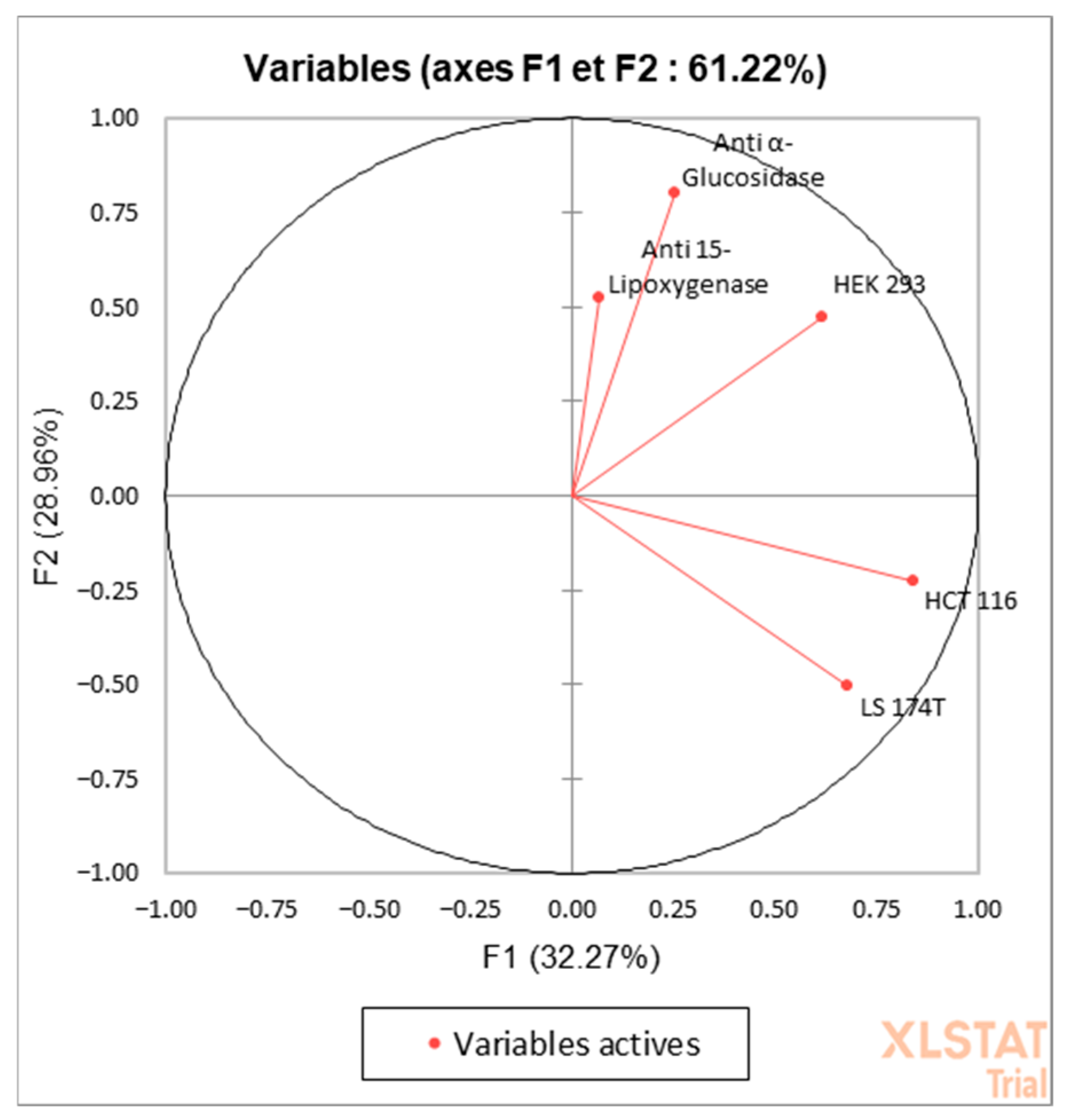
3.3. Principal Component Analysis (PCA)
3.4. Molecular Docking Analysis for Cytotoxic Activity (PDB: 1M17)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1H-NMR | proton nuclear magnetic resonance |
| 13C-NMR | carbon nuclear magnetic resonance |
| HRMS | high-resolution mass spectrometry |
| TNF-α | tumour necrosis factor α |
| IL-6 | Interleukin-6 |
| NDGA | nordihydroguaiaretic acid |
| PNP-G | p-nitrophenyl-α-D-glucopyranoside |
References
- Natalucci, V.; Marmondi, F.; Biraghi, M.; Bonato, M. The Effectiveness of Wearable Devices in Non-Communicable Diseases to Manage Physical Activity and Nutrition: Where We Are? Nutrients 2023, 15, 913. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Lawson, K.D.; Kolbe-Alexander, T.L.; Finkelstein, E.A.; Katzmarzyk, P.T.; Van Mechelen, W.; Pratt, M. The Economic Burden of Physical Inactivity: A Global Analysis of Major Non-Communicable Diseases. Lancet 2016, 388, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Asiminicesei, D.-M.; Fertu, D.I.; Gavrilescu, M. Impact of Heavy Metal Pollution in the Environment on the Metabolic Profile of Medicinal Plants and Their Therapeutic Potential. Plants 2024, 13, 913. [Google Scholar] [CrossRef]
- Ouedraogo, W.J.; Yerbanga, R.S.; Meda, R.; Ouedraogo, J.B.; Ouedraogo, A.G. Revue Des Plantes Médicinales à Potentiel Aromatique Du Burkina Faso. Health Res. Afr. 2024, 2, 1–14. [Google Scholar]
- Aly, S.H.; Elbadry, A.M.M.; Doghish, A.S.; El-Nashar, H.A.S. Unveiling the Pharmacological Potential of Plant Triterpenoids in Breast Cancer Management: An Updated Review. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 5571–5596. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yin, X.; Kou, C.; Thimmappa, R.; Hua, X.; Xue, Z. Classification, Biosynthesis and Biological Function of Triterpene Esters in Plants. Plant Commun. 2024, 5, 100845. [Google Scholar] [CrossRef]
- Triaa, N.; Znati, M.; Ben Jannet, H.; Bouajila, J. Biological Activities of Novel Oleanolic Acid Derivatives from Bioconversion and Semi-Synthesis. Molecules 2024, 29, 3091. [Google Scholar] [CrossRef]
