

Primary Metabolites and Microbial Diversity in Commercial Kombucha Products


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Acquisition
2.2. Physiochemical Characterization
2.3. Culture-Based Microbiological Analyses
2.4. 16S and ITS Next-Generation Amplicon Sequencing
2.4.1. DNA Extraction
2.4.2. Next-Generation Sequencing
2.5. Data Processing and Analysis
3. Results
3.1. Physiochemical and Microbiological Characterizations
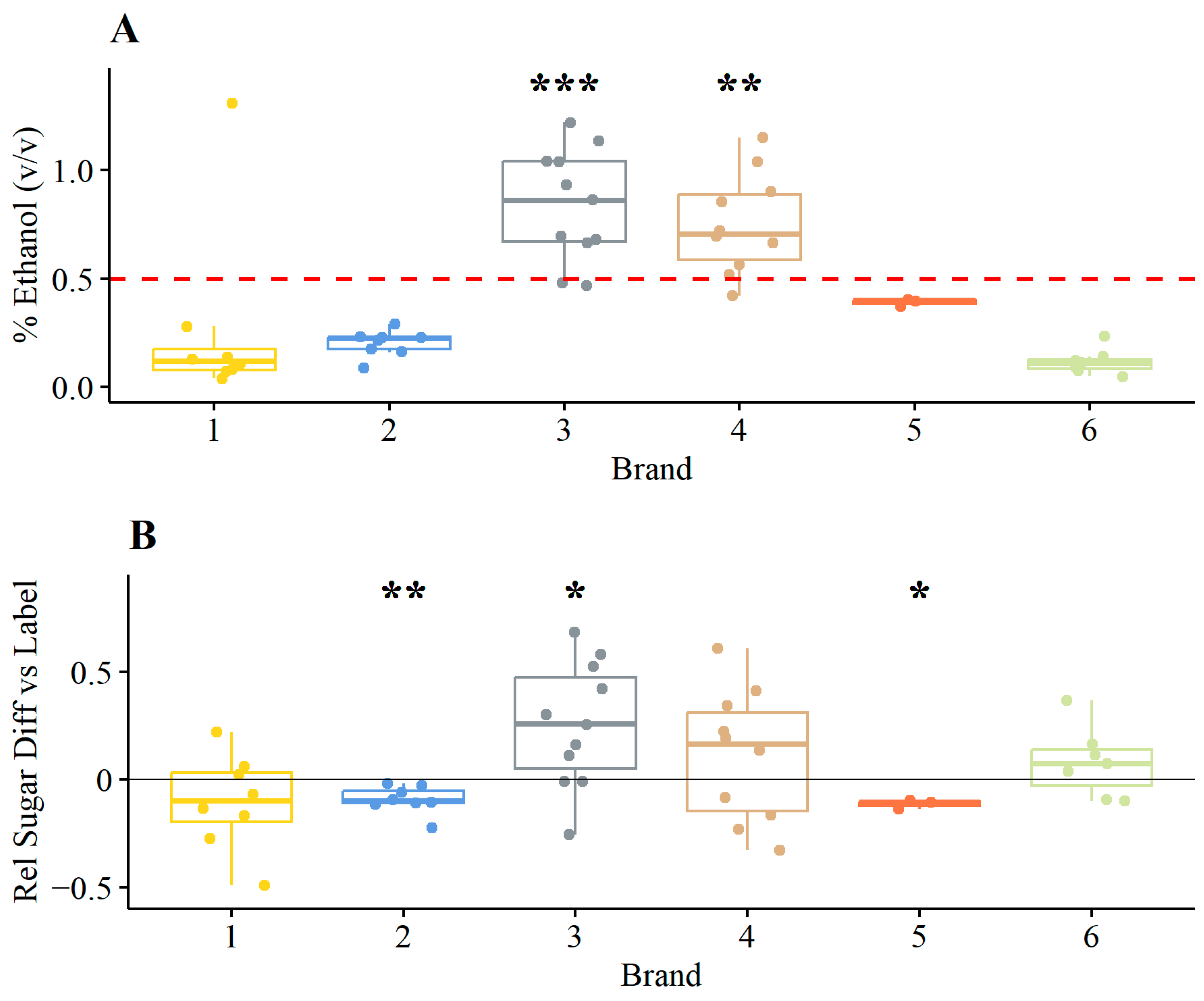
3.1.1. Ethanol and Relative Sugar Contents
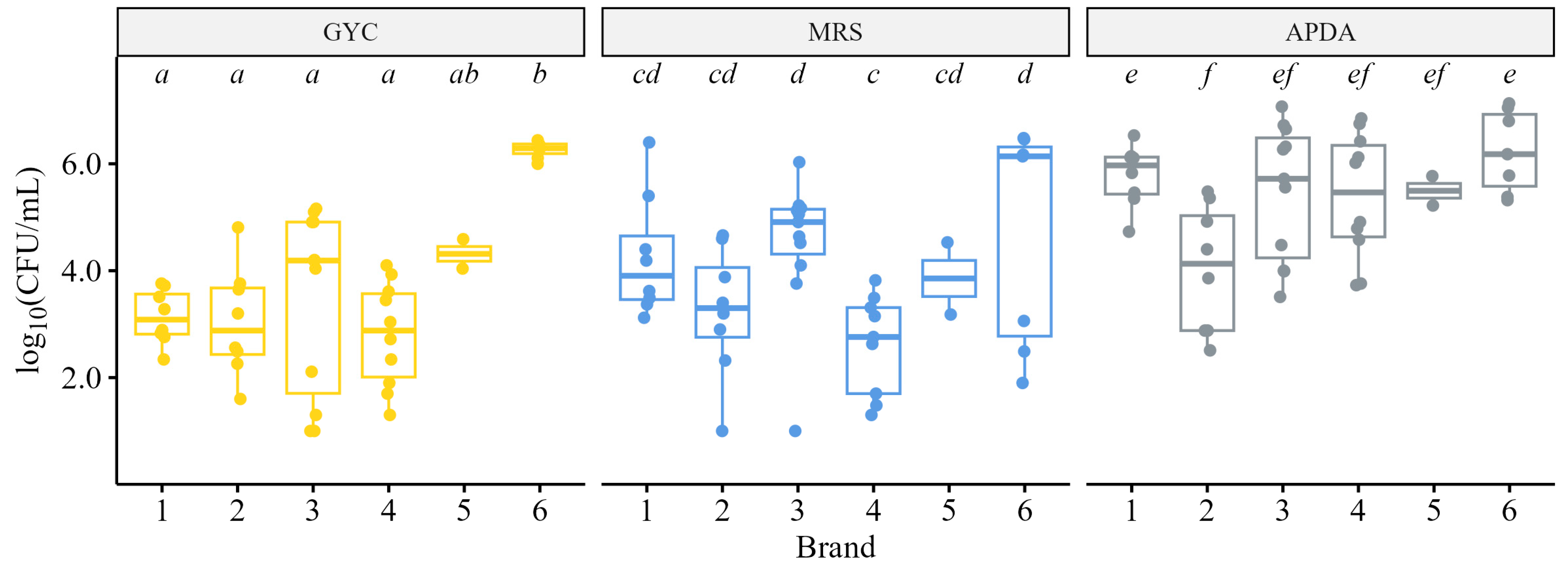
3.1.2. Culture-Based Microbiological Analyses
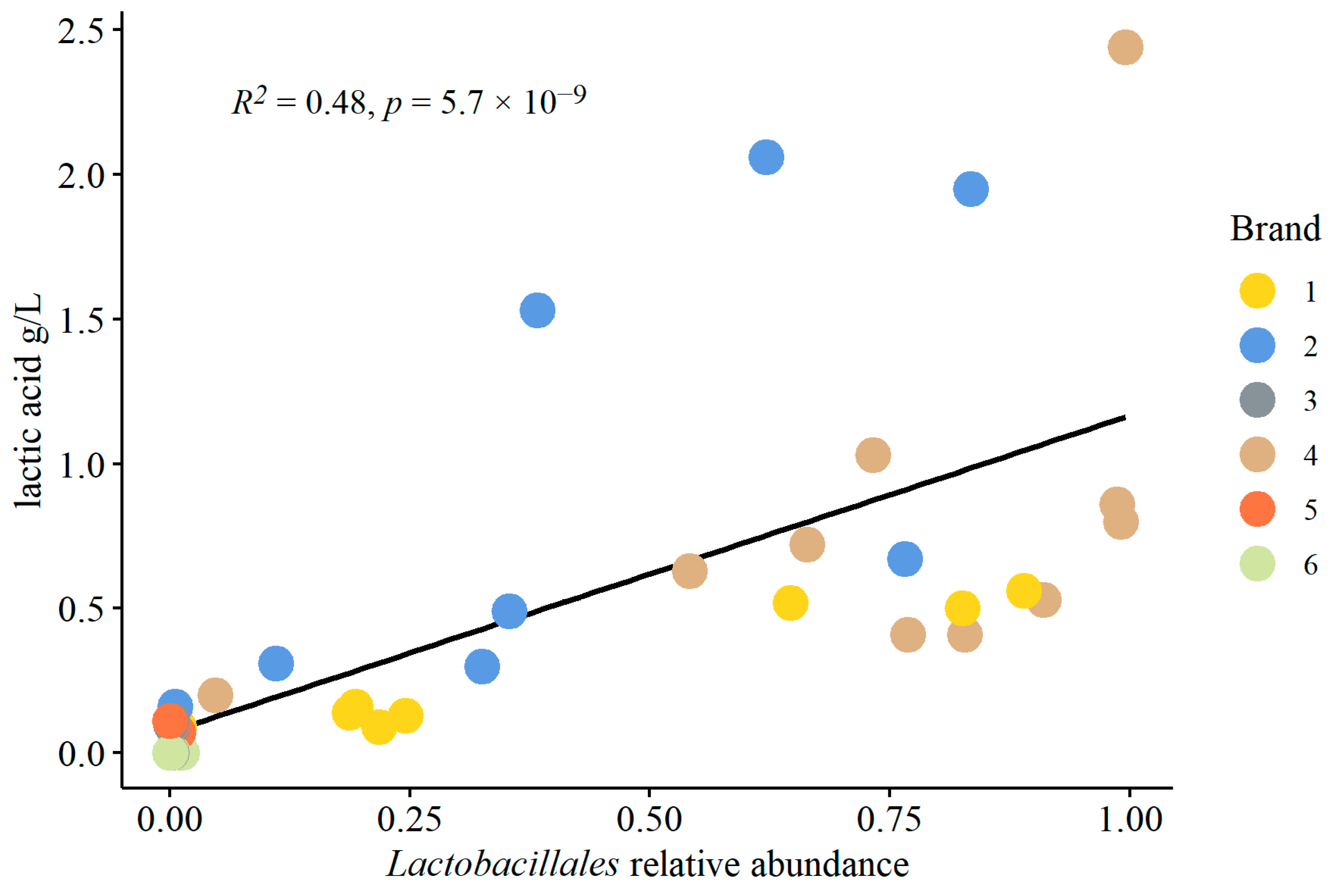
3.1.3. Physiochemical and Microbiological Multiple Linear Regressions
3.2. Next-Generation Amplicon Sequencing
3.2.1. Sequencing and Sequence Processing
3.2.2. Bacterial Community Analyses
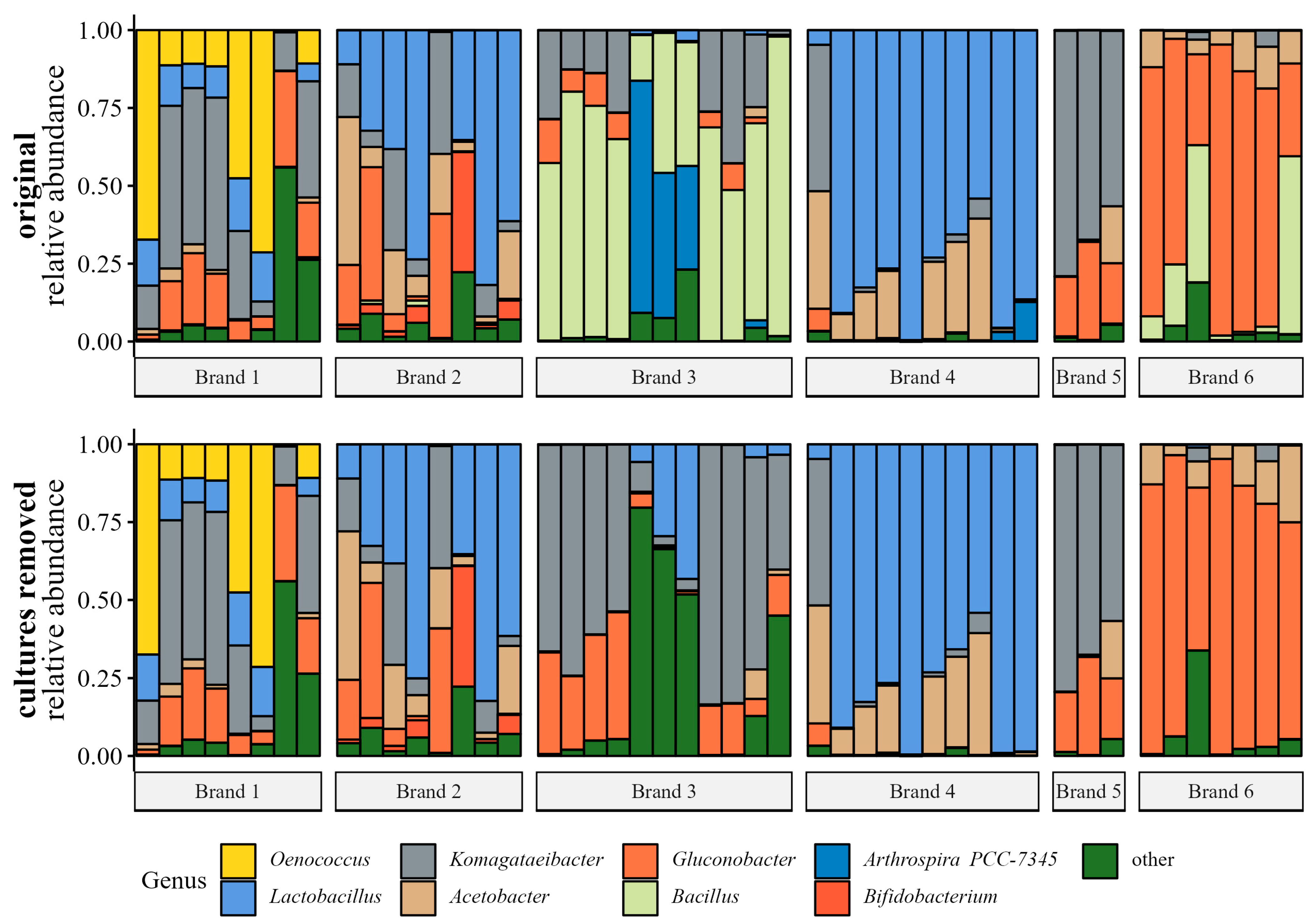
