Modulation of the Microtubule Network for Optimization of Nanoparticle Dynamics for the Advancement of Cancer Nanomedicine

 , , and
, , and 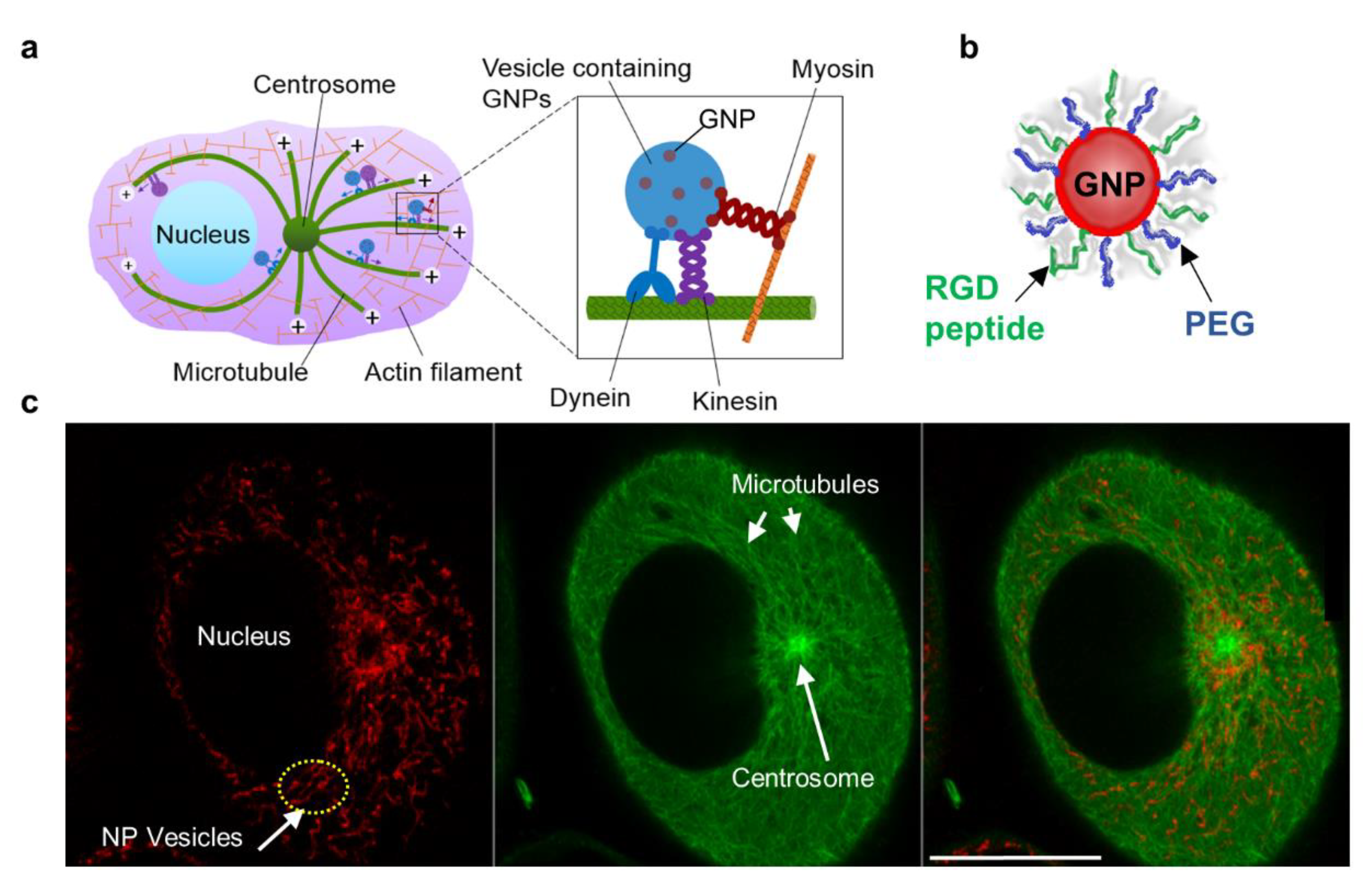{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- (a)
- Does NP size matter?
- (b)
- Will DTX concentration affect NP uptake, transport, and retention?
- (c)
- Does the relative timing of DTX and NP inoculation matter?