- Feng, A.; Yang, S.; Sun, Y.; Zhang, L.; Bo, F.; Li, L. Development and Evaluation of Oleanolic Acid Dosage Forms and Its Derivatives. BioMed Res. Int. 2020, 2020, 1308749. [Google Scholar] [CrossRef]
- Verma, N.; Raghuvanshi, D.S.; Singh, R.V. Recent Advances in the Chemistry and Biology of Oleanolic Acid and Its Derivatives. Eur. J. Med. Chem. 2024, 276, 116619. [Google Scholar] [CrossRef]
- Şenol, H.; Ghaffari-Moghaddam, M.; Bulut, Ş.; Akbaş, F.; Köse, A.; Topçu, G. Synthesis and Anticancer Activity of Novel Derivatives of α, β-Unsaturated Ketones Based on Oleanolic Acid: In Vitro and in Silico Studies against Prostate Cancer Cells. Chem. Biodivers. 2023, 20, e202301089. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Rubanik, L.V.; Petrova, A.V.; Smirnova, I.E.; Terekhova, A.V.; Baikova, I.P. Synthesis and Antichlamydial Activity of Oleanolic Acid Aza-Derivatives. Chem. Nat. Compd. 2024, 60, 84–91. [Google Scholar] [CrossRef]
- Xiaofei, J.; Mingqing, S.; Miao, S.; Yizhen, Y.; Shuang, Z.; Qinhua, X.; Kai, Z. Oleanolic Acid Inhibits Cervical Cancer Hela Cell Proliferation through Modulation of the ACSL4 Ferroptosis Signaling Pathway. Biochem. Biophys. Res. Commun. 2021, 545, 81–88. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Dai, S.-Y.; Deng, F.-H.; Peng, L.-H.; Li, C.; Pei, Y.-H. Recent Advances in Medicinal Chemistry of Oleanolic Acid Derivatives. Phytochemistry 2022, 203, 113397. [Google Scholar] [CrossRef]
- Gao, C.-X.; Tang, C.-H.; Wu, T.-J.; Hu, Y.; Peng, Y.-L.; Liu, M.-L.; Liu, Q.-W.; Chen, H.-F.; Yang, Z.-H.; Zheng, X. Anticancer Activity of Oleanolic Acid and Its Derivatives Modified at A-Ring and C-28 Position. J. Asian Nat. Prod. Res. 2023, 25, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.; Molath, A.; Choksi, H.; Kumar, S.; Mehra, R. Classifications of Polyphenols and Their Potential Application in Human Health and Diseases. Int. J. Physiol. Nutr. Phys. Educ. 2021, 6, 293–301. [Google Scholar] [CrossRef]
- Samanta, S.; Sarkar, T.; Chakraborty, R.; Rebezov, M.; Shariati, M.A.; Thiruvengadam, M.; Rengasamy, K.R. Dark Chocolate: An Overview of Its Biological Activity, Processing, and Fortification Approaches. Curr. Res. Food Sci. 2022, 5, 1916–1943. [Google Scholar] [CrossRef]