Bacterial Genera Relative Abundance
Bacterial Community Sample Ordination
Association between Variables and the Observed Bacterial Community Differences
3.2.3. Fungal Community Analyses
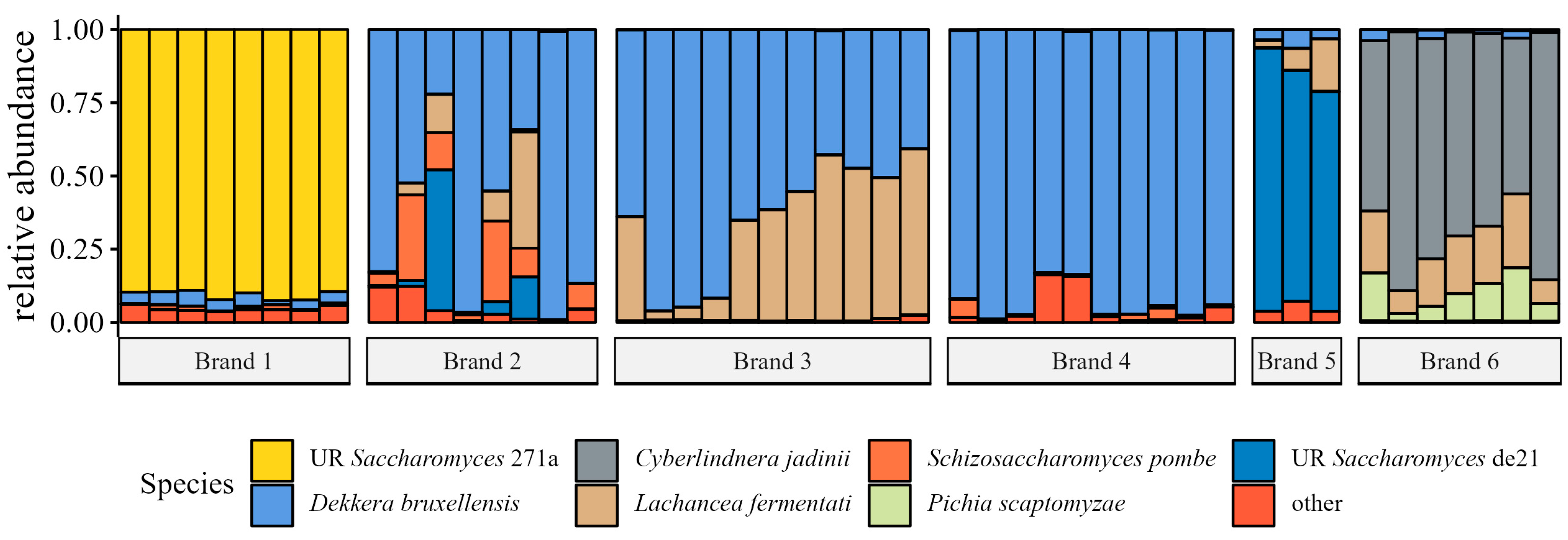
Fungal Species Relative Abundance
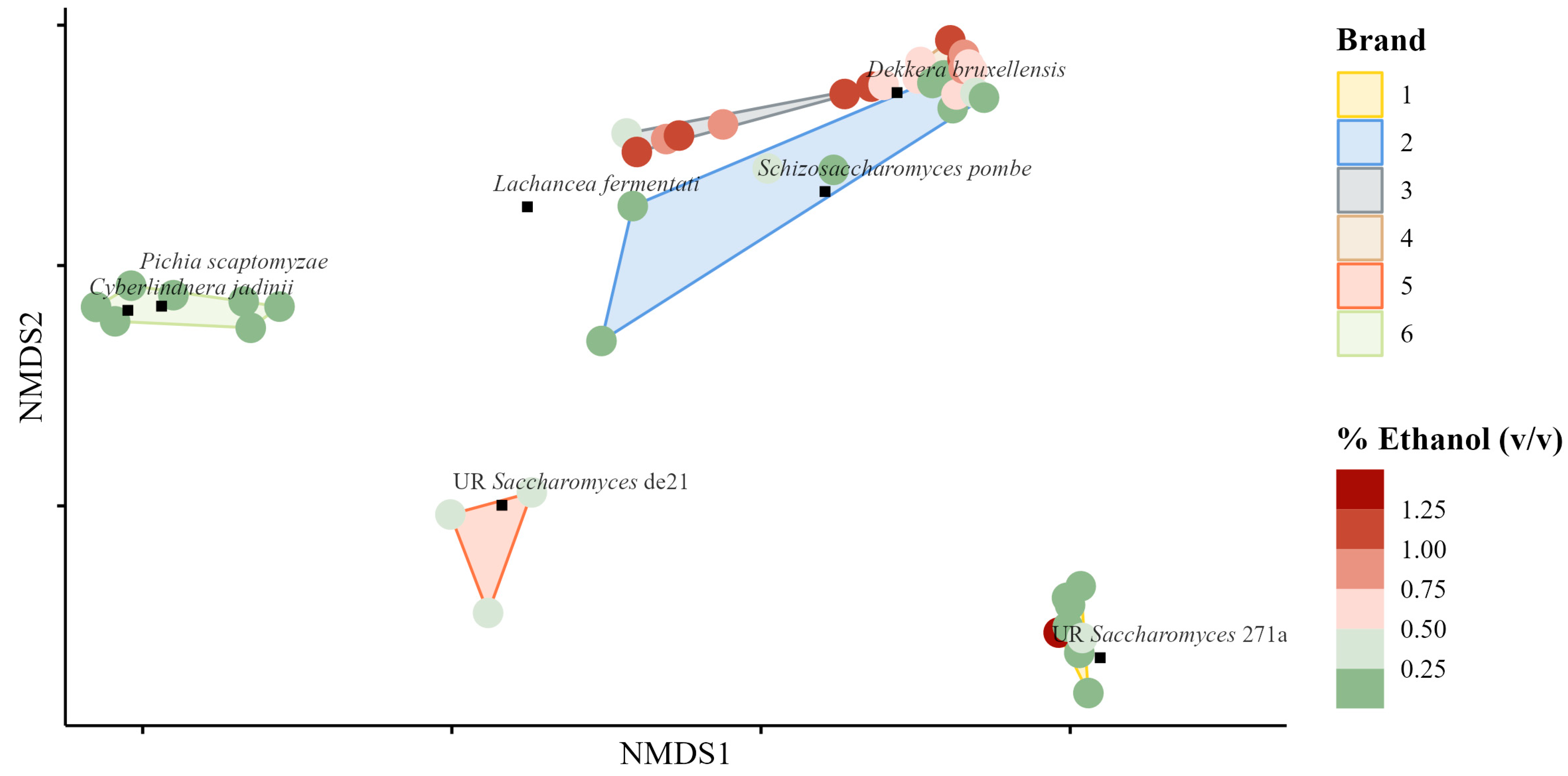
Fungal Community Sample Ordination
Association between Variables and the Observed Fungal Community Differences
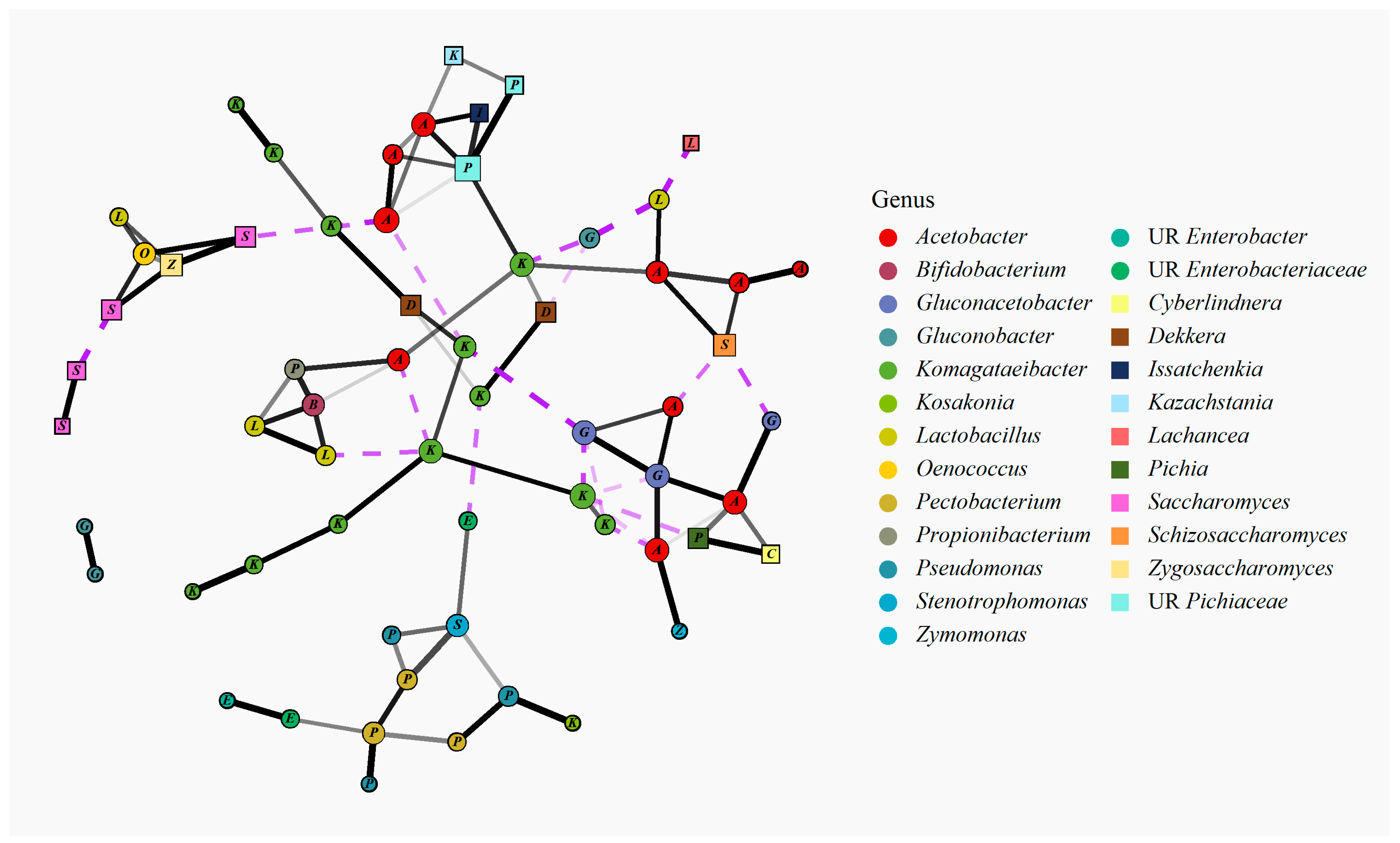
3.2.4. Bacterial and Fungal Species Co-Occurrence Analysis
4. Discussion
4.1. High Ethanol Content of Commercial Kombucha Products
4.2. High Sugar Contents Can Cause High Ethanol Contents
4.3. Brand Influences the Microbial Communities of Products
4.4. Lactic Acid Bacteria in Kombucha Fermentation