2. Materials and Methods
2.1. Cell Culture
2.2. GNP Preparation and Modification
2.3. Nanoparticle Surface Modification
2.4. Docetaxel (DTX) and GNP Inoculation
2.5. Cytotoxicity Assay
2.6. GNP Uptake and Retention Dynamics
2.7. Quantification of Uptake in Cells
2.8. Confocal Imaging
2.9. Measuring DNA Damage
2.10. Measuring Cell Survival Fraction
3. Results and Discussion
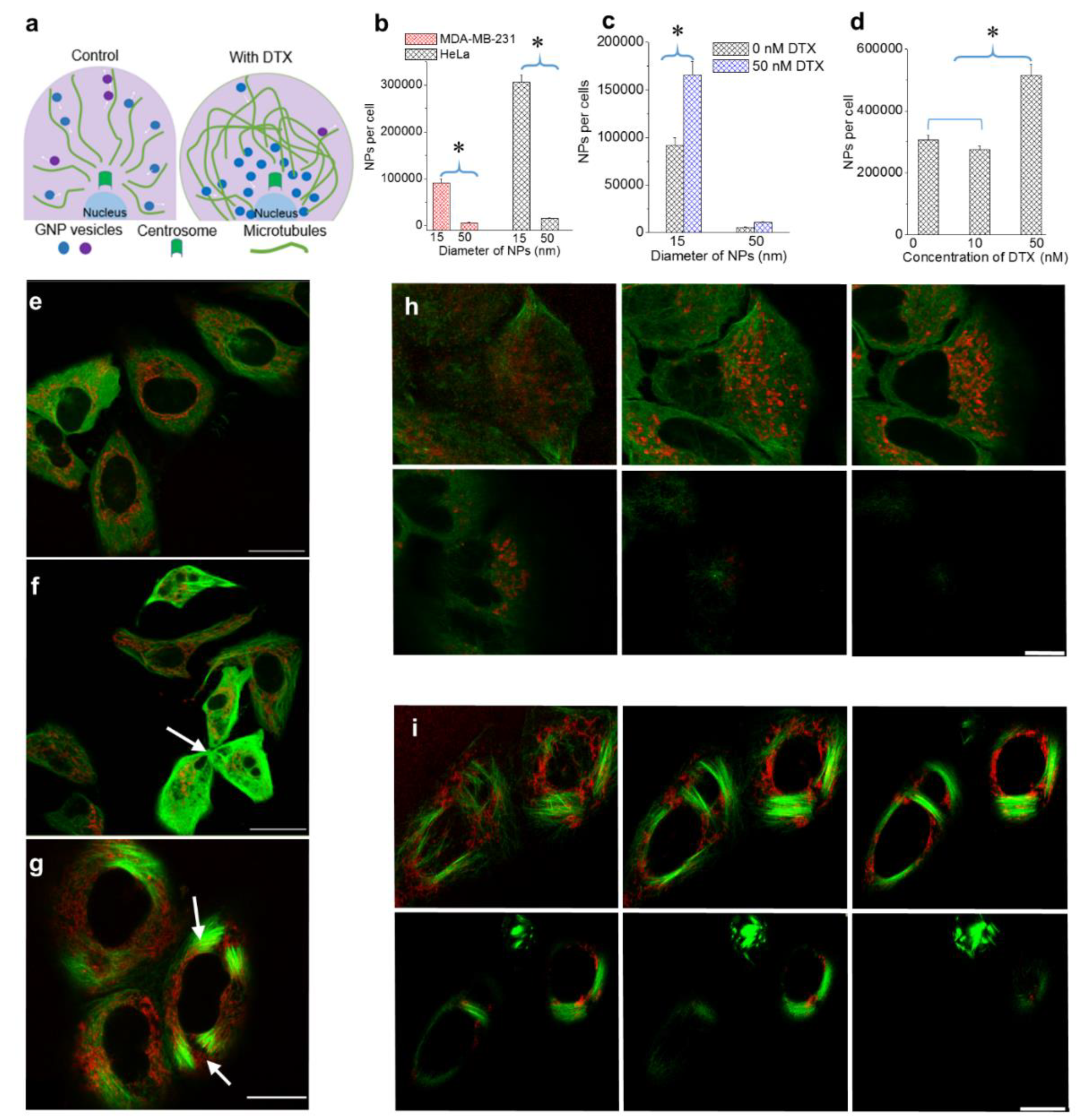
3.1. Effect of NP Size and Docetaxel Concentration on Intracellular Uptake of NPs
- (a)
- Following treatment with DTX, mitosis is arrested during metaphase. The prolonged time in M phase enables greater accumulation of NPs within cells. This led to the increase in uptake of NPs of both sizes.
