To Be or Not to Be: Environmental Factors that Drive Myelin Formation during Development and after CNS Trauma


{kind=link}
{kind=link}
Abstract
1. Introduction
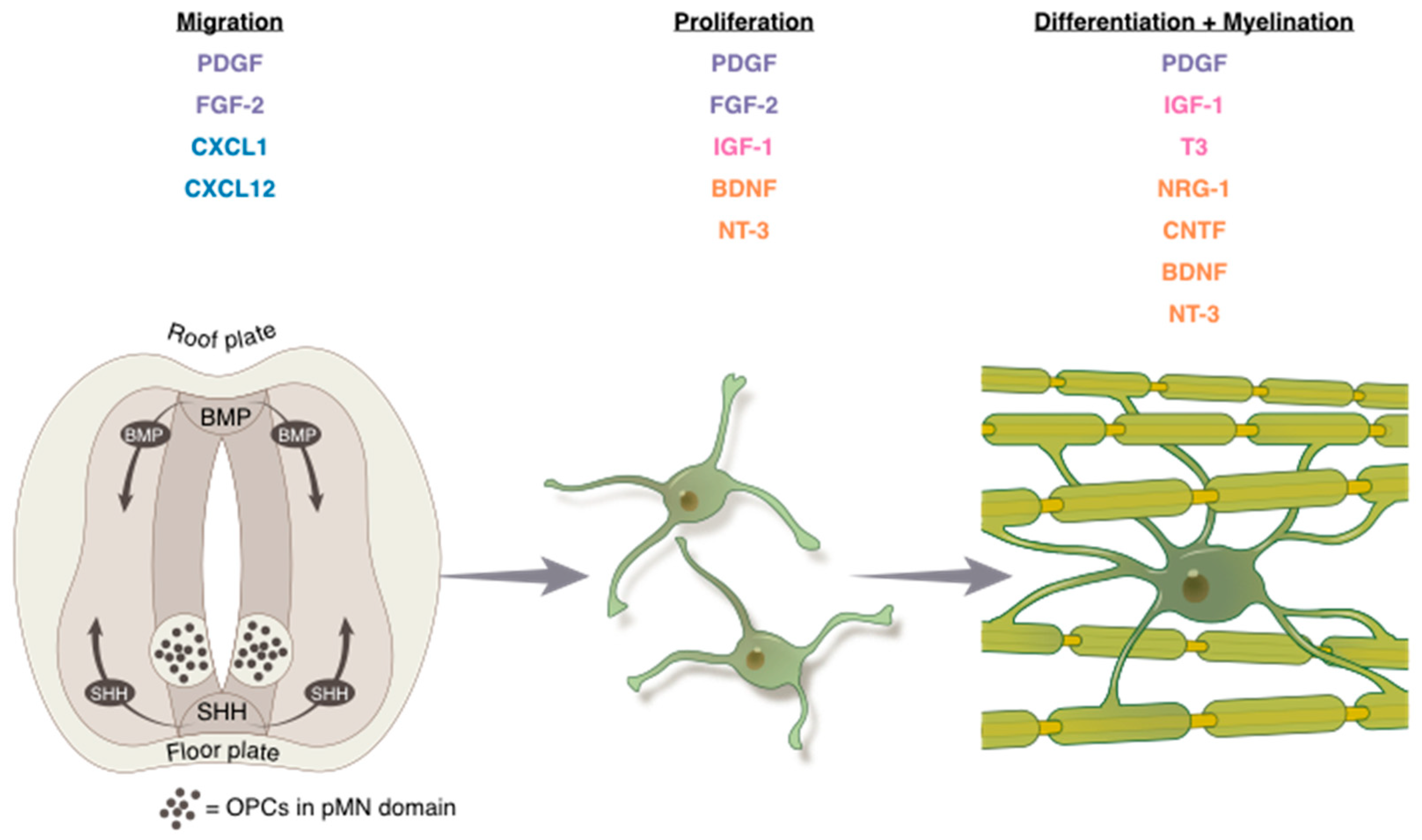
2. Development: OPC Migration, Proliferation, and Differentiation
2.1. Migration
2.1.1. Growth Factors
2.1.2. Chemokines
2.2. Proliferation
2.2.1. Three Major Mitogens
2.2.2. Neurotrophic Factors
2.3. Differentiation
2.3.1. Hormones
2.3.2. Neurotrophic Factors
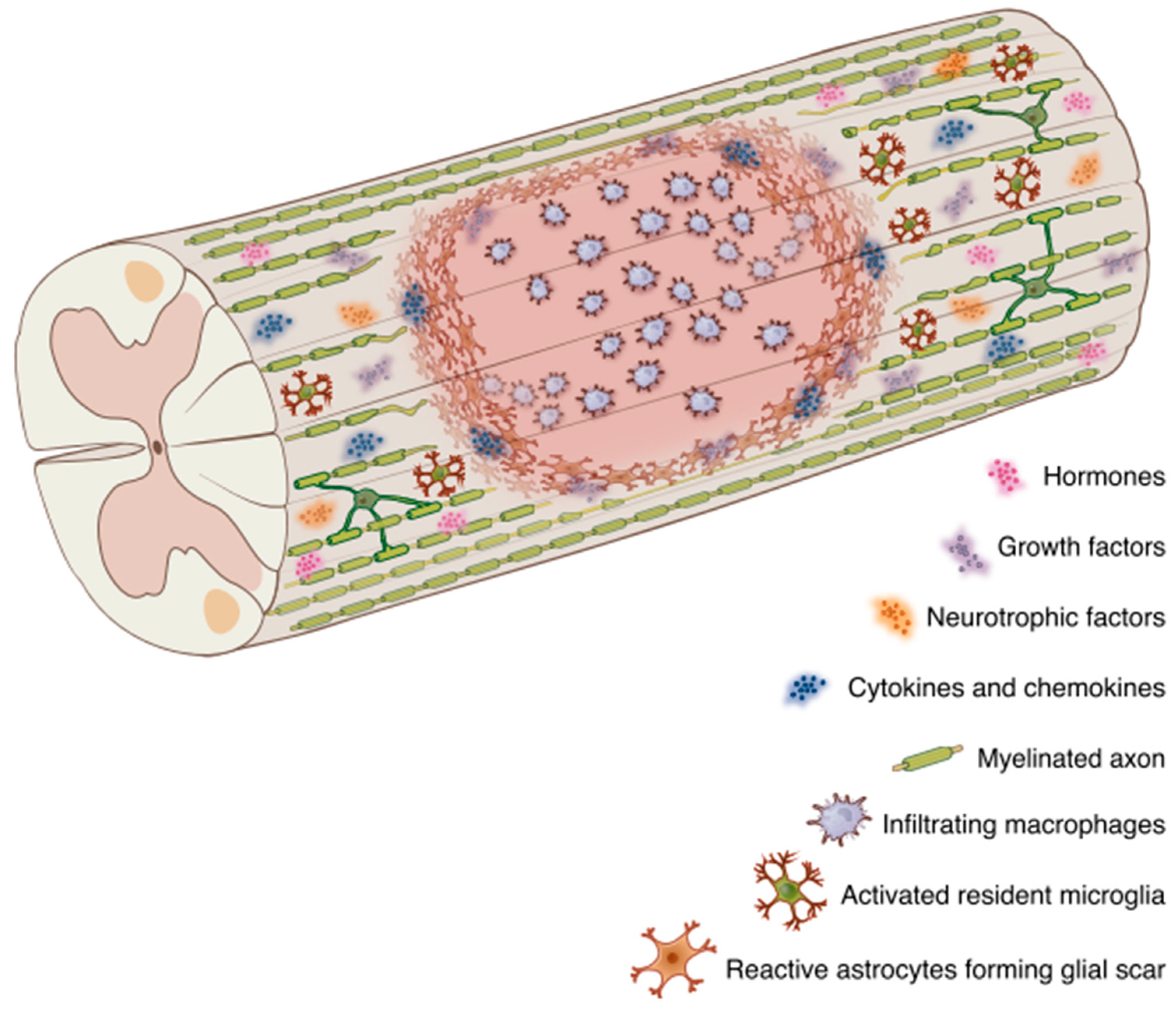
3. Central Nervous System Trauma
3.1. Growth Factors, Neurotrophic Factors, and Hormones Modulate Myelin Regeneration
3.1.1. Growth Factors
3.1.2. Neurotrophic Factors
3.1.3. Hormones
3.2. Components of the Injury-Induced Glial Scar Affect OL Lineage Cells
3.3. The Neuroimmune Axis
3.3.1. Cytokines and Microglia/Macrophage Signaling
3.3.2. Cytokine Signaling Regulators
3.3.3. Chemokines
4. Summary and Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Boullerne, A.I. The history of myelin. Exp. Neurol. 2016, 283, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Rasband, M.N.; Peles, E. The Nodes of Ranvier: Molecular Assembly and Maintenance. Cold Spring Harb. Perspect. Biol. 2015, 8, a020495. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Chan, J.R. Regulation and dysregulation of axon infrastructure by myelinating glia. J. Cell Biol. 2017, 216, 3903–3916. [Google Scholar] [CrossRef] [PubMed]
- Zalc, B. The acquisition of myelin: An evolutionary perspective. Brain Res. 2016, 1641, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.K.; Vieira, M.S.; Vasconcellos, R.; Goulart, V.A.M.; Kihara, A.H.; Resende, R.R. Decoding cell signalling and regulation of oligodendrocyte differentiation. Semin. Cell Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Boshans, L.; Goncalves, C.M.; Wegrzyn, J.; Patel, K.D. Lineage, fate, and fate potential of NG2-glia. Brain Res. 2016, 1638, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Toyama, Y.; Nishiyama, A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J. Neurosci. Res. 2002, 69, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Gans, C.; Northcutt, R.G. Neural crest and the origin of vertebrates: a new head. Science 1983, 220, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Noll, E.; Miller, R.H. Oligodendrocyte precursors originate at the ventral ventricular zone dorsal to the ventral midline region in the embryonic rat spinal cord. Development 1993, 118, 563–573. [Google Scholar] [PubMed]
- Rowitch, D.H. Glial specification in the vertebrate neural tube. Nat. Rev. Neurosci. 2004, 5, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Richardson, W.D.; Kessaris, N.; Pringle, N. Oligodendrocyte wars. Nat. Rev. Neurosci. 2006, 7, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.B.; Clarke, L.E.; Burzomato, V.; Kessaris, N.; Anderson, P.N.; Attwell, D.; Richardson, W.D. Dorsally and ventrally derived oligodendrocytes have similar electrical properties but myelinate preferred tracts. J. Neurosci. 2011, 31, 6809–6819. [Google Scholar] [CrossRef] [PubMed]
- Levison, S.W.; Goldman, J.E. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron 1993, 10, 201–212. [Google Scholar] [CrossRef]
- Lachapelle, F.; Gumpel, M.; Baulac, M.; Jacque, C.; Duc, P.; Baumann, N. Transplantation of CNS Fragments into the Brain of Shiverer Mutant Mice: Extensive Myelination by Implanted Oligodendrocytes. Dev. Neurosci. 1983, 6, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Qi, Y.; Hu, X.; Tan, M.; Liu, Z.; Zhang, J.; Li, Q.; Sander, M.; Qiu, M. Generation of Oligodendrocyte Precursor Cells from Mouse Dorsal Spinal Cord Independent of Nkx6 Regulation and Shh Signaling. Neuron 2005, 45, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Jessell, T.M. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 2000, 1, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Mekki-Dauriac, S.; Agius, E.; Kan, P.; Cochard, P. Bone morphogenetic proteins negatively control oligodendrocyte precursor specification in the chick spinal cord. Development 2002, 129, 5117–5130. [Google Scholar] [PubMed]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef] [PubMed]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.B.; Takada, N.; Latimer, A.J.; Shin, J.; Carney, T.J.; Kelsh, R.N.; Appel, B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat. Neurosci. 2006, 9, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.Y.C.; Rosenberg, S.S.; Fancy, S.P.J.; Zhao, C.; Shen, Y.-A.A.; Hahn, A.T.; McGee, A.W.; Xu, X.; Zheng, B.; Zhang, L.I.; et al. Neurite outgrowth inhibitor Nogo-A establishes spatial segregation and extent of oligodendrocyte myelination. Proc. Natl. Acad. Sci. USA 2012, 109, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Richardson, W.D.; Pringle, N.; Mosley, M.J.; Westermark, B.; Dubois-Dalcqt, M. A Role for Platelet-Derived Growth Factor in Normal Gliogenesis in the Central Nervous System. Cell 1988, 53, 309–319. [Google Scholar] [CrossRef]
- Pringle, N.P.; Mudhar, H.S.; Collarini, E.J.; Richardson, W.D. PDGF receptors in the rat CNS: during late neurogenesis, PDGF alpha- receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development 1992, 115, 535–551. [Google Scholar] [PubMed]
- Frost, E.E.; Zhou, Z.; Krasnesky, K.; Armstrong, R.C. Initiation of oligodendrocyte progenitor cell migration by a PDGF-A activated extracellular regulated kinase (ERK) signaling pathway. Neurochem. Res. 2009, 34, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.E.; Serafini, T.; de la Torre, J.; Tessier-Lavigne, M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell 1994, 78, 425–435. [Google Scholar] [CrossRef]
- Jarjour, A.A.; Manitt, C.; Moore, S.W.; Thompson, K.M.; Yuh, S.-J.; Kennedy, T.E. Netrin-1 is a chemorepellent for oligodendrocyte precursor cells in the embryonic spinal cord. J. Neurosci. 2003, 23, 3735–3744. [Google Scholar] [CrossRef] [PubMed]
- Hart, I.K.; Richardson, W.D.; Heldin, C.-H.; Westermark, B.; Raff, M.C. PDGF receptors on cells of the oligodendrocyte-type-2 astrocyte (O-2A) cell lineage. Development 1989, 105, 595–603. [Google Scholar] [PubMed]
- Tsai, H.-H.; Tessier-Lavigne, M.; Miller, R.H. Netrin 1 mediates spinal cord oligodendrocyte precursor dispersal. Development 2003, 130, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-H.; Macklin, W.B.; Miller, R.H. Netrin-1 is required for the normal development of spinal cord oligodendrocytes. J. Neurosci. 2006, 26, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Leferink, P.S.; Breeuwsma, N.; Bugiani, M.; van der Knaap, M.S.; Heine, V.M. Affected astrocytes in the spinal cord of the leukodystrophy vanishing white matter. Glia 2018, 66, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Baron, W.; Shattil, S.J.; ffrench-Constant, C. The oligodendrocyte precursor mitogen PDGF stimulates proliferation by activation of alpha(v)beta3 integrins. EMBO J. 2002, 21, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Ousley, F.; Gospodarowicz, D. Bovine brain astrocytes express basic fibroblast growth factor, a neurotropic and angiogenic mitogen. Brain Res. 1988, 462, 223–232. [Google Scholar] [CrossRef]
- Wolswijk, G.; Noble, M. Cooperation Between PDGF and FGF Converts Slowly Dividing O-2A adutt Progenitor Cells to Rapidly Dividing Cells with Characteristics of O_2A perinatal Progenitor Cells. J. Cell Biol. 1992, 118, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Milner, R.; Anderson, H.J.; Rippon, R.F.; McKay, J.S.; Franklin, R.J.M.; Marchionni, M.A.; Reynolds, R.; Ffrench-Constant, C. Contrasting effects of mitogenic growth factors on oligodendrocyte precursor cell migration. Glia 1997, 19, 85–90. [Google Scholar] [CrossRef]