4.5. Fungal Community Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenwalt, C.J.; Steinkraus, K.H.; Ledford, R.A. Kombucha, the Fermented Tea: Microbiology, Composition, and Claimed Health Effects. J. Food Prot. 2000, 63, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Sievers, M.; Lanini, C.; Weber, A.; Schuler-Schmid, U.; Teuber, M. Microbiology and Fermentation Balance in a Kombucha Beverage Obtained from a Tea Fungus Fermentation. Syst. Appl. Microbiol. 1995, 18, 590–594. [Google Scholar] [CrossRef]
- Le, T. Kombucha Production in the US; IBISWorld: New York, NY, USA, 2021. [Google Scholar]
- Srivastava, N. Patent Insights: Fermented Ingredients in Food & Drink; Mintel: London, UK, 2021. [Google Scholar]
- Srivastava, N. Patent Insights: Gut Health For Holistic Wellbeing; Mintel: London, UK, 2023. [Google Scholar]
- Kapp, J.M.; Sumner, W. Kombucha: A Systematic Review of the Empirical Evidence of Human Health Benefit. Ann. Epidemiol. 2019, 30, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J. Whole Foods, Millennium Sued Over Kombucha Drinks. Bloomberg Law News, 18 November 2015. [Google Scholar]
- Steinberg, J. Brew Dr. Kombucha Overstated Probiotic Content, Suit Says. Bloomberg Law News, 17 October 2019. [Google Scholar]
- Steinberg, J. O Organics Kombucha Deceptively Sold as Non-Alcoholic, Suit Says. Bloomberg Law News, 11 April 2019. [Google Scholar]
- Steinberg, J. Health-Ade Kombucha Suit Alleges Sugar Belies Healthful Name. Bloomberg Law News, 8 October 2021. [Google Scholar]
- Steinberg, J. Tribucha ‘Non-Alcoholic’ Kombucha Over the Limit, Suit Says (1). Bloomberg Law News, 7 October 2022. [Google Scholar]
- Talebi, M.; Frink, L.A.; Patil, R.A.; Armstrong, D.W. Examination of the Varied and Changing Ethanol Content of Commercial Kombucha Products. Food Anal. Methods 2017, 10, 4062–4067. [Google Scholar] [CrossRef]