- (b)
- The presence of DTX did not significantly affect the endocytosis process since it is a process largely governed by the actin cytoskeleton closer to the cell periphery [39]. The cells’ ability to maintain efficient endocytosis enabled significantly higher accumulation of smaller NPs in DTX-treated cells (seen in Figure 2c).
3.2. Distribution of Nanoparticles During Cell Division (Mitosis)
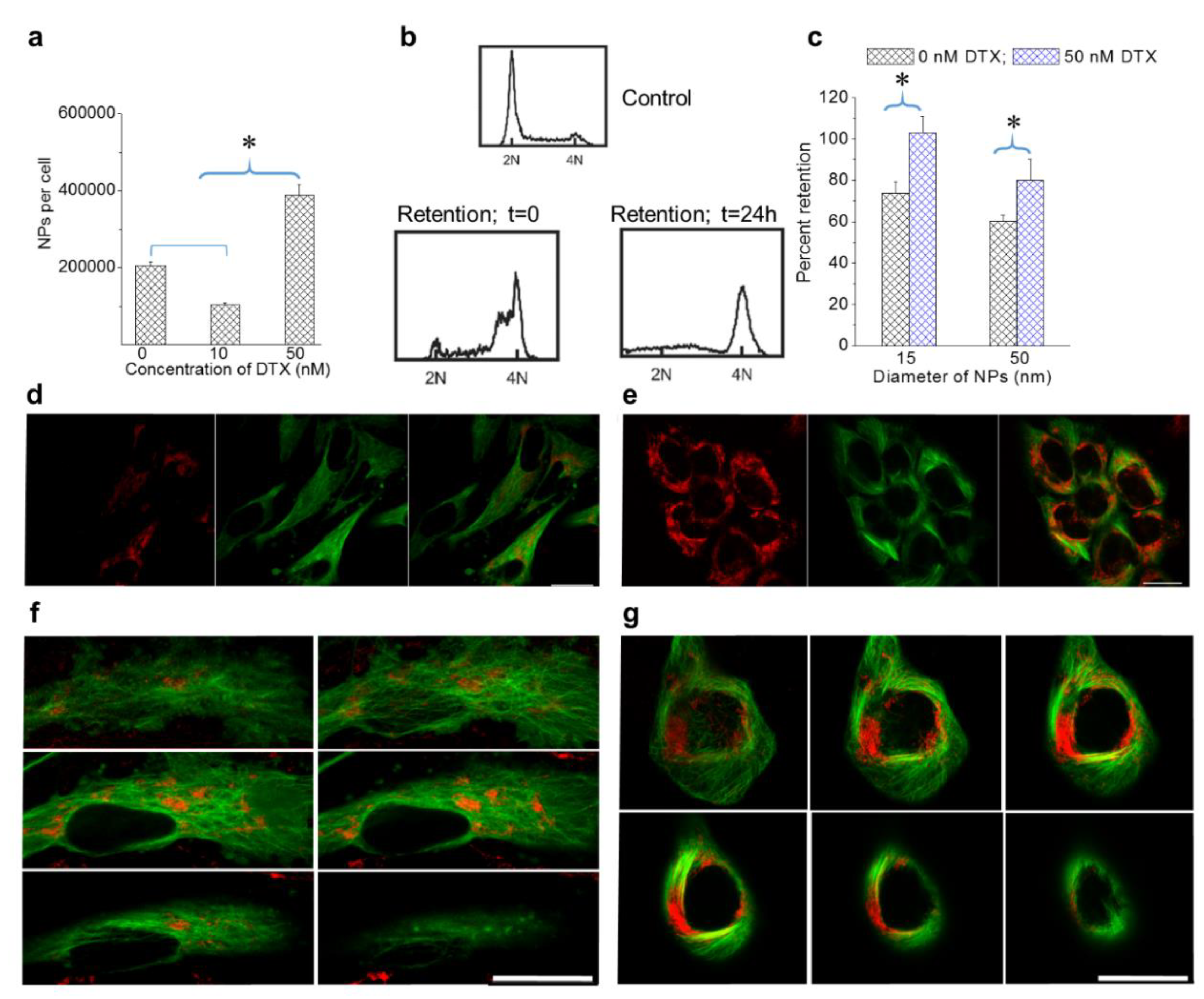
3.3. The Effect of the Docetaxel Concentration on the Intracellular Retention of NPs
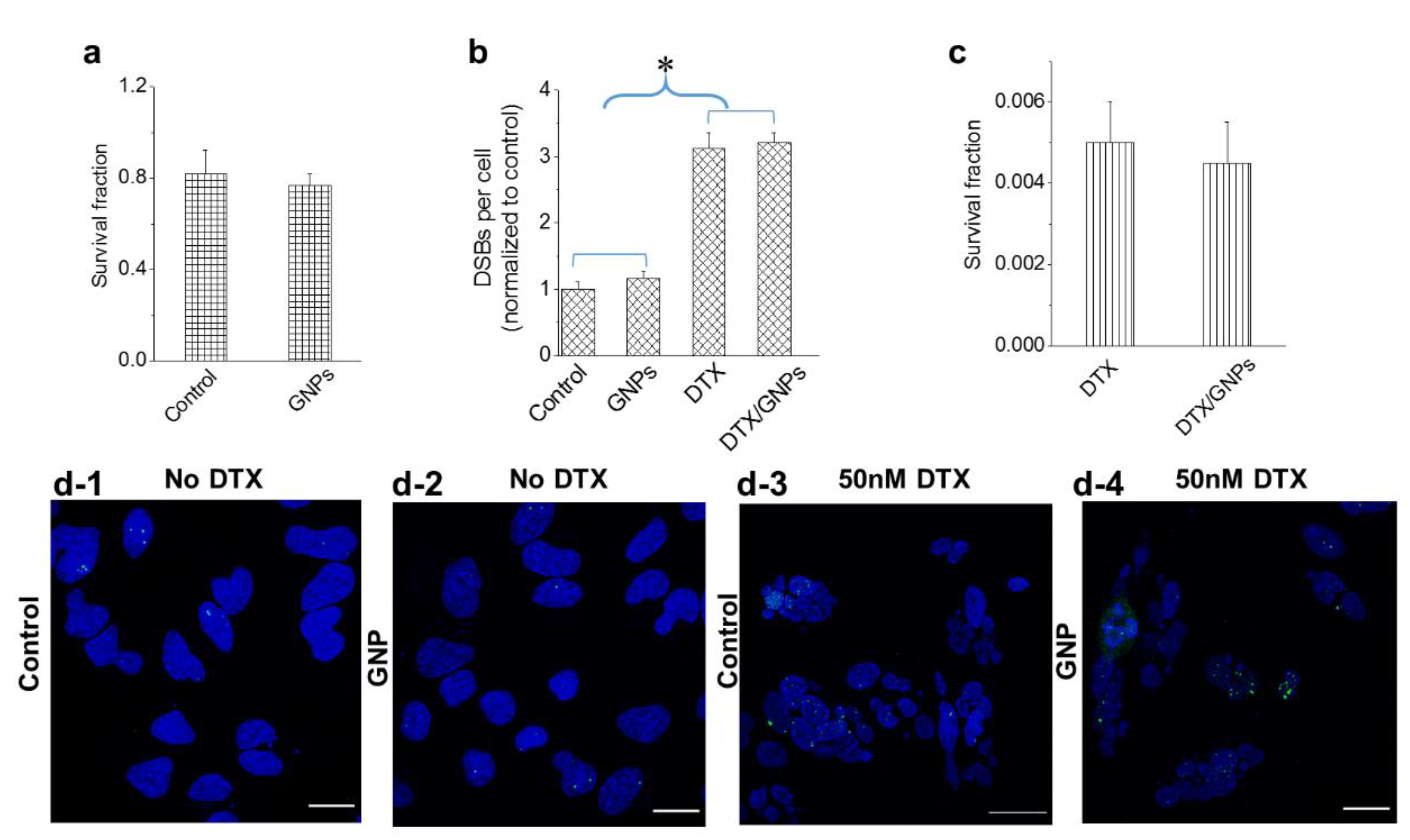
3.4. Effect of GNPs on the Action of the Drug Docetaxel
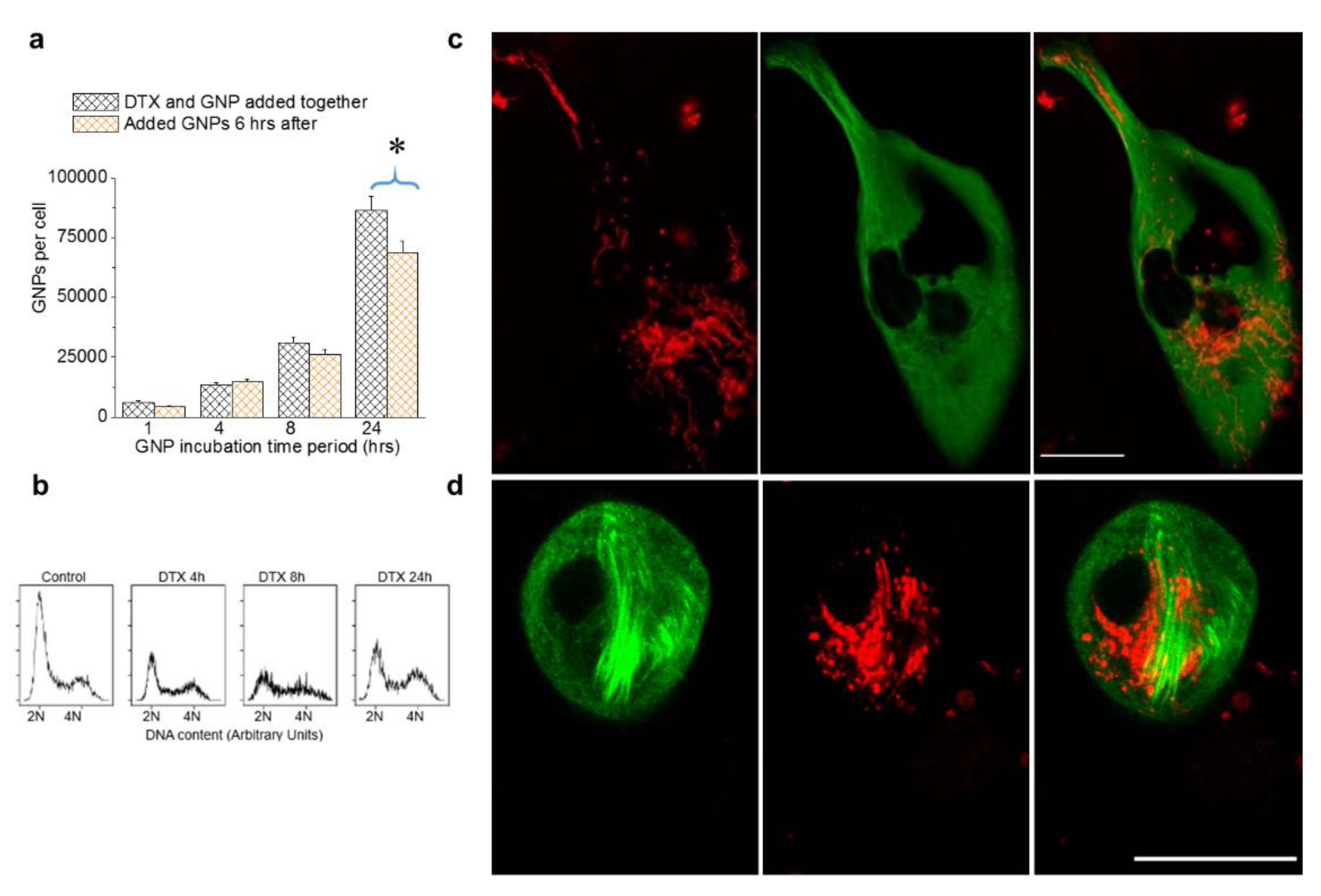
3.5. The Relative Timing of DTX and GNP Inoculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. PTA Peer Rev. J. Formul. Manag. 2017, 42, 742–755. [Google Scholar]
- Paciotti, G.F.; Zhao, J.; Cao, S.; Brodie, P.J.; Tamarkin, L.; Huhta, M.; Myer, L.D.; Friedman, J.; Kingston, D.G.I. Synthesis and Evaluation of Paclitaxel-Loaded Gold Nanoparticles for Tumor-Targeted Drug Delivery. Bioconj. Chem. 2016, 27, 2646–2657. [Google Scholar] [CrossRef]
- Paciotti, G.F.; Myer, L.; Weinreich, D.; Goia, D.; Pavel, N.; McLaughlin, R.E.; Tamarkin, L. Colloidal gold: A novel nanoparticle vector for tumor directed drug delivery. Drug Deliv. 2004, 11, 169–183. [Google Scholar] [CrossRef]
- Yang, C.; Uertz, J.; Chithrani, D.B. Colloidal Gold-Mediated Delivery of Bleomycin for Improved Outcome in Chemotherapy. Nanomaterials 2016, 6, 48. [Google Scholar] [CrossRef]
- Retif, P.; Pinel, S.; Toussaint, M.; Frochot, C.; Chouikrat, R.; Bastogne, T.; Barberi-Heyob, M. Nanoparticles for Radiation Therapy Enhancement: The Key Parameters. Theranostics 2015, 5, 1030–1044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Berg, A.; Levanon, H.; Fessenden, R.W.; Meisel, D. On the interactions of free radicals with gold nanoparticles. J. Am. Chem. Soc. 2003, 125, 7959–7963. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sanche, L. Low energy electrons in nanoscale radiation physics: Relationship to radiosensitization and chemoradiation therapy. Rev. Nanosci. Nanotechnol. 2013, 2, 1–28. [Google Scholar] [CrossRef]