- Vora, P.; Pillai, P.P.; Zhu, W.; Mustapha, J.; Namaka, M.P.; Frost, E.E. Differential effects of growth factors on oligodendrocyte progenitor migration. Eur. J. Cell Biol. 2011, 90, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Zhang, J.-X.; Shen, L.; Qi, Q.; Cheng, X.-X.; Zhong, Z.-R.; Jiang, Z.-Q.; Wang, R.; Lü, H.-Z.; Hu, J.-G. Schwann cells induce Proliferation and Migration of Oligodendrocyte Precursor Cells Through Secretion of PDGF-AA and FGF-2. J. Mol. Neurosci. 2015, 56, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Murcia-Belmonte, V.; Medina-Rodríguez, E.M.; Bribián, A.; de Castro, F.; Esteban, P.F. ERK1/2 signaling is essential for the chemoattraction exerted by human FGF2 and human anosmin-1 on newborn rat and mouse OPCs via FGFR1. Glia 2014, 62, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Osterhout, D.J.; Ebner, S.; Xu, J.; Ornitz, D.M.; Zazanis, G.A.; McKinnon, R.D. Transplanted oligodendrocyte progenitor cells expressing a dominant-negative FGF receptor transgene fail to migrate in vivo. J. Neurosci. 1997, 17, 9122–9132. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Tani, M.; Strieter, R.M.; Ransohoff, R.M.; Miller, R.H. The chemokine growth-regulated oncogene-alpha promotes spinal cord oligodendrocyte precursor proliferation. J. Neurosci. 1998, 18, 10457–10463. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Stangel, M. Expression of the chemokine receptors CXCR1 and CXCR2 in rat oligodendroglial cells. Brain Res. Dev. Brain Res. 2001, 128, 77–81. [Google Scholar] [CrossRef]
- Padovani-Claudio, D.A.; Liu, L.; Ransohoff, R.M.; Miller, R.H. Alterations in the oligodendrocyte lineage, myelin, and white matter in adult mice lacking the chemokine receptor CXCR2. Glia 2006, 54, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Tirotta, E.; Ransohoff, R.M.; Lane, T.E. CXCR2 signaling protects oligodendrocyte progenitor cells from IFN-γ/CXCL10-mediated apoptosis. Glia 2011, 59, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-H.; Frost, E.; To, V.; Robinson, S.; ffrench-Constant, C.; Geertman, R.; Ransohoff, R.M.; Miller, R.H. The Chemokine Receptor CXCR2 Controls Positioning of Oligodendrocyte Precursors in Developing Spinal Cord by Arresting Their Migration. Cell 2002, 110, 373–383. [Google Scholar] [CrossRef]
- Li, M.; Ransohoff, R.M. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog. Neurobiol. 2008, 84, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Lavi, E.; Strizki, J.M.; Ulrich, A.M.; Zhang, W.; Fu, L.; Wang, Q.; O’Connor, M.; Hoxie, J.A.; González-Scarano, F. CXCR-4 (Fusin), a co-receptor for the type 1 human immunodeficiency virus (HIV-1), is expressed in the human brain in a variety of cell types, including microglia and neurons. Am. J. Pathol. 1997, 151, 1035–1042. [Google Scholar] [PubMed]
- McGrath, K.E.; Koniski, A.D.; Maltby, K.M.; McGann, J.K.; Palis, J. Embryonic Expression and Function of the Chemokine SDF-1 and Its Receptor, CXCR4. Dev. Biol. 1999, 213, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Dziembowska, M.; Tham, T.N.; Lau, P.; Vitry, S.; Lazarini, F.; Dubois-Dalcq, M.; Lau, P. A Role for CXCR4 Signaling in Survival and Migration of Neural and Oligodendrocyte Precursors. Glia 2005, 50, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Kadi, L.; Selvaraju, R.; de Lys, P.; Proudfoot, A.E.I.; Wells, T.N.C.; Boschert, U. Differential effects of chemokines on oligodendrocyte precursor proliferation and myelin formation in vitro. J. Neuroimmunol. 2006, 174, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; David, S.; Patel, R.; Abney, E.R.; Raff, M.C. A quantitative immunohistochemical study of macroglial cell development in the rat optic nerve: in vivo evidence for two distinct astrocyte lineages. Dev. Biol. 1985, 111, 35–41. [Google Scholar] [CrossRef]
- Hill, R.A.; Patel, K.D.; Medved, J.; Reiss, A.M.; Nishiyama, A. NG2 cells in white matter but not gray matter proliferate in response to PDGF. J. Neurosci. 2013, 33, 14558–14566. [Google Scholar] [CrossRef] [PubMed]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.-J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Rivers, L.E.; Young, K.M.; Rizzi, M.; Jamen, F.; Psachoulia, K.; Wade, A.; Kessaris, N.; Richardson, W.D. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 2008, 11, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Accetta, R.; Damiano, S.; Morano, A.; Mondola, P.; Paternò, R.; Avvedimento, E.V.; Santillo, M. Reactive Oxygen Species Derived from NOX3 and NOX5 Drive Differentiation of Human Oligodendrocytes. Front. Cell. Neurosci. 2016, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.; Smith, J.; Ladi, E.; Mayer-Proschel, M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc. Natl. Acad. Sci. USA 2000, 97, 10032–10037. [Google Scholar] [CrossRef]
- Clarke, L.E.; Young, K.M.; Hamilton, N.B.; Li, H.; Richardson, W.D.; Attwell, D. Properties and fate of oligodendrocyte progenitor cells in the corpus callosum, motor cortex, and piriform cortex of the mouse. J. Neurosci. 2012, 32, 8173–8185. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.; Murray, K. Purified astrocytes promote the in vitro division of a bipotential glial progenitor cell. EMBO J. 1984, 3, 2243–2247. [Google Scholar] [PubMed]
- Raff, M.C.; Lillien, L.E.; Richardson, W.D.; Burne, J.F.; Noble, M.D. Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature 1988, 333, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-G.; Fu, S.-L.; Wang, Y.-X.; Li, Y.; Jiang, X.-Y.; Wang, X.-F.; Qiu, M.-S.; Lu, P.-H.; Xu, X.-M. Platelet-derived growth factor-AA mediates oligodendrocyte lineage differentiation through activation of extracellular signal-regulated kinase signaling pathway. Neuroscience 2008, 151, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta - Mol. Cell Res. 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, R.D.; Matsui, T.; Dubois-Dalcq, M.; Aaronson, S.A. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron 1990, 5, 603–614. [Google Scholar] [CrossRef]
- Raff, M.C.; Miller, R.H.; Noble, M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 1983, 303, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 1997, 124, 2691–2700. [Google Scholar] [PubMed]
- Fruttiger, M.; Karlsson, L.; Hall, A.C.; Abramsson, A.; Calver, A.R.; Bostrom, H.; Willetts, K.; Bertold, C.H.; Heath, J.K.; Betsholtz, C.; et al. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development 1999, 126, 457–467. [Google Scholar] [PubMed]
- Calver, A.R.; Hall, A.C.; Yu, W.-P.; Walsh, F.S.; Heath, J.K.; Betsholtz, C.; Richardson, W.D. Oligodendrocyte Population Dynamics and the Role of PDGF In Vivo. Neuron 1998, 20, 869–882. [Google Scholar] [CrossRef]
- Hu, J.-G.; Wang, X.-F.; Deng, L.-X.; Liu, N.-K.; Gao, X.; Chen, J.; Zhou, F.C.; Xu, X.-M. Cotransplantation of Glial Restricted Precursor Cells and Schwann Cells Promotes Functional Recovery after Spinal Cord Injury. Cell Transplant. 2013, 22, 2219–2236. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, R.D.; Matsui, T.; Aranda, M.; Dubois-Dalcq, M. A Role for Fibroblast Growth Factor in Oligodendrocyte Development. Ann. N. Y. Acad. Sci. 1991, 638, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Gard, A.L.; Pfeiffer, S.E. Glial Cell Mitogens bFGF and PDGF Differentially Regulate Development of O4+GalC- Oligodendrocyte Progenitors. Dev. Biol. 1993, 159, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Bögler, O.; Wren, D.; Barnett, S.C.; Land, H.; Noble, M. Cooperation between two growth factors promotes extended self-renewal and inhibits differentiation of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proc. Natl. Acad. Sci. USA 1990, 87, 6368–6372. [Google Scholar] [CrossRef] [PubMed]
- Eccleston, P.A.; Silberberg, D.H. Fibroblast growth factor is a mitogen for oligodendrocytes in vitro. Brain Res. 1985, 353, 315–318. [Google Scholar] [CrossRef]