- Hano, C.; Tungmunnithum, D. Plant Polyphenols, More than Just Simple Natural Antioxidants: Oxidative Stress, Aging and Age-Related Diseases. Medicines 2020, 7, 26. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef]
- Mustafa, Y.F.; Bashir, M.K.; Oglah, M.K. Original and Innovative Advances in the Synthetic Schemes of Coumarin-Based Derivatives: A Review. Syst. Rev. Pharm. 2020, 11, 598–612. [Google Scholar]
- Khursheed, A.; Jain, V. Medicinal Research Progress of Natural Coumarin and Its Derivatives. Nat. Prod. J. 2021, 11, 648–662. [Google Scholar] [CrossRef]
- Li, H.; Li, M.; Xu, R.; Wang, S.; Zhang, Y.; Zhang, L.; Zhou, D.; Xiao, S. Synthesis, Structure Activity Relationship and in Vitro Anti-Influenza Virus Activity of Novel Polyphenol-Pentacyclic Triterpene Conjugates. Eur. J. Med. Chem. 2019, 163, 560–568. [Google Scholar] [CrossRef]
- Vega-Granados, K.; Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Martinez, A.; Alvarez De Cienfuegos, L.; Lupiañez, J.A.; Parra, A. Synthesis and Biological Activity of Triterpene–Coumarin Conjugates. J. Nat. Prod. 2021, 84, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signalling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Mustafi, R.; Dougherty, U.; Shah, H.; Dehghan, H.; Gliksberg, A.; Wu, J.; Zhu, H.; Joseph, L.; Hart, J.; Dive, C.; et al. Both stromal cell and colonocyte epidermal growth factor receptors control HCT116 colon cancer cell growth in tumor xenografts. Carcinogenesis 2012, 33, 1930–1939. [Google Scholar] [CrossRef]
- Geißler, A.L.; Geißler, M.; Kottmann, D.; Lutz, L.; Fichter, C.D.; Fritsch, R.; Weddeling, B.; Makowiec, F.; Werner, M.; Lassmann, S. ATM mutations and E-cadherin expression define sensitivity to EGFR-targeted therapy in colorectal cancer. Oncotarget 2017, 8, 17164–17190. [Google Scholar] [CrossRef]
- Mac, L. Egfr in the hct-116 line of human colorectal cancer cells. Adv. Cell Sci. Tissue Cult. 2022, 6, 122. [Google Scholar]