- Townley, H.E.; Kim, J.; Dobson, P.J. In vivo demonstration of enhanced radiotherapy using rare earth doped titania nanoparticles. Nanoscale 2012, 4, 5043–5050. [Google Scholar] [CrossRef]
- Mirjolet, C.; Papa, A.L.; Crehange, G.; Raguin, O.; Seignez, C.; Paul, C.; Truc, G.; Maingon, P.; Millot, N. The radiosensitization effect of titanate nanotubes as a new tool in radiation therapy for glioblastoma: A proof-of-concept. Radiother. Oncol. 2013, 108, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.; Misawa, M. Analysis of Potential Radiosensitizing Materials for X-Ray-Induced Photodynamic Therapy. NanoBiotechnology 2007, 3, 116–126. [Google Scholar] [CrossRef]
- Yang, W.; Read, P.W.; Mi, J.; Baisden, J.M.; Reardon, K.A.; Larner, J.M.; Helmke, B.P.; Sheng, K. Semiconductor Nanoparticles as Energy Mediators for Photosensitizer-Enhanced Radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2008, 72, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Jelveh, S.; Jalali, F.; van Prooijen, M.; Allen, C.; Bristow, R.G.; Hill, R.P.; Jaffray, D.A. Gold nanoparticles as radiation sensitizers in cancer therapy. Radiat. Res. 2010, 173, 719–728. [Google Scholar] [CrossRef]
- Le Duc, G.; Miladi, I.; Alric, C.; Mowat, P.; Brauer-Krisch, E.; Bouchet, A.; Khalil, E.; Billotey, C.; Janier, M.; Lux, F.; et al. Toward an image-guided microbeam radiation therapy using gadolinium-based nanoparticles. ACS Nano 2011, 5, 9566–9574. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Huang, Z.; Chen, Z.; Xu, R.; Wu, H.; Zang, F.; Wang, C.; Gu, N. Silver nanoparticles: A novel radiation sensitizer for glioma? Nanoscale 2013, 5, 11829. [Google Scholar] [CrossRef]
- Hainfeld, J.F.; Dilmanian, F.A.; Slatkin, D.N.; Smilowitz, H.M. Radiotherapy enhancement with gold nanoparticles. J. Pharm. Pharmacol. 2008, 60, 977–985. [Google Scholar] [CrossRef]