- Vaccarino, F.M.; Schwartz, M.L.; Raballo, R.; Nilsen, J.; Rhee, J.; Zhou, M.; Doetschman, T.; Coffin, J.D.; Wyland, J.J.; Hung, Y.-T.E. Erratum: Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat. Neurosci. 1999, 2, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Murtie, J.C.; Zhou, Y.-X.; Le, T.Q.; Armstrong, R.C. In vivo analysis of oligodendrocyte lineage development in postnatal FGF2 null mice. Glia 2005, 49, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Kumar, M.; Murray, K.; Morrison, R.S.; Pfeiffer, S.E. Regulation of FGF Receptors in the Oligodendrocyte Lineage. Mol. Cell. Neurosci. 1996, 7, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.; Rom, E.; Sun, H.; Yayon, A.; Bansal, R. Distinct Fibroblast Growth Factor (FGF)/FGF Receptor Signaling Pairs Initiate Diverse Cellular Responses in the Oligodendrocyte Lineage. J. Neurosci. 2005, 25, 7470–7479. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-X.; Flint, N.C.; Murtie, J.C.; Le, T.Q.; Armstrong, R.C. Retroviral lineage analysis of fibroblast growth factor receptor signaling in FGF2 inhibition of oligodendrocyte progenitor differentiation. Glia 2006, 54, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Goddard, D.R.; Berry, M.; Kirvell, S.L.; Butt, A.M. Fibroblast Growth Factor-2 Inhibits Myelin Production by Oligodendrocytes in Vivo. Mol. Cell. Neurosci. 2001, 18, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Fressinaud, C.; Vallat, J.M.; Labourdette, G. Basic fibroblast growth factor down-regulates myelin basic protein gene expression and alters myelin compaction of mature oligodendrocytes in vitro. J. Neurosci. Res. 1995, 40, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Colognato, H.; ffrench-Constant, C. Contrasting effects of mitogenic growth factors on myelination in neuron–oligodendrocyte co-cultures. Glia 2007, 55, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Frederick, T.J.; Wood, T.L. IGF-I Synergizes with FGF-2 to Stimulate Oligodendrocyte Progenitor Entry into the Cell Cycle. Dev. Biol. 2001, 232, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Shemer, J.; Raizada, M.K.; Masters, B.A.; Ota, A.; Leroith, D. Insulin-like Growth Factor I Receptors in Neuronal and Glial Cells CHARACTERIZATION AND BIOLOGICAL EFFECTS IN PRIMARY CULTURE*. J. Biol. Chem. 1987, 262, 7693–7699. [Google Scholar] [PubMed]
- Jones, J.I.; Clemmons, D.R. Insulin-Like Growth Factors and Their Binding Proteins: Biological Actions*. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [CrossRef] [PubMed]
- McMorris, F.A.; Smith, T.M.; DeSalvo, S.; Furlanetto, R.W. Insulin-like growth factor I/somatomedin C: a potent inducer of oligodendrocyte development. Proc. Natl. Acad. Sci. USA 1986, 83, 822–826. [Google Scholar] [CrossRef] [PubMed]
- McMorris, F.A.; Dubois-Dalcq, M. Insulin-like growth factor I promotes cell proliferation and oligodendroglial commitment in rat glial progenitor cells developing in vitro. J. Neurosci. Res. 1988, 21, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Popken, G.J.; Hodge, R.D.; Ye, P.; Zhang, J.; Ng, W.; O’Kusky, J.R.; D’Ercole, A.J. In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur. J. Neurosci. 2004, 19, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.D.; D’Ercole, A.J.; O’Kusky, J.R. Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J. Neurosci. 2004, 24, 10201–10210. [Google Scholar] [CrossRef] [PubMed]
- Frederick, T.J.; Min, J.; Altieri, S.C.; Mitchell, N.E.; Wood, T.L. Synergistic induction of cyclin D1 in oligodendrocyte progenitor cells by IGF-I and FGF-2 requires differential stimulation of multiple signaling pathways. Glia 2007, 55, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.-L.; Almazan, G. IGF-I-induced oligodendrocyte progenitor proliferation requires PI3K/Akt, MEK/ERK, and Src-like tyrosine kinases. J. Neurochem. 2007, 100, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Palacios, N.; Sanchez-Franco, F.; Fernandez, M.; Sanchez, I.; Cacicedo, L. Intracellular events mediating insulin-like growth factor I-induced oligodendrocyte development: modulation by cyclic AMP. J. Neurochem. 2005, 95, 1091–1107. [Google Scholar] [CrossRef] [PubMed]
- Frederick, T.J.; Wood, T.L. IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol. Cell. Neurosci. 2004, 25, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Mairet-Coello, G.; Tury, A.; DiCicco-Bloom, E. Insulin-like growth factor-1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J. Neurosci. 2009, 29, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Hart, I.K.; Coles, H.S.R.; Burne, J.F.; Voyvodic, J.T.; Richardson, W.D.; Raff, M.C. Cell death in the oligodendrocyte lineage. J. Neurobiol. 1992, 23, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Schmid, R.; Sendnter, M.; Raff, M.C. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development 1993, 118, 283–295. [Google Scholar] [PubMed]
- Pang, Y.; Zheng, B.; Fan, L.-W.; Rhodes, P.G.; Cai, Z. IGF-1 protects oligodendrocyte progenitors against TNFα-induced damage by activation of PI3K/Akt and interruption of the mitochondrial apoptotic pathway. Glia 2007, 55, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V. The p75 neurotrophin receptor. J. Neurobiol. 1994, 25, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Kilpatrick, T.J.; Murray, S.S. The Role of Neurotrophins in the Regulation of Myelin Development. Neurosignals 2009, 17, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Barbacid, M. Neurotrophic factors and their receptors. Curr. Opin. Cell Biol. 1995, 7, 148–155. [Google Scholar] [CrossRef]
- Chao, M.V.; Hempstead, B.L. p75 and Trk: a two-receptor system. Trends Neurosci. 1995, 18, 321–326. [Google Scholar] [CrossRef]
- Van’t Veer, A.; Du, Y.; Fischer, T.Z.; Boetig, D.R.; Wood, M.R.; Dreyfus, C.F. Brain-derived neurotrophic factor effects on oligodendrocyte progenitors of the basal forebrain are mediated through trkB and the MAP kinase pathway. J. Neurosci. Res. 2009, 87, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Fischer, T.Z.; Clinton-Luke, P.; Lercher, L.D.; Dreyfus, C.F. Distinct effects of p75 in mediating actions of neurotrophins on basal forebrain oligodendrocytes. Mol. Cell. Neurosci. 2006, 31, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Chu, A.K.; Sato-Bigbee, C. Possible role of CREB in the stimulation of oligodendrocyte precursor cell proliferation by neurotrophin-3. J. Neurochem. 2000, 74, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Raff, M.C.; Gaese, F.; Bartke, I.; Dechant, G.; Barde, Y.-A. A crucial role for neurotrophin-3 in oligodendrocyte development. Nature 1994, 367, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.A.; Kumar, S.; Liebl, D.; Chang, R.; Parada, L.F.; De Vellis, J. Mice lacking NT-3, and its receptor TrkC, exhibit profound deficiencies in CNS glial cells. Glia 1999, 26, 153–165. [Google Scholar] [CrossRef]
- Olguín-Albuerne, M.; Morán, J. Redox Signaling Mechanisms in Nervous System Development. Antioxid. Redox Signal. 2018, 28, 1603–1625. [Google Scholar] [CrossRef] [PubMed]
- Caillava, C.; Baron-Van Evercooren, A. Differential requirement of cyclin-dependent kinase 2 for oligodendrocyte progenitor cell proliferation and differentiation. Cell Div. 2012, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Belachew, S.; Aguirre, A.A.; Wang, H.; Vautier, F.; Yuan, X.; Anderson, S.; Kirby, M.; Gallo, V. Cyclin-dependent kinase-2 controls oligodendrocyte progenitor cell cycle progression and is downregulated in adult oligodendrocyte progenitors. J. Neurosci. 2002, 22, 8553–8562. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jolson, D.M.; Cuta, K.K.; Mariash, C.N.; Anderson, G.W. Triiodothyronine is a survival factor for developing oligodendrocytes. Mol. Cell. Endocrinol. 2003, 199, 49–60. [Google Scholar] [CrossRef]
- Ahlgren, S.C.; Wallace, H.; Bishop, J.; Neophytou, C.; Raff, M.C. Effects of Thyroid Hormone on Embryonic Oligodendrocyte Precursor Cell Development in Vivo and in Vitro. Mol. Cell. Neurosci. 1997, 9, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.-B.; Apperly, J.; Raff, M. Cell-Intrinsic Timers and Thyroid Hormone Regulate the Probability of Cell-Cycle Withdrawal and Differentiation of Oligodendrocyte Precursor Cells. Dev. Biol. 1998, 197, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Lazar, M.A.; Raff, M.C. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 1994, 120, 1097–1108. [Google Scholar] [PubMed]
- Sarliève, L.L.; Rodríguez-Peña, A.; Langley, K. Expression of Thyroid Hormone Receptor Isoforms in the Oligodendrocyte Lineage. Neurochem. Res. 2003, 29, 903–922. [Google Scholar] [CrossRef]
- Baas, D.; Fressinaud, C.; Ittel, M.E.; Reeber, A.; Dalençon, D.; Puymirat, J.; Sarliève, L.L. Expression of thyroid hormone receptor isoforms in rat oligodendrocyte cultures. Effect of 3,5,3′-triiodo-l-thyronine. Neurosci. Lett. 1994, 176, 47–51. [Google Scholar] [CrossRef]
- Carré, J.-L.; Demerens, C.; Rodríguez-Peñ, A.; Floch, H.H.; Vincendon, G.; Sarliève, L.L. Thyroid Hormone Receptor Isoforms Are Sequentially Expressed in Oligodendrocyte Lineage Cells During Rat Cerebral Development. J. Neurosci. Res 1998, 54, 584–594. [Google Scholar] [CrossRef]