- Ayadi, J.; Debouba, M.; Rahmani, R.; Bouajila, J. The Phytochemical Screening and Biological Properties of Brassica napus L. Var. Napobrassica (Rutabaga) Seeds. Molecules 2023, 28, 6250. [Google Scholar] [CrossRef]
- Ben Khadher, T.; Sassi-Aydi, S.; Aydi, S.; Mars, M.; Bouajila, J. Phytochemical Profiling and Biological Potential of Prunus Dulcis Shell Extracts. Plants 2023, 12, 2733. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Vardanega, R.; Santos, D.T.; Meireles, M.A.A. Intensification of Bioactive Compounds Extraction from Medicinal Plants Using Ultrasonic Irradiation. Pharmacogn. Rev. 2014, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Gadjalova, A.V.; Mihaylova, D. Ultrasound-Assisted Extraction of Medicinal Plants and Evaluation of Their Biological Activity. Food Res. 2019, 3, 530–536. [Google Scholar] [CrossRef]
- Xie, P.; Huang, L.; Zhang, C.; Deng, Y.; Wang, X.; Cheng, J. Enhanced Extraction of Hydroxytyrosol, Maslinic Acid and Oleanolic Acid from Olive Pomace: Process Parameters, Kinetics and Thermodynamics, and Greenness Assessment. Food Chem. 2019, 276, 662–674. [Google Scholar] [CrossRef]
- Yang, K.; Wang, S.-B.; Pei, D.; Pu, L.-M.; Huang, X.-Y. Effective Separation of Maslinic Acid and Oleanolic Acid from Olive Pomace Using High-Speed Shear off-Line Coupled with High-Speed Countercurrent Chromatography and Their Antibacterial Activity Test. J. Chromatogr. B 2024, 1236, 124069. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Su, C.; Huang, J.; Liu, J.; Zheng, Y.; Chen, Z. Conjugation of Uridine with Oleanolic Acid Derivatives as Potential Antitumor Agents. Chem. Biol. Drug Des. 2016, 88, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhu, Z.F.; Meng, F.; Xu, C.S.; Zhang, Y.H. Synthesis and Cytotoxicity of Oleanolic Acid/N-Aryl-N’-Hydroxyguanidine Hybrids. Chin. J. Nat. Med. 2010, 8, 436–440. [Google Scholar] [CrossRef]
- Bednarczyk-Cwynar, B.; Günther, A. Advances in Chemistry and Pharmacology of Triterpenoid Synthetic Dimers. Curr. Med. Chem. 2017, 24, 2205–2240. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, Y.-Y.; Hu, C.-M.; Wu, X.-Z.; Lin, J.; Xiong, Z.; Zhang, K.; Xu, X.-T. Synthesis and Biological Evaluation of Coumarin Derivatives Containing Oxime Ester as α-Glucosidase Inhibitors. Arab. J. Chem. 2022, 15, 104072. [Google Scholar] [CrossRef]