- Hainfeld, J.F.; Slatkin, D.N.; Smilowitz, H.M. The use of gold nanoparticles to enhance radiotherapy in mice. Phys. Med. Biol. 2004, 49, N309. [Google Scholar] [CrossRef]
- Zheng, Y.; Sanche, L. Gold nanoparticles enhance DNA damage induced by anti-cancer drugs and radiation. Radiat. Res. 2009, 172, 114–119. [Google Scholar] [CrossRef]
- Rieck, K.; Bromma, K.; Sung, W.; Bannister, A.; Schuemann, J.; Chithrani, D.B. Modulation of gold nanoparticle mediated radiation dose enhancement through synchronization of breast tumor cell population. Br. J. Radiol. 2019, 92, 20190283. [Google Scholar] [CrossRef]
- Kim, J.A.; Åberg, C.; Salvati, A.; Dawson, K.A. Role of cell cycle on the cellular uptake and dilution of nanoparticles in a cell population. Nat. Nanotechnol. 2011, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, A.; Giocanti, N.; Favaudon, V.; Bornens, M. Pulse treatment of interphasic HeLa cells with nanomolar doses of docetaxel affects centrosome organization and leads to catastrophic exit of mitosis. J. Cell Sci. 1997, 110, 2403–2415. [Google Scholar] [PubMed]
- Granger, E.; McNee, G.; Allan, V.; Woodman, P. The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin. Cell Dev. Biol. 2014, 31, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D. Intracellular uptake, transport, and processing of gold nanostructures. Mol. Membr. Biol. 2010, 27, 299–311. [Google Scholar] [CrossRef]
- Vale, R.D.; Milligan, R.A. The Way Things Move: Looking Under the Hood of Molecular Motor Proteins. Science 2000, 288, 88. [Google Scholar] [CrossRef]
- Barlan, K.; Gelfand, V.I. Microtubule-Based Transport and the Distribution, Tethering, and Organization of Organelles. Cold Spring Harb. Perspect. Biol. 2017, 9, a025817. [Google Scholar] [CrossRef]
- Shen, Y.; Ma, Z.; Chen, F.; Dong, Q.; Hu, Q.; Bai, L.; Chen, J. Effective photothermal chemotherapy with docetaxel-loaded gold nanospheres in advanced prostate cancer. J. Drug Target. 2015, 23, 568–576. [Google Scholar] [CrossRef]