- Almazan, G.; Honegger, P.; Matthieu, J.M. Triiodothyronine stimulation of oligodendroglial differentiation and myelination. A developmental study. Dev. Neurosci. 1985, 7, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Baas, D.; Bourbeau, D.; Sarli Ve, L.L.; Ittel, M.-E.; Dussault, J.H.; Puymirat, J. Oligodendrocyte Maturation and Progenitor Cell Proliferation Are Independently Regulated by Thyroid Hormone. Glia 1997, 19, 324–332. [Google Scholar] [CrossRef]
- Almeida, R.; Lyons, D. Oligodendrocyte Development in the Absence of Their Target Axons In Vivo. PLoS ONE 2016, 11, e0164432. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.D.; Powell-Braxton, L.; Widmer, H.R.; Valverde, J.; Hefti, F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 1995, 14, 717–730. [Google Scholar] [CrossRef]
- Zeger, M.; Popken, G.; Zhang, J.; Xuan, S.; Lu, Q.R.; Schwab, M.H.; Nave, K.-A.; Rowitch, D.; D’Ercole, A.J.; Ye, P. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normalin vivo oligodendrocyte development and myelination. Glia 2007, 55, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Behringer, R.R.; Brinster, R.L.; McMorris, F.A. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron 1993, 10, 729–740. [Google Scholar] [CrossRef]
- Ye, P.; Carson, J.; D’Ercole, A.J. In vivo actions of insulin-like growth factor-I (IGF-I) on brain myelination: studies of IGF-I and IGF binding protein-1 (IGFBP-1) transgenic mice. J. Neurosci. 1995, 15, 7344–7356. [Google Scholar] [CrossRef] [PubMed]
- Mozell, R.L.; McMorris, F.A. Insulin-like growth factor I stimulates oligodendrocyte development and myelination in rat brain aggregate cultures. J. Neurosci. Res. 1991, 30, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Luzi, P.; Zaka, M.; Rao, H.Z.; Curtis, M.; Rafi, M.A.; Wenger, D.A. Generation of transgenic mice expressing insulin-like growth factor-1 under the control of the myelin basic protein promoter: increased myelination and potential for studies on the effects of increased IGF-1 on experimentally and genetically induced demyelination. Neurochem. Res. 2004, 29, 881–889. [Google Scholar] [PubMed]
- Adler, R.; Landa, K.B.; Manthorpe, M.; Varon, S. Cholinergic neuronotrophic factors: intraocular distribution of trophic activity for ciliary neurons. Science 1979, 204, 1434–1436. [Google Scholar] [CrossRef] [PubMed]
- Stöckli, K.A.; Lillien, L.E.; Näher-Noé, M.; Breitfeld, G.; Hughes, R.A.; Raff, M.C.; Thoenen, H.; Sendtner, M. Regional Distribution, Developmental Changes, and Cellular Localization of CNTF-mRNA and Protein in the Rat Brain. J. Cell Biol. 1991, 115, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Burne, J.F.; Holtmann, B.; Thoenen, H.; Sendtner, M.; Raff, M.C. Ciliary Neurotrophic Factor Enhances the Rate of Oligodendrocyte Generation. Mol. Cell. Neurosci. 1996, 8, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Marmur, R.; Kessler, J.A.; Zhu, G.; Gokhan, S.; Mehler, M.F. Differentiation of oligodendroglial progenitors derived from cortical multipotent cells requires extrinsic signals including activation of gp130/LIFbeta receptors. J. Neurosci. 1998, 18, 9800–9811. [Google Scholar] [CrossRef] [PubMed]
- Stankoff, B.; Aigrot, M.-S.; Noël, F.; Wattilliaux, A.; Zalc, B.; Lubetzki, C. Ciliary neurotrophic factor (CNTF) enhances myelin formation: A novel role for CNTF and CNTF-related molecules. J. Neurosci. 2002, 22, 9221–9227. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola, N.; Mayer-Pröschel, M.; Rodriguez-Peña, A.; Noble, M. Evidence for the Existence of at Least Two Timing Mechanisms That Contribute to Oligodendrocyte Generationin Vitro. Dev. Biol. 1996, 180, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Bhakoo, K.; Noble, M. Ciliary neurotrophic factor and leukemia inhibitory factor promote the generation, maturation and survival of oligodendrocytes in vitro. Development 1994, 120, 143–153. [Google Scholar] [PubMed]
- Salehi, Z.; Hadiyan, S.P.; Navidi, R. Ciliary Neurotrophic Factor Role in Myelin Oligodendrocyte Glycoprotein Expression in Cuprizone-Induced Multiple Sclerosis Mice. Cell. Mol. Neurobiol. 2013, 33, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Mäurer, M.; Gaupp, S.; Martini, R.; Holtmann, B.; Giess, R.; Rieckmann, P.; Lassmann, H.; Toyka, K.V.; Sendtner, M.; et al. CNTF is a major protective factor in demyelinating CNS disease: A neurotrophic cytokine as modulator in neuroinflammation. Nat. Med. 2002, 8, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Ernfors, P.; Ibáñez, C.F.; Ebendal, T.; Olson, L.; Persson, H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5454–5458. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Gorath, M.; Richter-Landsberg, C. Neurotrophin-3 (NT-3) Modulates Early Differentiation of Oligodendrocytes in Rat Brain Cortical Cultures. Glia 1999, 28, 244–255. [Google Scholar] [CrossRef]
- Rubio, N.; Rodriguez, R.; Arevalo, M.A. In vitro myelination by oligodendrocyte precursor cells transfected with the neurotrophin-3 gene. Glia 2004, 47, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Lercher, L.D.; Zhou, R.; Dreyfus, C.F. Mitogen-activated protein kinase pathway mediates effects of brain-derived neurotrophic factor on differentiation of basal forebrain oligodendrocytes. J. Neurosci. Res. 2006, 84, 1692–1702. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Wong, A.W.; Willingham, M.M.; van den Buuse, M.; Kilpatrick, T.J.; Murray, S.S. Brain-Derived Neurotrophic Factor Promotes Central Nervous System Myelination via a Direct Effect upon Oligodendrocytes. Neurosignals 2010, 18, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Cosgaya, J.M.; Chan, J.R.; Shooter, E.M. The Neurotrophin Receptor p75NTR as a Positive Modulator of Myelination. Science 2002, 298, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Cellerino, A.; Carroll, P.; Thoenen, H.; Barde, Y.-A. Reduced Size of Retinal Ganglion Cell Axons and Hypomyelination in Mice Lacking Brain-Derived Neurotrophic Factor. Mol. Cell. Neurosci. 1997, 9, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Djalali, S.; Holtje, M.; Grosse, G.; Rothe, T.; Stroh, T.; Grosse, J.; Deng, D.R.; Hellweg, R.; Grantyn, R.; Hortnagl, H.; et al. Effects of brain-derived neurotrophic factor (BDNF) on glial cells and serotonergic neurones during development. J. Neurochem. 2005, 92, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Vondran, M.W.; Clinton-Luke, P.; Honeywell, J.Z.; Dreyfus, C.F. BDNF+/- mice exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia 2010, 58, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.W.; Xiao, J.; Kemper, D.; Kilpatrick, T.J.; Murray, S.S. Oligodendroglial expression of TrkB independently regulates myelination and progenitor cell proliferation. J. Neurosci. 2013, 33, 4947–4957. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Fischer, T.Z.; Lee, L.N.; Lercher, L.D.; Dreyfus, C.F. Regionally Specific Effects of BDNF on Oligodendrocytes. Dev. Neurosci. 2003, 25, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Nave, K.-A. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 2014, 83, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Canoll, P.D.; Kraemer, R.; Teng, K.K.; Marchionni, M.A.; Salzer, J.L. GGF/Neuregulin Induces a Phenotypic Reversion of Oligodendrocytes. Mol. Cell. Neurosci. 1999, 13, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.-K.; Kosciuczyk, K.; Tapley, L.; Karimi-Abdolrezaee, S. Dysregulation of the neuregulin-1-ErbB network modulates endogenous oligodendrocyte differentiation and preservation after spinal cord injury. Eur. J. Neurosci. 2013, 38, 2693–2715. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.I.; Mallon, B.S.; Matsui, T.; Ogawa, W.; Rosenzweig, A.; Okamoto, T.; Macklin, W.B. Akt-mediated survival of oligodendrocytes induced by neuregulins. J. Neurosci. 2000, 20, 7622–7630. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.A.; Tang, D.G.; Cheng, L.; Prochiantz, A.; Mudge, A.W.; Raff, M.C. Evidence that axon-derived neuregulin promotes oligodendrocyte survival in the developing rat optic nerve. Neuron 2000, 28, 81–90. [Google Scholar] [CrossRef]