- Xu, X.-T.; Deng, X.-Y.; Chen, J.; Liang, Q.-M.; Zhang, K.; Li, D.-L.; Wu, P.-P.; Zheng, X.; Zhou, R.-P.; Jiang, Z.-Y. Synthesis and Biological Evaluation of Coumarin Derivatives as α-Glucosidase Inhibitors. Eur. J. Med. Chem. 2020, 189, 112013. [Google Scholar] [CrossRef]
- Günther, A.; Zalewski, P.; Sip, S.; Ruszkowski, P.; Bednarczyk-Cwynar, B. Acetylation of Oleanolic Acid Dimers as a Method of Synthesis of Powerful Cytotoxic Agents. Molecules 2024, 29, 4291. [Google Scholar] [CrossRef]
- Günther, A.; Zalewski, P.; Sip, S.; Ruszkowski, P.; Bednarczyk-Cwynar, B. Oleanolic Acid Dimers with Potential Application in Medicine—Design, Synthesis, Physico-Chemical Characteristics, Cytotoxic and Antioxidant Activity. Int. J. Mol. Sci. 2024, 25, 6989. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, A.; Doan, P.; Chandraseelan, J.G.; Sandberg, O.; Yli-Harja, O.; Candeias, N.R.; Kandhavelu, M. Synthesis of Phenol-Derivatives and Biological Screening for Anticancer Activity. AntiCancer Agents Med. Chem. 2017, 17, 1710–1720. [Google Scholar] [CrossRef]
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef] [PubMed]
- Phutdhawong, W.; Chuenchid, A.; Taechowisan, T.; Sirirak, J.; Phutdhawong, W.S. Synthesis and Biological Activity Evaluation of Coumarin-3-Carboxamide Derivatives. Molecules 2021, 26, 1653. [Google Scholar] [CrossRef]
- Martínez-Lara, R.I.; Cobos-Ontiveros, L.A.; Meza-Ireta, S.A.; Martínez-Montiel, M.; Colín-Lozano, B.; Puerta, A.; Padrón, J.M.; López, Ó.; Vega-Báez, J.L.; Merino-Montiel, P.; et al. Novel Coumarin-steroid/Terpenoid Hybrids: In Vitro and in Silico Anticancer Studies. Chem. Biodivers. 2024, 21, e202401315. [Google Scholar] [CrossRef] [PubMed]
- Herrera-R, A.; Naranjo, T.W.; Maldonado, M.E.; Moreno-Q, G.; Yepes, A.; Cardona-G, W. Styrylcoumarin 7-SC2 Induces Apoptosis in SW480 Human Colon Adenocarcinoma Cells and Inhibits Azoxymethane-Induced Aberrant Crypt Foci Formation in BALB/c Mice. Med. Chem. Res. 2020, 29, 377–395. [Google Scholar] [CrossRef]