- DOT TTB Beer: Meaning of Terms, 27 CFR § 25.11. Code of Federal Regulations. Alcohol 2021. Available online: https://www.ecfr.gov/current/title-27/chapter-I/subchapter-A/part-25/subpart-B/section-25.11 (accessed on 30 June 2024).
- DOT TTB Beer: Materials for the Production of Beer, 27 CFR § 25.15. Code of Federal Regulations. Alcohol 2021. Available online: https://www.ecfr.gov/current/title-27/chapter-I/subchapter-A/part-25/subpart-B/section-25.15 (accessed on 30 June 2024).
- Alcohol and Tobacco Tax and Trade Bureau. Kombucha. Available online: https://www.ttb.gov/kombucha (accessed on 3 April 2023).
- Anderson, P.; Kokole, D.; Llopis, E.J. Production, Consumption, and Potential Public Health Impact of Low- and No-Alcohol Products: Results of a Scoping Review. Nutrients 2021, 13, 3153. [Google Scholar] [CrossRef] [PubMed]
- Faruque, S.; Tong, J.; Lacmanovic, V.; Agbonghae, C.; Minaya, D.; Czaja, K. The Dose Makes the Poison: Sugar and Obesity in the United States—A Review. Pol. J. Food Nutr. Sci. 2019, 69, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Després, J.-P.; Hu, F.B. Sugar-Sweetened Beverages, Obesity, Type 2 Diabetes Mellitus, and Cardiovascular Disease Risk. Circulation 2010, 121, 1356–1364. [Google Scholar] [CrossRef]
- Laquatra, I.; Sollid, K.; Smith Edge, M.; Pelzel, J.; Turner, J. Including “Added Sugars” on the Nutrition Facts Panel: How Consumers Perceive the Proposed Change. J. Acad. Nutr. Diet. 2015, 115, 1758–1763. [Google Scholar] [CrossRef]
- DHHS FDA Food Labeling, 21 CFR § 101. Code of Federal Regulations. In Food for Human Consumption; 2022. Available online: https://www.ecfr.gov/current/title-21/chapter-I/subchapter-B/part-101 (accessed on 30 June 2024).