- Colognato, H.; Baron, W.; Avellana-Adalid, V.; Relvas, J.B.; Evercooren, A.B.-V.; Georges-Labouesse, E.; ffrench-Constant, C. CNS integrins switch growth factor signalling to promote target-dependent survival. Nat. Cell Biol. 2002, 4, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Colognato, H.; Ramachandrappa, S.; Olsen, I.M.; ffrench-Constant, C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J. Cell Biol. 2004, 167, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Sun, Q.; Oglesbee, M.; Yoon, S.O. The role of ErbB2 signaling in the onset of terminal differentiation of oligodendrocytes in vivo. J. Neurosci. 2003, 23, 5561–5571. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Murtie, J.C.; El-Khodor, B.F.; Edgar, N.; Sardi, S.P.; Hooks, B.M.; Benoit-Marand, M.; Chen, C.; Moore, H.; O’Donnell, P.; Brunner, D.; Corfas, G. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 8131–8136. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Miller, R.; Krane, I.; Vartanian, T. The erbB2 gene is required for the development of terminally differentiated spinal cord oligodendrocytes. J. Cell Biol. 2001, 154, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Küspert, M.; Wegner, M. SomethiNG 2 talk about—Transcriptional regulation in embryonic and adult oligodendrocyte precursors. Brain Res. 2016, 1638, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Traiffort, E.; Zakaria, M.; Laouarem, Y.; Ferent, J. Hedgehog: A Key Signaling in the Development of the Oligodendrocyte Lineage. J. Dev. Biol. 2016, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Crowe, M.J.; Bresnahan, J.C.; Shuman, S.L.; Masters, J.N.; Crowe, M.S. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 1997, 3, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Lytle, J.M.; Vicini, S.; Wrathall, J.R. Phenotypic changes in NG2+ cells after spinal cord injury. J. Neurotrauma 2006, 23, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.D.; Rosenberg, L.J.; Wrathall, J.R. Temporal-spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp. Neurol. 2001. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Wei, P.; Stokes, B.T. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J. Neurosci. 2001, 21, 3392–3400. [Google Scholar] [CrossRef] [PubMed]
- Lotocki, G.; de Rivero Vaccari, J.; Alonso, O.; Molano, J.S.; Nixon, R.; Dietrich, W.D.; Bramlett, H.M. Oligodendrocyte Vulnerability Following Traumatic Brain Injury in Rats: Effect of Moderate Hypothermia. Ther. Hypothermia Temp. Manag. 2011, 1, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Gensert, J.M.; Goldman, J.E. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 1997, 19, 197–203. [Google Scholar] [CrossRef]
- Hesp, Z.C.; Goldstein, E.Z.; Goldstein, E.A.; Miranda, C.J.; Kaspar, B.K.; Kaspar, B.K.; McTigue, D.M. Chronic oligodendrogenesis and remyelination after spinal cord injury in mice and rats. J. Neurosci. 2015, 35, 1274–1290. [Google Scholar] [CrossRef] [PubMed]
- Sellers, D.L.; Maris, D.O.; Horner, P.J. Postinjury Niches Induce Temporal Shifts in Progenitor Fates to Direct Lesion Repair after Spinal Cord Injury. J. Neurosci. 2009, 29, 6722–6733. [Google Scholar] [CrossRef] [PubMed]
- Zai, L.J.; Wrathall, J.R. Cell proliferation and replacement following contusive spinal cord injury. Glia 2005, 50, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Barnabé-Heider, F.; Göritz, C.; Sabelström, H.; Takebayashi, H.; Pfrieger, F.W.; Meletis, K.; Frisén, J. Origin of New Glial Cells in Intact and Injured Adult Spinal Cord. Cell Stem Cell 2010, 7, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Meletis, K.; Barnabé-Heider, F.; Carlén, M.; Evergren, E.; Tomilin, N.; Shupliakov, O.; Frisén, J. Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 2008, 6, e182. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Abdolrezaee, S.; Billakanti, R. Reactive Astrogliosis after Spinal Cord Injury—Beneficial and Detrimental Effects. Mol. Neurobiol. 2012, 46, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, S.; Hamilton, L.K.; Vaugeois, A.; Beaudoin, S.; Breault-Dugas, C.; Pineau, I.; Lévesque, S.A.; Grégoire, C.-A.; Fernandes, K.J.L. Central Canal Ependymal Cells Proliferate Extensively in Response to Traumatic Spinal Cord Injury but Not Demyelinating Lesions. PLoS ONE 2014, 9, e85916. [Google Scholar] [CrossRef] [PubMed]
- BUNGE, M.B.; BUNGE, R.P.; RIS, H. Ultrastructural study of remyelination in an experimental lesion in adult cat spinal cord. J. Biophys. Biochem. Cytol. 1961, 10, 67–94. [Google Scholar] [CrossRef] [PubMed]
- Gledhill, R.F.; Harrison, B.M.; McDonald, W.I. Demyelination and Remyelination after Acute Spinal Cord Compression. Exp. Neurol. 1973, 38, 472–487. [Google Scholar] [CrossRef]
- Rasminsky, M.; Sears, T.A. Internodal conduction in undissected demyelinated nerve fibres. J. Physiol. 1972, 227, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Mcdonald, W.I. Pathophysiology in multiple sclerosis. Brain 1974, 97, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Wrathall, J.R.; Li, W.; Hudson, L.D. Myelin gene expression after experimental contusive spinal cord injury. J. Neurosci. 1998, 18, 8780–8793. [Google Scholar] [CrossRef] [PubMed]
- Totoiu, M.O.; Keirstead, H.S. Spinal cord injury is accompanied by chronic progressive demyelination. J. Comp. Neurol. 2005, 486, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Guest, J.D.; Hiester, E.D.; Bunge, R.P. Demyelination and Schwann cell responses adjacent to injury epicenter cavities following chronic human spinal cord injury. Exp. Neurol. 2005, 192, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Blight, A.R. Delayed Demyelination and Macrophage Invasion: A Candidate for Secondary Cell Damage in Spinal Cord Injury. Cent. Nerv. Syst. Trauma 1985, 2, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Lee, W.T.; Bokara, K.K.; Seo, S.K.; Park, S.H.; Kim, J.H.; Yenari, M.A.; Park, K.A.; Lee, J.E. The Multifaceted Effects of Agmatine on Functional Recovery after Spinal Cord Injury through Modulations of BMP-2/4/7 Expressions in Neurons and Glial Cells. PLoS ONE 2013, 8, e53911. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, T.; Yone, K.; Matsuoka, E.; Takenouchi, H.; Nakashima, K.; Sakou, T.; Komiya, S.; Izumo, S. Traumatic injury-induced BMP7 expression in the adult rat spinal cord. Brain Res. 2001, 921, 219–225. [Google Scholar] [CrossRef]
- Grinspan, J.B.; Edell, E.; Carpio, D.F.; Beesley, J.S.; Lavy, L.; Pleasure, D.; Golden, J.A. Stage-specific effects of bone morphogenetic proteins on the oligodendrocyte lineage. J. Neurobiol. 2000, 43, 1–17. [Google Scholar] [CrossRef]
- Sabo, J.K.; Aumann, T.D.; Merlo, D.; Kilpatrick, T.J.; Cate, H.S. Remyelination Is Altered by Bone Morphogenic Protein Signaling in Demyelinated Lesions. J. Neurosci. 2011, 31, 4504–4510. [Google Scholar] [CrossRef] [PubMed]
- See, J.; Zhang, X.; Eraydin, N.; Golden, J.A.; Grinspan, J.B. Oligodendrocyte maturation is inhibited by bone morphogenetic protein. Mol. Cell. Neurosci. 2004, 26, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Cate, H.S.; Sabo, J.K.; Merlo, D.; Kemper, D.; Aumann, T.D.; Robinson, J.; Merson, T.D.; Emery, B.; Perreau, V.M.; Kilpatrick, T.J. Modulation of bone morphogenic protein signalling alters numbers of astrocytes and oligodendroglia in the subventricular zone during cuprizone-induced demyelination. J. Neurochem. 2010, 115, 11–22. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, C.; Fernández-Martos, C.M.; Shields, S.D.; Arenas, E.; Javier Rodríguez, F. Wnts are expressed in the spinal cord of adult mice and are differentially induced after injury. J. Neurotrauma 2014, 31, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Martos, C.M.; González-Fernández, C.; González, P.; Maqueda, A.; Arenas, E.; Rodríguez, F.J. Differential expression of Wnts after spinal cord contusion injury in adult rats. PLoS ONE 2011, 6, e27000. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.P.; Coulter, M.; Miotke, J.; Meyer, R.L.; Takemaru, K.-I.; Levine, J.M. Abrogation of β-catenin signaling in oligodendrocyte precursor cells reduces glial scarring and promotes axon regeneration after CNS injury. J. Neurosci. 