- Ghalandari, B.; Asadollahi, K.; Shakerizadeh, A.; Komeili, A.; Riazi, G.; Kamrava, S.K.; Attaran, N. Microtubule network as a potential candidate for targeting by gold nanoparticle-assisted photothermal therapy. J. Photochem. Photobiol. B Biol. 2019, 192, 131–140. [Google Scholar] [CrossRef]
- Yohan, D.; Chithrani, B.D. Applications of nanoparticles in nanomedicine. J. Biomed. Nanotechnol. 2014, 10, 2371–2392. [Google Scholar] [CrossRef]
- Li, C.; Li, D.; Wan, G.; Xu, J.; Hou, W. Facile synthesis of concentrated gold nanoparticles with low size-distribution in water: Temperature and pH controls. Nanoscale Res. Lett. 2011, 6, 440. [Google Scholar] [CrossRef]
- Cruje, C.; Yang, C.; Uertz, J.; van Prooijen, M.; Chithrani, B.D. Optimization of PEG coated nanoscale gold particles for enhanced radiation therapy. RSC Adv. 2015, 5, 101525–101532. [Google Scholar] [CrossRef]
- Wu, P.H.; Onodera, Y.; Ichikawa, Y.; Rankin, E.B.; Giaccia, A.J.; Watanabe, Y.; Qian, W.; Hashimoto, T.; Shirato, H.; Nam, J.M. Targeting integrins with RGD-conjugated gold nanoparticles in radiotherapy decreases the invasive activity of breast cancer cells. Int. J. Nanomed. 2017, 12, 5069–5085. [Google Scholar] [CrossRef]
- Yang, C.; Bromma, K.; Chithrani, D. Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models. Cancers 2018, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Stoeva, S.I.; Prasad, B.L.V.; Uma, S.; Stoimenov, P.K.; Zaikovski, V.; Sorensen, C.M.; Klabunde, K.J. Face-Centered Cubic and Hexagonal Closed-Packed Nanocrystal Superlattices of Gold Nanoparticles Prepared by Different Methods. J. Phys. Chem. B 2003, 107, 7441–7448. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Stewart, J.; Allen, C.; Jaffray, D.A. Intracellular uptake, transport, and processing of nanostructures in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 118–127. [Google Scholar] [CrossRef]