- Holbro, T.; Hynes, N.E. ErbB Receptors: Directing Key Signaling Networks Throughout Life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Guérin, O.; Fischel, J.L.; Ferrero, J.-M.; Bozec, A.; Milano, G. EGFR Targeting in Hormone-Refractory Prostate Cancer: Current Appraisal and Prospects for Treatment. Pharmaceuticals 2010, 3, 2238–2247. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Alkhoja, O.A.; Mohamed, W.R. Design, Synthesis and Antitumor Activity of Novel Pyrazolo [3, 4-d] Pyrimidine Derivatives as EGFR-TK Inhibitors. Bioorganic Chem. 2016, 66, 88–96. [Google Scholar] [CrossRef]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers 2019, 11, 1826. [Google Scholar] [CrossRef]
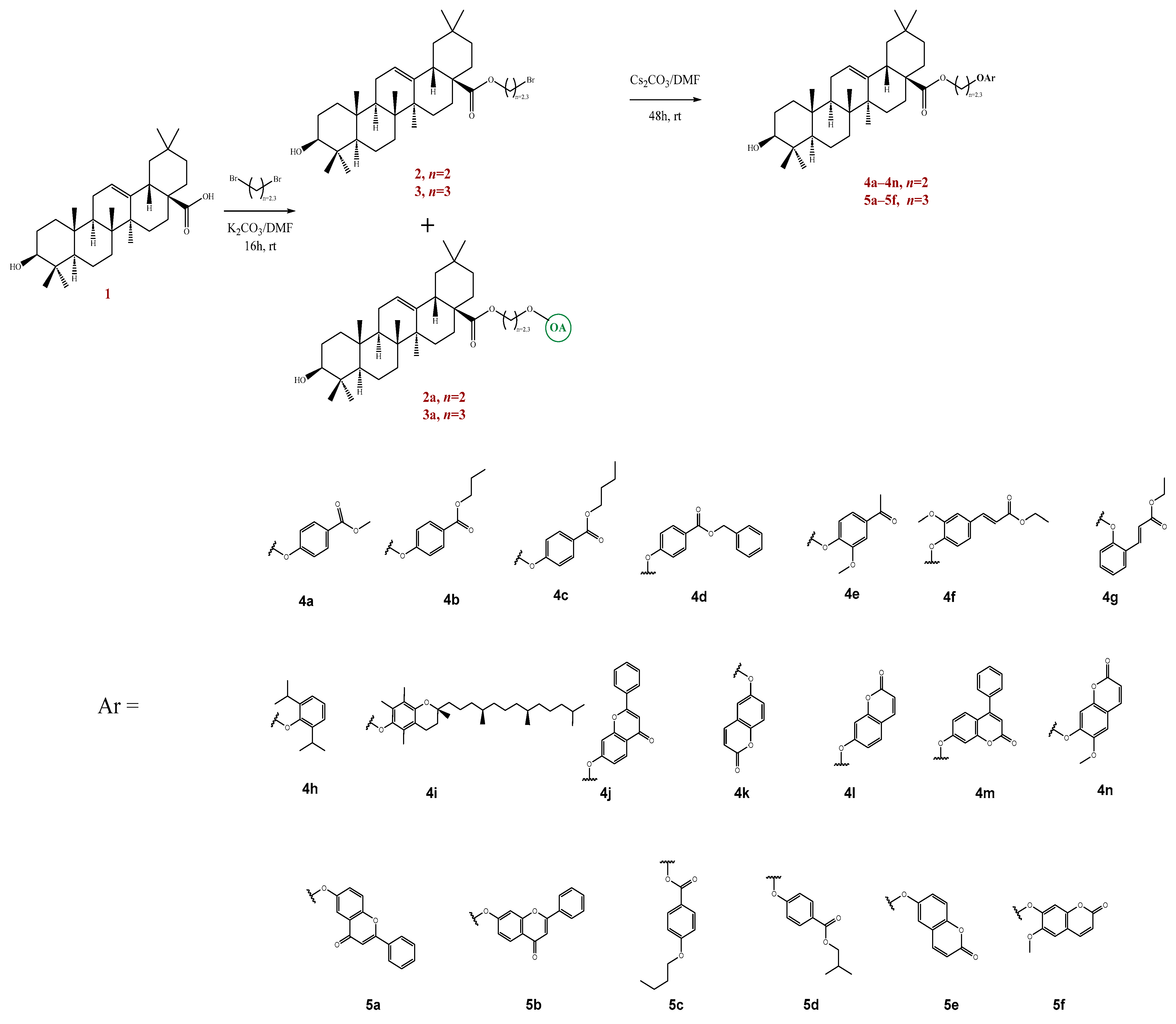


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Anti-15-Lipoxygenase | Anti-α-Glucosidase | ||
|---|---|---|---|---|
| Inhibition (%) | IC50 (μM) | Inhibition (%) | IC50 (μM) | |
| 1 (OA) | na | - | 11.5 ± 3.6 g | - |
| 2 | 22.6 ± 15.7 b | - | 10.9 ± 1.1 g | - |
| 2a | 83.1 ± 18.3 a | 52.4 ± 16.1 | 61.5 ± 4.1 a | 59.5 ± 10.8 |
| 3 | 86.7 ± 14.1 a | 29.0 ± 14.7 | 13.8 ± 2.5 g | - |
| 3a | 39.8 ± 9.6 b | - | 54.3 ± 12.5 a | - |
| 4a | 11.5 ± 1.1 d | - | 11.5 ± 1.1 g | - |
| 4b | na | - | 23.0 ± 1.4 f | - |
| 4c | na | - | na | - |
| 4d | na | - | 4.1 ± 4.5 h | - |
| 4e | na | - | na | - |
| 4f | 7.0 ± 2.0 d | - | na | - |
| 4g | na | - | 4.5 ± 3.1 h | - |
| 4h | na | - | 39.9 ± 7.1 c | - |
| 4i | na | - | na | - |
| 4j | 18.8 ± 6.6 c | - | 12.5 ± 4.6 g | - |
| 4k | na | - | 35.5 ± 3.5 e | - |
| 4l | 7.8 ± 5.3 d | - | 11.2 ± 1.6 g | - |
| 4m | na | - | 43.7 ± 2.1 b | - |
| 4n | na | - | 10.7 ± 4.0 g | - |
| 5a | na | - | 2.7 ± 5.5 h | - |
| 5b | na | - | 57.7 ± 11.5 a | - |
| 5c | na | - | 39.8 ± 5.8 d | - |
| 5d | na | - | na | - |
| 5e | 24.0 ± 4.6 b | - | 38.0 ± 3.0 e | - |
| 5f | na | - | 54.6 ± 3.4 a | - |
| NDGA | 84.7 ± 1.8 | - | - | - |
| Acarbose | - | - | 75.3 ± 0.4 | - |
| Compounds | IC50 (µM) | ||
|---|---|---|---|
| HCT-116 | LS-174T | HEK-293 | |
| 1 (OA) | - | - | - |
| 2 | - | 55.3 ± 5.0 | - |
| 2a | - | - | - |
| 3 | - | 55.0 ± 4.0 | 80.7 ± 2.6 |
| 3a | - | - | - |
| 4a | - | - | 79.1 ± 0.6 |