- Degnan, F.H. The US Food and Drug Administration and Probiotics: Regulatory Categorization. Clin. Infect. Dis. 2008, 46, S133–S136. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. The International Scientific Association for Probiotics and Prebiotics Consensus Statement on the Scope and Appropriate Use of the Term Probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Marco, M.L.; Sanders, M.E.; Gänzle, M.; Arrieta, M.C.; Cotter, P.D.; De Vuyst, L.; Hill, C.; Holzapfel, W.; Lebeer, S.; Merenstein, D.; et al. The International Scientific Association for Probiotics and Prebiotics (ISAPP) Consensus Statement on Fermented Foods. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Coton, M.; Pawtowski, A.; Taminiau, B.; Burgaud, G.; Deniel, F.; Coulloumme-Labarthe, L.; Fall, A.; Daube, G.; Coton, E. Unraveling Microbial Ecology of Industrial-Scale Kombucha Fermentations by Metabarcoding and Culture-Based Methods. FEMS Microbiol. Ecol. 2017, 93, fix048. [Google Scholar] [CrossRef]
- Harrison, K.; Curtin, C. Microbial Composition of SCOBY Starter Cultures Used by Commercial Kombucha Brewers in North America. Microorganisms 2021, 9, 1060. [Google Scholar] [CrossRef]
- Yang, J.; Lagishetty, V.; Kurnia, P.; Henning, S.M.; Ahdoot, A.I.; Jacobs, J.P. Microbial and Chemical Profiles of Commercial Kombucha Products. Nutrients 2022, 14, 670. [Google Scholar] [CrossRef]
- Drysdale, G.S.; Fleet, G.H. Acetic Acid Bacteria in Winemaking: A Review. Am. J. Enol. Vitic. 1988, 39, 143–154. [Google Scholar] [CrossRef]
- De Man, J.C.; Rogosa, M.; Sharpe, M.E. A Medium for the Cultivation of Lactobacilli. J. Appl. Bacteriol. 1960, 23, 130–135. [Google Scholar] [CrossRef]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in Bacterial Communities along the 2000 Km Salinity Gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols; Elsevier: Amsterdam, Netherlands, 1990; pp. 315–322. [Google Scholar]
- Tedersoo, L.; Lindahl, B. Fungal Identification Biases in Microbiome Projects. Environ. Microbiol. Rep. 2016, 8, 774–779. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome Diversity: High-Throughput Sequencing and Identification of Fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Posit team RStudio: Integrated Development Environment for R 2023. Available online: https://support.posit.co/hc/en-us/articles/206212048-Citing-RStudio (accessed on 30 June 2024).
- Ushey, K. Renv: Project Environments 2022. Available online: https://rstudio.github.io/renv/authors.html#citation (accessed on 30 June 2024).
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous Inference in General Parametric Models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible Sequence Taxonomy Reference Database Management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE Database for Molecular Identification of Fungi: Handling Dark Taxa and Parallel Taxonomic Classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Huzurbazar, S.; Jafari, F. PERFect: PERmutation Filtering Test for Microbiome Data. Biostatistics 2019, 20, 615–631. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Package ‘vegan’. Community Ecology Package, Version 2.6-6.1. 2022. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 30 June 2024).
- Lahti, L.; Shetty, S. Microbiome R Package. Bioconductor 2019, 10, B9. [Google Scholar] [CrossRef]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015, 11, 1004226. [Google Scholar] [CrossRef]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating Microbiome Composition with Environmental Covariates Using Generalized UniFrac Distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Yang, L.; Chen, J. A Comprehensive Evaluation of Microbial Differential Abundance Analysis Methods: Current Status and Potential Solutions. Microbiome 2022, 10, 130. [Google Scholar] [CrossRef]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots 2023. Available online: https://rpkgs.datanovia.com/ggpubr/authors.html#citation (accessed on 30 June 2024).
- Wilke, C.O.; Wiernik, B.M. Ggtext: Improved Text Rendering Support for “Ggplot2” 2022. Available online: https://cran.r-project.org/web/packages/ggtext/index.html (accessed on 30 June 2024).
- Campitelli, E. Ggnewscale: Multiple Fill and Colour Scales in “Ggplot2” 2022. Available online: https://cran.r-project.org/web/packages/ggnewscale/index.html (accessed on 30 June 2024).
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. Inter. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Briatte, F. Ggnetwork: Geometries to Plot Networks with “Ggplot2” 2023. Available online: https://cran.r-project.org/web/packages/ggnetwork/index.html (accessed on 30 June 2024).