- Wolfe, T.; Chatterjee, D.; Lee, J.; Grant, J.D.; Bhattarai, S.; Tailor, R.; Goodrich, G.; Nicolucci, P.; Krishnan, S. Targeted gold nanoparticles enhance sensitization of prostate tumors to megavoltage radiation therapy in vivo. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Brunsvig, P.F.R.; Andersen, A.; Aamdal, S.; Kristensen, V.; Olsen, H. Pharmacokinetic analysis of two different docetaxel dose levels in patients with non-small cell lung cancer treated with docetaxel as monotherapy or with concurrent radiotherapy. BMC Cancer 2007, 7, 197. [Google Scholar] [CrossRef]
- Smythe, E.; Ayscough, K.R. Actin regulation in endocytosis. J. Cell Sci. 2006, 119, 4589. [Google Scholar] [CrossRef]
- Miyanaga, S.; Ninomiya, I.; Tsukada, T.; Okamoto, K.; Harada, S.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; et al. Concentration-dependent radiosensitizing effect of docetaxel in esophageal squamous cell carcinoma cells. Int. J. Oncol. 2016, 48, 517–524. [Google Scholar] [CrossRef]
- Bannister, A.H.; Bromma, K.; Sung, W.; Monica, M.; Cicon, L.; Howard, P.; Chow, R.L.; Scheumann, J.; Chithrani, D.B. Modulation of nanoparticle uptake, intracellular distribution, and retention with docetaxel to enhance radiotherapy. Br. J. Radiol. 2019, 93, 20190742. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Chan, W.C.W. Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Kumar Maity, T.; Robey, R.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Kiura, K.; Ueoka, H.; Segawa, Y.; Tabata, M.; Kamei, H.; Takigawa, N.; Hiraki, S.; Watanabe, Y.; Bessho, A.; Eguchi, K.; et al. Phase I/II study of docetaxel and cisplatin with concurrent thoracic radiation therapy for locally advanced non-small-cell lung cancer. Br. J. Cancer 2003, 89, 795–802. [Google Scholar] [CrossRef]
- Varveris, H.; Mazonakis, M.; Vlachaki, M.; Kachris, S.; Lyraraki, E.; Zoras, O.; Maris, T.; Froudarakis, M.; Velegrakis, J.; Perysinakis, C.; et al. A phase I trial of weekly docetaxel and cisplatinum combined to concurrent hyperfractionated radiotherapy for non-small cell lung cancer and squamous cell carcinoma of head and neck. Oncol. Rep. 2003, 10, 185–195. [Google Scholar] [CrossRef]