| 4b | - | - | - |
| 4c | - | - | - |
| 4d | 38.5 ± 1.7 | - | - |
| 4e | - | 44.3 ± 3.9 | 78.2 ± 1.3 |
| 4f | - | - | - |
| 4g | - | - | - |
| 4h | - | - | - |
| 4i | - | - | - |
| 4j | - | - | 69.3 ± 2.3 |
| 4k | 39.3 ± 2.9 | - | 59.2 ± 3.5 |
| 4l | 43.0 ± 14.4 | 44.0 ± 5.6 | 53.5 ± 5.3 |
| 4m | 40.0 ± 0.1 | - | 66.7 ± 3.8 |
| 4n | 71.5 ± 4.8 | 37.2 ± 2.5 | - |
| 5a | - | - | 78.6 ± 8.2 |
| 5b | - | - | 42.2 ± 6.6 |
| 5c | - | - | - |
| 5d | - | 38.0 ± 1.5 | - |
| 5e | - | 54.9 ± 7.8 | 69.9 ± 0.7 |
| 5f | - | 41.8 ± 1.5 | 81.1 ± 5.4 |
| Tamoxifen | 35.0 ± 3.0 | 30.9 ± 1.6 | 39.6 ± 5.6 |
| F1 | F2 | |
|---|---|---|
| Anti-15-Lipoxygenase | 0.298 | 18.814 |
| Anti-α-Glucosidase | 3.910 | 44.534 |
| HCT-116 | 43.586 | 3.445 |
| LS-174T | 28.653 | 17.621 |
| HEK-293 | 23.553 | 15.586 |
| F1 | F2 | |
|---|---|---|
| Anti-15-Lipoxygenase | 0.069 | 0.522 |
| Anti-α-Glucosidase | 0.251 | 0.803 |
| HCT-116 | 0.839 | −0.223 |
| LS-174T | 0.680 | −0.505 |
| HEK-293 | 0.616 | 0.475 |
| Compounds | Binding Energy (kcal/mol) | Interaction Detail: NI/NIAA: IAA |
|---|---|---|
| 2 | −8.9 | 13/6: LEU-694, PHE-699, VAL-702, ALA-719,CYS-773*, LEU-820 |
| 3 | −8.4 | 16/9: LEU-694, PHE-699, VAL-702, ALA-719, LYS-721, THR-766*, CYS-773, LEU-820, THR-830 |
| 4d | −10.1 | 15/9: PHE-699, VAL-702, LYS-721, ILE-735, GLU-738, THR-766*, CYS-773, LEU-820, ASP-831 |
| 4e | −9.5 | 17/10: LEU-694, PHE-699, VAL-702, ALA-719, LYS-721, THR-766*, GLY-772, CYS-773, LEU-820, THR-830 |
| 4k | −10.2 | 15/7: PHE-699, VAL-702, LYS-721, THR-766*, ARG-817, LEU-820, ASP-831 |
| 4l | −10.5 | 16/9: LEU-694, PHE-699, VAL-702, ALA-719, LYS-721, THR-766*, MET-769*, CYS-773, LEU-820 |
| 4m | −11.6 | 19/12: LEU-694, PHE-699, VAL-702, ALA-719, LYS-721, MET-769*, CYS-773, LEU-775, ARG-817, LEU-820, LEU-834, LYS-851* |
| 4n | −9.2 | 16/8: PHE-699, VAL-702, LYS-721*, THR-766*, CYS-773, ARG-817*, LEU-820, ASP-831 |
| 5d | −9.6 | 11/6: PHE-699, VAL-702, LYS-721, THR-766*, LEU-820, LEU-834 |
| 5e | −9.0 | 11/8: LEU-694, PHE-699, VAL-702, MET-769*, CYS-773, LEU-775, ARG-817, LEU-820 |
| 5f | −9.4 | 16/10: PHE-699, VAL-702, ALA-719, MET-769*, CYS-773, LEU-775, ARG-817, LEU-820, ASP-831, LYS-851* |