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; Wiley: New York, NY, USA, 2017; pp. 1–15. [Google Scholar] [CrossRef]
- Kaashyap, M.; Cohen, M.; Mantri, N. Microbial Diversity and Characteristics of Kombucha as Revealed by Metagenomic and Physicochemical Analysis. Nutrients 2021, 13, 4446. [Google Scholar] [CrossRef]
- Marsh, A.J.; O’Sullivan, O.; Hill, C.; Ross, R.P.; Cotter, P.D. Sequence-Based Analysis of the Bacterial and Fungal Compositions of Multiple Kombucha (Tea Fungus) Samples. Food Microbiol. 2014, 38, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Arıkan, M.; Mitchell, A.L.; Finn, R.D.; Gürel, F. Microbial Composition of Kombucha Determined Using Amplicon Sequencing and Shotgun Metagenomics. J. Food Sci. 2020, 85, 455–464. [Google Scholar] [CrossRef]
- May, A.; Narayanan, S.; Alcock, J.; Varsani, A.; Maley, C.; Aktipis, A. Kombucha: A Novel Model System for Cooperation and Conflict in a Complex Multi-Species Microbial Ecosystem. PeerJ 2019, 7, e7565. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Rutherfurd-Markwick, K.; Zhang, X.-X.; Mutukumira, A.N. Kombucha: Production and Microbiological Research. Foods 2022, 11, 3456. [Google Scholar] [CrossRef] [PubMed]
- Galafassi, S.; Merico, A.; Pizza, F.; Hellborg, L.; Molinari, F.; Piškur, J.; Compagno, C. Dekkera/Brettanomyces Yeasts for Ethanol Production from Renewable Sources under Oxygen-Limited and Low-PH Conditions. J. Ind. Microbiol. Biotechnol. 2011, 38, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Passoth, V.; Blomqvist, J.; Schnürer, J. Dekkera Bruxellensis and Lactobacillus Vini Form a Stable Ethanol-Producing Consortium in a Commercial Alcohol Production Process. Appl. Environ. Microbiol. 2007, 73, 4354–4356. [Google Scholar] [CrossRef] [PubMed]
- Menoncin, M.; Bonatto, D. Molecular and Biochemical Aspects of Brettanomyces in Brewing. J. Inst. Brew. 2019, 125, 402–411. [Google Scholar] [CrossRef]
- Aguilar Uscanga, M.G.; Délia, M.-L.; Strehaiano, P. Brettanomyces Bruxellensis: Effect of Oxygen on Growth and Acetic Acid Production. Appl. Microbiol. Biotechnol. 2003, 61, 157–162. [Google Scholar] [CrossRef]
- Schifferdecker, A.J.; Siurkus, J.; Andersen, M.R.; Joerck-Ramberg, D.; Ling, Z.; Zhou, N.; Blevins, J.E.; Sibirny, A.A.; Piškur, J.; Ishchuk, O.P. Alcohol Dehydrogenase Gene ADH3 Activates Glucose Alcoholic Fermentation in Genetically Engineered Dekkera Bruxellensis Yeast. Appl. Microbiol. Biotechnol. 2016, 100, 3219–3231. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blinded Brand | n | % Ethanol (v/v) | pH | Lactic Acid (g/L) | Acetic Acid (g/L) | Total Sugar (g/L) |
|---|---|---|---|---|---|---|
| 1 | 8 | 0.27 a [−0.086–0.63] b | 3.3 [3.1–3.5] | 0.27 [0.097–0.45] | 2.1 [1.6–2.7] | 23 [19–28] |
| 2 | 8 | 0.20 [0.15–0.25] | 3.1 [3.0–3.3] | 0.93 [0.28–1.6] | 2.7 [−0.39–5.7] | 27 [25–29] |
| 3 | 11 | 0.84 [0.66–1.0] | 3.0 [2.8–3.1] | 0.097 [0.075–0.12] | 2.4 [2.1–2.8] | 36 [32–40] |
| 4 | 10 | 0.75 [0.58–0.92] | 3.1 [2.9–3.2] | 0.80 [0.36–1.3] | 1.3 [0.97–1.6] | 32 [27–37] |
| 5 | 3 | 0.39 [0.35–0.43] | 3.7 [3.4–4.0] | 0.093 [0.042–0.15] | 0.32 [0.12–0.52] | 20 [19–21] |
| 6 | 7 | 0.12 [0.064–0.17] | 3.1 [2.9–3.3] | n.d. c | 1.1 [0.87–1.3] | 42 [37–47] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sogin, J.H.; Worobo, R.W. Primary Metabolites and Microbial Diversity in Commercial Kombucha Products. Fermentation 2024, 10, 385. https://doi.org/10.3390/fermentation10080385
Sogin JH, Worobo RW. Primary Metabolites and Microbial Diversity in Commercial Kombucha Products. Fermentation. 2024; 10(8):385. https://doi.org/10.3390/fermentation10080385
Chicago/Turabian StyleSogin, Jonathan H., and Randy W. Worobo. 2024. "Primary Metabolites and Microbial Diversity in Commercial Kombucha Products" Fermentation 10, no. 8: 385. https://doi.org/10.3390/fermentation10080385
APA StyleSogin, J. H., & Worobo, R. W. (2024). Primary Metabolites and Microbial Diversity in Commercial Kombucha Products. Fermentation, 10(8), 385. https://doi.org/10.3390/fermentation10080385