2014, 34, 10285–10297. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.-F.; Wang, Y.; Lin, Y.-H.; Wu, Y.; Zhu, A.-Y.; Wang, R.; Shen, L.; Xi, J.; Qi, Q.; Jiang, Z.-Q.; Lü, H.-Z.; Hu, J.-G. Transplantation of PDGF-AA-Overexpressing Oligodendrocyte Precursor Cells Promotes Recovery in Rat Following Spinal Cord Injury. Front. Cell. Neurosci. 2017, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Parikh, Z.S.; Vora, P.; Frost, E.E.; Pillai, P.P. pERK1/2 Peripheral Recruitment and Filopodia Protrusion Augment Oligodendrocyte Progenitor Cell Migration: Combined Effects of PDGF-A and Fibronectin. Cell. Mol. Neurobiol. 2017, 37, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cheng, X.; Qi, J.; Xie, B.; Zhao, X.; Zheng, K.; Zhang, Z.; Qiu, M. EGF Enhances Oligodendrogenesis from Glial Progenitor Cells. Front. Mol. Neurosci. 2017, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.B.; McTigue, D.M. Chronically increased ciliary neurotrophic factor and fibroblast growth factor-2 expression after spinal contusion in rats. J. Comp. Neurol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Louis, J.C.; Magal, E.; Takayama, S.; Varon, S. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science 1993, 259, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Oyesiku, N.M.; Wilcox, J.N.; Wigston, D.J. Changes in expression of ciliary neurotrophic factor (CNTF) and CNTF-receptor alpha after spinal cord injury. J. Neurobiol. 1997, 32, 251–261. [Google Scholar] [CrossRef]
- Oyesiku, N.M.; Evans, C.-O.; Houston, S.; Darrell, R.S.; Smith, J.S.; Fulop, Z.L.; Dixon, C.E.; Stein, D.G. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999, 833, 161–172. [Google Scholar] [CrossRef]
- Cao, Q.; He, Q.; Wang, Y.; Cheng, X.; Howard, R.M.; Zhang, Y.; DeVries, W.H.; Shields, C.B.; Magnuson, D.S.K.; Xu, X.-M.; et al. Transplantation of ciliary neurotrophic factor-expressing adult oligodendrocyte precursor cells promotes remyelination and functional recovery after spinal cord injury. J. Neurosci. 2010, 30, 2989–3001. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Horner, P.J.; Stokes, B.T.; Gage, F.H. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J. Neurosci. 1998, 18, 5354–5365. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.M.; Guénard, V.; Kleitman, N.; Aebischer, P.; Bunge, M.B. A Combination of BDNF and NT-3 Promotes Supraspinal Axonal Regeneration into Schwann Cell Grafts in Adult Rat Thoracic Spinal Cord. Exp. Neurol. 1995, 134, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Seidlits, S.K.; Goodman, A.G.; Kukushliev, T.V.; Hassani, D.M.; Cummings, B.J.; Anderson, A.J.; Shea, L.D. Sonic hedgehog and neurotrophin-3 increase oligodendrocyte numbers and myelination after spinal cord injury. Integr. Biol. (Camb). 2014, 6, 694–705. [Google Scholar] [CrossRef] [PubMed]
- McCreedy, D.A.; Wilems, T.S.; Xu, H.; Butts, J.C.; Brown, C.R.; Smith, A.W.; Sakiyama-Elbert, S.E. Survival, Differentiation, and Migration of High-Purity Mouse Embryonic Stem Cell-derived Progenitor Motor Neurons in Fibrin Scaffolds after Sub-Acute Spinal Cord Injury. Biomater. Sci. 2014, 2, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Bartholdi, D.; Schwab, M.E. Oligodendroglial reaction following spinal cord injury in rat: transient upregulation of MBP mRNA. Glia 1998, 23, 278–284. [Google Scholar] [CrossRef]
- Dougherty, K.D.; Dreyfus, C.F.; Black, I.B. Brain-Derived Neurotrophic Factor in Astrocytes, Oligodendrocytes, and Microglia/Macrophages after Spinal Cord Injury. Neurobiol. Dis. 2000, 7, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, O.; Murakami, M.; Ino, H.; Yamazaki, M.; Nemoto, T.; Koda, M.; Nakayama, C.; Moriya, H. Acute up-regulation of brain-derived neurotrophic factor expression resulting from experimentally induced injury in the rat spinal cord. Acta Neuropathol. 2001, 102, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, O.; Murakami, M.; Ino, H.; Yamazaki, M.; Koda, M.; Nakayama, C.; Moriya, H. Effects of Brain-Derived Neurotrophic Factor (BDNF) on Compression-Induced Spinal Cord Injury: BDNF Attenuates Down-Regulation of Superoxide Dismutase Expression and Promotes Up-Regulation of Myelin Basic Protein Expression. J. Neuropathol. Exp. Neurol. 2002, 61, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, W.; Cho, H.M.; Ouyang, H.; Li, W.; Lin, Y.; Do, J.; Zhang, L.; Ding, S.; Liu, Y.; et al. Integration and long distance axonal regeneration in the central nervous system from transplanted primitive neural stem cells. J. Biol. Chem. 2013, 288, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.S.; Nyberg, F.; Westman, J.; Alm, P.; Gordh, T.; Lindholm, D. Brain derived neurotrophic factor and insulin like growth factor-1 attenuate upregulation of nitric oxide synthase and cell injury following trauma to the spinal cord. Amino Acids 1998, 14, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.K.; Kim, J.W.; Kang, K.S.; Lee, Y.H.; Hua, Q.H.; Park, T.I.; Park, J.Y.; Sohn, Y.K. The Role of Inducible Nitric Oxide Synthase Following Spinal Cord Injury in Rat. J. Korean Med. Sci. 2005, 20, 663. [Google Scholar] [CrossRef] [PubMed]
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, Q.; Wu, Y.; Hu, W.; Gu, P.; Fu, Z. Combined therapy of methylprednisolone and brain-derived neurotrophic factor promotes axonal regeneration and functional recovery after spinal cord injury in rats. Chin. Med. J. (Engl). 2003, 116, 414–418. [Google Scholar] [PubMed]
- Hu, J.-G.; Shen, L.; Wang, R.; Wang, Q.-Y.; Zhang, C.; Xi, J.; Ma, S.-F.; Zhou, J.-S.; Lü, H.-Z. Effects of Olig2-overexpressing neural stem cells and myelin basic protein-activated T cells on recovery from spinal cord injury. Neurotherapeutics 2012, 9, 422–445. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cejudo, J.; Gutierrez-Fernandez, M.; Otero-Ortega, L.; Rodriguez-Frutos, B.; Fuentes, B.; Vallejo-Cremades, M.T.; Hernanz, T.N.; Cerdan, S.; Diez-Tejedor, E. Brain-Derived Neurotrophic Factor Administration Mediated Oligodendrocyte Differentiation and Myelin Formation in Subcortical Ischemic Stroke. Stroke 2015, 46, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Kataria, H.; Alizadeh, A.; Shahriary, G.M.; Saboktakin Rizi, S.; Henrie, R.; Santhosh, K.T.; Thliveris, J.A.; Karimi-Abdolrezaee, S. Neuregulin-1 promotes remyelination and fosters a pro-regenerative inflammatory response in focal demyelinating lesions of the spinal cord. Glia 2018, 66, 538–561. [Google Scholar] [CrossRef] [PubMed]
- Bartus, K.; Galino, J.; James, N.D.; Hernandez-Miranda, L.R.; Dawes, J.M.; Fricker, F.R.; Garratt, A.N.; McMahon, S.B.; Ramer, M.S.; Birchmeier, C.; et al. Neuregulin-1 controls an endogenous repair mechanism after spinal cord injury. Brain 2016, 139, 1394–1416. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.; Karimi-Abdolrezaee, S. Microenvironmental regulation of oligodendrocyte replacement and remyelination in spinal cord injury. J. Physiol. 2016, 594, 3539–3552. [Google Scholar] [CrossRef] [PubMed]
- Cheville, A.L.; Kirshblum, S.C. Thyroid hormone changes in chronic spinal cord injury. J. Spinal Cord Med. 1995, 18, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Bugaresti, J.M.; Tator, C.H.; Silverberg, J.D.; Szalai, J.P.; Malkin, D.G.; Malkin, A.; Tay, S.K. Changes in thyroid hormones, thyroid stimulating hormone and cortisol in acute spinal cord injury. Spinal Cord 1992, 30, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Shultz, R.B.; Wang, Z.; Nong, J.; Zhang, Z.; Zhong, Y. Local delivery of thyroid hormone enhances oligodendrogenesis and myelination after spinal cord injury. J. Neural Eng. 2017, 14, 36014. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, M.; Granata, R.; Ghigo, E. The IGF system. Acta Diabetol. 2011, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Estrada, J.; Garcia-Segura, L.M.; Torres-Aleman, I. Expression of insulin-like growth factor I by astrocytes in response to injury. Brain Res. 1992, 592, 343–347. [Google Scholar] [CrossRef]