| Tamoxifen | −7.3 | 9/8: LEU-694, PHE-699, VAL-702, GLU-738, CYS-773, LEU-820, THR-830*,ASP-831 |
| Compounds | 3D Binding Interactions | 3D Pocket Positioning |
|---|---|---|
| 4d | 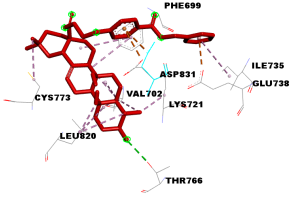 | 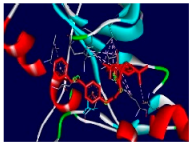 |
| 4e | 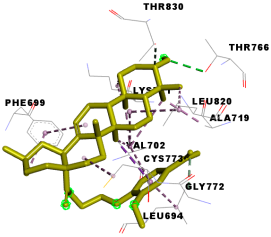 | 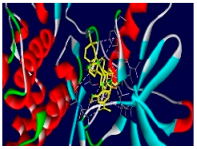 |
| 4k | 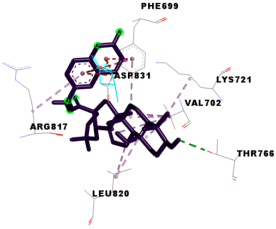 | 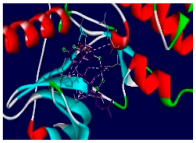 |
| 4l | 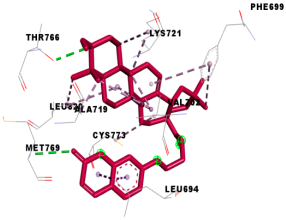 | 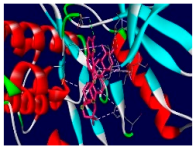 |
| 4m | 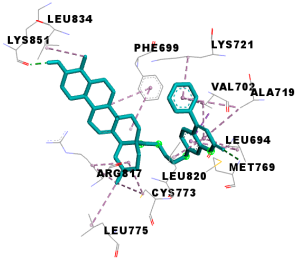 | 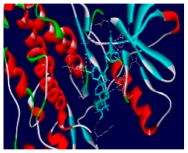 |
| 5d | 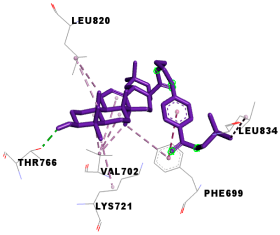 | 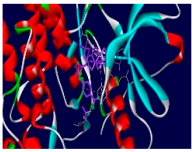 |
| Tamoxifen | 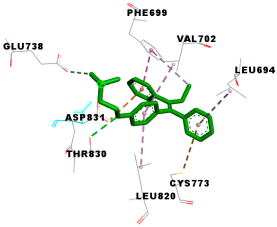 | 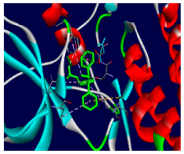 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Triaa, N.; Jlizi, S.; Znati, M.; Ben Jannet, H.; Bouajila, J. New Derivatives of Oleanolic Acid: Semi-Synthesis and Evaluation of Their Anti-15-LOX, Anti-α-Glucosidase and Anticancer Activities and Molecular Docking Studies. Chemistry 2025, 7, 36. https://doi.org/10.3390/chemistry7020036
Triaa N, Jlizi S, Znati M, Ben Jannet H, Bouajila J. New Derivatives of Oleanolic Acid: Semi-Synthesis and Evaluation of Their Anti-15-LOX, Anti-α-Glucosidase and Anticancer Activities and Molecular Docking Studies. Chemistry. 2025; 7(2):36. https://doi.org/10.3390/chemistry7020036
Chicago/Turabian StyleTriaa, Nahla, Salma Jlizi, Mansour Znati, Hichem Ben Jannet, and Jalloul Bouajila. 2025. "New Derivatives of Oleanolic Acid: Semi-Synthesis and Evaluation of Their Anti-15-LOX, Anti-α-Glucosidase and Anticancer Activities and Molecular Docking Studies" Chemistry 7, no. 2: 36. https://doi.org/10.3390/chemistry7020036
APA StyleTriaa, N., Jlizi, S., Znati, M., Ben Jannet, H., & Bouajila, J. (2025). New Derivatives of Oleanolic Acid: Semi-Synthesis and Evaluation of Their Anti-15-LOX, Anti-α-Glucosidase and Anticancer Activities and Molecular Docking Studies. Chemistry, 7(2), 36. https://doi.org/10.3390/chemistry7020036