- O’Donnell, S.L.; Frederick, T.J.; Krady, J.K.; Vannucci, S.J.; Wood, T.L. IGF-I and microglia/macrophage proliferation in the ischemic mouse brain. Glia 2002, 39, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Latov, N.; Nilaver, G.; Zimmerman, E.A.; Johnson, W.G.; Silverman, A.J.; Defendini, R.; Cote, L. Fibrillary astrocytes proliferate in response to brain injury: a study combining immunoperoxidase technique for glial fibrillary acidic protein and radioautography of tritiated thymidine. Dev. Biol. 1979, 72, 381–384. [Google Scholar] [CrossRef]
- Dusart, I.; Schwab, M.E. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur. J. Neurosci. 1994, 6, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Hinks, G.L.; Franklin, R.J.M. Distinctive Patterns of PDGF-A, FGF-2, IGF-I, and TGF-β1 Gene Expression during Remyelination of Experimentally-Induced Spinal Cord Demyelination. Mol. Cell. Neurosci. 1999, 14, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.-L.; West, N.R.; Bondy, C.A.; Brenner, M.; Hudson, L.D.; Zhou, J.; Collins, G.H.; Webster, H.D. Cryogenic spinal cord injury induces astrocytic gene expression of insulin-like growth factor I and insulin-like growth factor binding protein 2 during myelin regeneration. J. Neurosci. Res. 1995, 40, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Kazanis, I.; Giannakopoulou, M.; Philippidis, H.; Stylianopoulou, F. Alterations in IGF-I, BDNF and NT-3 levels following experimental brain trauma and the effect of IGF-I administration. Exp. Neurol. 2004, 186, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, A.; Sperl, A.; Heller, R.; Kunzmann, K.; Graeser, V.; Akbar, M.; Gerner, H.J.; Biglari, B. Elevated Serum Insulin-Like Growth Factor 1 Levels in Patients with Neurological Remission after Traumatic Spinal Cord Injury. PLoS ONE 2016, 11, e0159764. [Google Scholar] [CrossRef] [PubMed]
- Feeney, C.; Sharp, D.J.; Hellyer, P.J.; Jolly, A.E.; Cole, J.H.; Scott, G.; Baxter, D.; Jilka, S.; Ross, E.; Ham, T.E.; et al. Serum insulin-like growth factor-I levels are associated with improved white matter recovery after traumatic brain injury. Ann. Neurol. 2017, 82, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.J.; Nockels, R.P.; Pan, H.Q.; Shaffrey, C.I.; Chopp, M. Progesterone is Neuroprotective After Acute Experimental Spinal Cord Trauma in Rats. Spine (Phila. Pa. 1976). 1999, 24, 2134. [Google Scholar] [CrossRef]
- Labombarda, F.; González, S.L.; Lima, A.; Roig, P.; Guennoun, R.; Schumacher, M.; de Nicola, A.F. Effects of progesterone on oligodendrocyte progenitors, oligodendrocyte transcription factors, and myelin proteins following spinal cord injury. Glia 2009, 57, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Choi, G.; Anderson, D.J. The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron 2001, 31, 791–807. [Google Scholar] [CrossRef]
- Arnett, H.A.; Fancy, S.P.J.; Alberta, J.A.; Zhao, C.; Plant, S.R.; Kaing, S.; Raine, C.S.; Rowitch, D.H.; Franklin, R.J.M.; Stiles, C.D. bHLH Transcription Factor Olig1 Is Required to Repair Demyelinated Lesions in the CNS. Science 2004, 306, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Galani, R.; Hoffman, S.W.; Stein, D.G. Effects of the duration of progesterone treatment on the resolution of cerebral edema induced by cortical contusions in rats. Restor. Neurol. Neurosci. 2001, 18, 161–166. [Google Scholar] [PubMed]
- Cutler, S.M.; Cekic, M.; Miller, D.M.; Wali, B.; VanLandingham, J.W.; Stein, D.G. Progesterone Improves Acute Recovery after Traumatic Brain Injury in the Aged Rat. J. Neurotrauma 2007, 24, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Asher, R.A. The glial scar and central nervous system repair. Brain Res. Bull. 1999, 49, 377–391. [Google Scholar] [CrossRef]
- Siebert, J.R.; Osterhout, D.J. The inhibitory effects of chondroitin sulfate proteoglycans on oligodendrocytes. J. Neurochem. 2011, 119, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.C.; Shamblott, M.J.; Gary, D.S.; Belegu, V.; Hurtado, A.; Malone, M.L.; McDonald, J.W. Chondroitin sulfate proteoglycans inhibit oligodendrocyte myelination through PTPσ. Exp. Neurol. 2013, 247, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Jahan, N.; Hannila, S.S. Transforming growth factor β-induced expression of chondroitin sulfate proteoglycans is mediated through non-Smad signaling pathways. Exp. Neurol. 2015, 263, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef] [PubMed]
- Susarla, B.T.S.; Laing, E.D.; Yu, P.; Katagiri, Y.; Geller, H.M.; Symes, A.J. Smad proteins differentially regulate transforming growth factor-β-mediated induction of chondroitin sulfate proteoglycans. J. Neurochem. 2011, 119, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Döring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann. Neurol. 2012, 72, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Abdolrezaee, S.; Eftekharpour, E.; Wang, J.; Schut, D.; Fehlings, M.G. Synergistic Effects of Transplanted Adult Neural Stem/Progenitor Cells, Chondroitinase, and Growth Factors Promote Functional Repair and Plasticity of the Chronically Injured Spinal Cord. J. Neurosci. 2010, 30, 1657–1676. [Google Scholar] [CrossRef] [PubMed]
- Siebert, J.R.; Stelzner, D.J.; Osterhout, D.J. Chondroitinase treatment following spinal contusion injury increases migration of oligodendrocyte progenitor cells. Exp. Neurol. 2011, 231, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-Derived Endothelin-1 Inhibits Remyelination through Notch Activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-F.; Liu, X.-K.; Li, R.; Zhang, P.; Chu, Z.; Wang, C.-L.; Liu, H.-R.; Qi, J.; Lv, G.-Y.; Wang, G.-Y.; et al. Effect of glial cells on remyelination after spinal cord injury. Neural Regen. Res. 2017, 12, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P.-Y. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Vela, J.M.; Molina-Holgado, E.; Arévalo-Martı́n, Á.; Almazán, G.; Guaza, C. Interleukin-1 Regulates Proliferation and Differentiation of Oligodendrocyte Progenitor Cells. Mol. Cell. Neurosci. 2002, 20, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Kroner, A.; David, S. Iron efflux from astrocytes plays a role in remyelination. J. Neurosci. 2012, 32, 4841–4847. [Google Scholar] [CrossRef] [PubMed]
- Kıray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283, 541–549. [Google Scholar] [CrossRef]
- Tripathi, R.; McTigue, D.M. Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia 2007. [Google Scholar] [CrossRef] [PubMed]
- Almad, A.; McTigue, D.M. Chronic expression of PPAR-δ by oligodendrocyte lineage cells in the injured rat spinal cord. J. Comp. Neurol. 2010, 518, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Hesp, Z.C.; Yoseph, R.Y.; Suzuki, R.; Jukkola, P.; Wilson, C.; Nishiyama, A.; McTigue, D.M. Proliferating NG2-Cell-Dependent Angiogenesis and Scar Formation Alter Axon Growth and Functional Recovery After Spinal Cord Injury in Mice. J. Neurosci. 2018, 38, 1366–1382. [Google Scholar] [CrossRef] [PubMed]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Melik Parsadaniantz, S.; Franklin, R.J.M.; et al. Demyelination Causes Adult CNS Progenitors to Revert to an Immature State and Express Immune Cues That Support Their Migration. J. Neurosci. 2015, 35, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Levine, J. The reactions and role of NG2 glia in spinal cord injury. Brain Res. 2016, 1638, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.Z.; Church, J.S.; Hesp, Z.C.; Popovich, P.G.; McTigue, D.M. A silver lining of neuroinflammation: Beneficial effects on myelination. Exp. Neurol. 2016. [CrossRef] [PubMed]
- Setzu, A.; Lathia, J.D.; Zhao, C.; Wells, K.; Rao, M.S.; Ffrench-Constant, C.; Franklin, R.J.M. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia 2006, 54, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Azari, M.F.; Profyris, C.; Karnezis, T.; Bernard, C.C.; Small, D.H.; Cheema, S.S.; Ozturk, E.; Hatzinisiriou, I.; Petratos, S. Leukemia Inhibitory Factor Arrests Oligodendrocyte Death and Demyelination in Spinal Cord Injury. J. Neuropathol. Exp. Neurol. 2006, 65, 914–929. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.J.; Patterson, P.H. Leukemia inhibitory factor promotes oligodendrocyte survival after spinal cord injury. Glia 2005, 51, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zang, D.W.; Cheema, S.S. Leukemia Inhibitory Factor Promotes Recovery of Locomotor Function following Spinal Cord Injury in the Mouse. J. Neurotrauma 2003, 20, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Rittchen, S.; Boyd, A.; Burns, A.; Park, J.; Fahmy, T.M.; Metcalfe, S.; Williams, A. Myelin repair in vivo is increased by targeting oligodendrocyte precursor cells with nanoparticles encapsulating leukaemia inhibitory factor (LIF). Biomaterials 2015, 56, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Letterio, J.J.; Roberts, A.B. REGULATION OF IMMUNE RESPONSES BY TGF-β. Annu. Rev. Immunol. 1998, 16, 137–161. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Popovich, P.G.; Morgan, T.E.; Stokes, B.T. Localization of Transforming Growth Factor-β1 and Receptor mRNA after Experimental Spinal Cord Injury. Exp. Neurol. 2000, 163, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011, 331, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Kohta, M.; Kohmura, E.; Yamashita, T. Inhibition of TGF-β1 promotes functional recovery after spinal cord injury. Neurosci. Res. 2009, 65, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Biol. 2017, 101, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Popovich, P.G.; McTigue, D.M. Oligodendrocyte Generation Is Differentially Influenced by Toll-Like Receptor (TLR) 2 and TLR4-Mediated Intraspinal Macrophage Activation. J. Neuropathol. Exp. Neurol. 2007, 66, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.Z.; Church, J.S.; Pukos, N.; Gottipati, M.K.; Popovich, P.G.; McTigue, D.M. Intraspinal TLR4 activation promotes iron storage but does not protect neurons or oligodendrocytes from progressive iron-mediated damage. Exp. Neurol. 2017, 298, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; McTigue, D.M. Iron is essential for oligodendrocyte genesis following intraspinal macrophage activation. Exp. Neurol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Goldstein, E.Z.; Sahinkaya, F.R.; Wei, P.; Popovich, P.G.; McTigue, D.M. Ferritin Stimulates Oligodendrocyte Genesis in the Adult Spinal Cord and Can Be Transferred from Macrophages to NG2 Cells In Vivo. J. Neurosci. 2012, 32, 5374–5384. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Lai, W.; Rivest, S.; Hart, R.P.; Satoskar, A.R.; Popovich, P.G. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J. Neurochem. 2007, 102, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Church, J.S.; Kigerl, K.A.; Lerch, J.K.; Popovich, P.G.; McTigue, D.M. TLR4 Deficiency Impairs Oligodendrocyte Formation in the Injured Spinal Cord. J. Neurosci. 2016, 36, 6352–6364. [Google Scholar] [CrossRef] [PubMed]
- Sauerbeck, A.; Schonberg, D.L.; Laws, J.L.; McTigue, D.M. Systemic iron chelation results in limited functional and histological recovery after traumatic spinal cord injury in rats. Exp. Neurol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sahinkaya, F.R.; Milich, L.M.; McTigue, D.M. Changes in NG2 cells and oligodendrocytes in a new model of intraspinal hemorrhage. Exp. Neurol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rathore, K.I.; Kerr, B.J.; Redensek, A.; López-Vales, R.; Jeong, S.Y.; Ponka, P.; David, S. Ceruloplasmin protects injured spinal cord from iron-mediated oxidative damage. J. Neurosci. 2008, 28, 12736–12747. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Weissenbach, M.; Haan, S.; Heinrich, P.C.; Schaper, F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000, 275, 12848–12856. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Lin, C.-Y.; Li, K.; Lee, Y.-S. Effects of Reducing Suppressors of Cytokine Signaling-3 (SOCS3) Expression on Dendritic Outgrowth and Demyelination after Spinal Cord Injury. PLoS ONE 2015, 10, e0138301. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and Autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.S.; Bishop, E.T.; Rückerl, D.; Gaspar-Pereira, S.; Barker, R.N.; Allen, J.E.; Rees, A.J.; Wilson, H.M. Suppressor of cytokine signaling ( SOCS ) 1 is a key determinant of differential macrophage activation and function. J. Leukoc. Biol 2011, 90, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.J.; Girolami, E.I.; Ghasemlou, N.; Jeong, S.Y.; David, S. The protective effects of 15-deoxy-Δ-12,14-prostaglandin J2 in spinal cord injury. Glia 2008, 56, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.E.; Whyte, C.S.; Gordon, P.; Barker, R.N.; Rees, A.J.; Wilson, H.M. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology 2014, 141, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Nakamura, M.; Katoh, H.; Miyao, T.; Shimazaki, T.; Ishii, K.; Yamane, J.; Yoshimura, A.; Iwamoto, Y.; Toyama, Y.; et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 2006, 12, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Hackett, A.R.; Lee, J.K. Understanding the NG2 Glial Scar after Spinal Cord Injury. Front. Neurol. 2016, 7, 199. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.J.; Longbrake, E.E.; Shawler, T.M.; Kigerl, K.A.; Lai, W.; Tovar, C.A.; Ransohoff, R.M.; Popovich, P.G. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J. Neurosci. 2011, 31, 9910–9922. [Google Scholar] [CrossRef] [PubMed]
- Freria, C.M.; Hall, J.C.E.; Wei, P.; Guan, Z.; McTigue, D.M.; Popovich, P.G. Deletion of the Fractalkine Receptor, CX3CR1, Improves Endogenous Repair, Axon Sprouting, and Synaptogenesis after Spinal Cord Injury in Mice. J. Neurosci. 2017, 37, 3568–3587. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; McCandless, E.E.; Dorsey, D.; Klein, R.S. CXCR4 promotes differentiation of oligodendrocyte progenitors and remyelination. Proc. Natl. Acad. Sci. USA 2010, 107, 11062–11067. [Google Scholar] [CrossRef] [PubMed]
- Jaerve, A.; Müller, H.W. Chemokines in CNS injury and repair. Cell Tissue Res. 2012, 349, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Williams, J.L.; Muccigrosso, M.M.; Liu, L.; Sun, T.; Rubin, J.B.; Klein, R.S. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012, 124, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Tirotta, E.; Kirby, L.A.; Hatch, M.N.; Lane, T.E. IFN-γ-induced apoptosis of human embryonic stem cell derived oligodendrocyte progenitor cells is restricted by CXCR2 signaling. Stem Cell Res. 2012, 9, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Omari, K.M.; Lutz, S.E.; Santambrogio, L.; Lira, S.A.; Raine, C.S. Neuroprotection and Remyelination after Autoimmune Demyelination in Mice that Inducibly Overexpress CXCL1. Am. J. Pathol. 2009, 174, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, R.; Zecevic, N. The effect of CXCL1 on human fetal oligodendrocyte progenitor cells. Glia 2008, 56, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Omari, K.M.; John, G.R.; Sealfon, S.C.; Raine, C.S. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain 2005, 128, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Omari, K.M.; John, G.; Lango, R.; Raine, C.S. Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia 2006, 53, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Zeng, X. The Application of Stem Cell Based Tissue Engineering in Spinal Cord Injury Repair. J. Tissue Sci. Eng. 2015, 6. [Google Scholar] [CrossRef]
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.W.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B.; et al. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 403–422. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pukos, N.; Yoseph, R.; M. McTigue, D. To Be or Not to Be: Environmental Factors that Drive Myelin Formation during Development and after CNS Trauma. Neuroglia 2018, 1, 63-90. https://doi.org/10.3390/neuroglia1010007
Pukos N, Yoseph R, M. McTigue D. To Be or Not to Be: Environmental Factors that Drive Myelin Formation during Development and after CNS Trauma. Neuroglia. 2018; 1(1):63-90. https://doi.org/10.3390/neuroglia1010007
Chicago/Turabian StylePukos, Nicole, Rim Yoseph, and Dana M. McTigue. 2018. "To Be or Not to Be: Environmental Factors that Drive Myelin Formation during Development and after CNS Trauma" Neuroglia 1, no. 1: 63-90. https://doi.org/10.3390/neuroglia1010007
APA StylePukos, N., Yoseph, R., & M. McTigue, D. (2018). To Be or Not to Be: Environmental Factors that Drive Myelin Formation during Development and after CNS Trauma. Neuroglia, 1(1), 63-90. https://doi.org/10.3390/neuroglia1010007