- Alvarez, E.A.; Wolfson, A.H.; Pearson, J.M.; Crisp, M.P.; Mendez, L.E.; Lambrou, N.C.; Lucci, J.A., 3rd. A phase I study of docetaxel as a radio-sensitizer for locally advanced squamous cell cervical cancer. Gynecol. Oncol. 2009, 113, 195–199. [Google Scholar] [CrossRef]
- Kumar, P. A new paradigm for the treatment of high-risk prostate cancer: Radiosensitization with docetaxel. Rev. Urol. 2003, 5, S71–S77. [Google Scholar]
- Kumar, P.; Weiss, R. Radiosensitization with docetaxel and 3-D CRT. Results of a completed phase I trial. Proc. Am. Soc. Clin. Oncol. 2003, 22, 404. [Google Scholar]
- Karasawa, K.; Ito, K.; Takada, T.; Matsumoto, F.; Haruyama, T.; Ito, S.; Ikeda, K. 2434: Hyperfractionated Radiotherapy With Concurrent Docetaxel for Advanced Head and Neck Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, S450–S451. [Google Scholar] [CrossRef]
- Harari, P.M.; Harris, J.; Kies, M.S.; Myers, J.N.; Jordan, R.C.; Gillison, M.L.; Foote, R.L.; Machtay, M.; Rotman, M.; Khuntia, D.; et al. Postoperative Chemoradiotherapy and Cetuximab for High-Risk Squamous Cell Carcinoma of the Head and Neck: Radiation Therapy Oncology Group RTOG-0234. J. Clin. Oncol. 2014, 32, 2486–2495. [Google Scholar] [CrossRef]
- Jelveh, S.; Chithrani, D.B. Gold Nanostructures as a Platform for Combinational Therapy in Future Cancer Therapeutics. Cancers 2011, 3, 1081–1110. [Google Scholar] [CrossRef] [PubMed]
- Schuemann, J.; Berbeco, R.; Chithrani, D.B.; Cho, S.H.; Kumar, R.; McMahon, S.J.; Sridhar, S.; Krishnan, S. Roadmap to Clinical Use of Gold Nanoparticles for Radiation Sensitization. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 189–205. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bannister, A.; Dissanayake, D.; Kowalewski, A.; Cicon, L.; Bromma, K.; Chithrani, D.B. Modulation of the Microtubule Network for Optimization of Nanoparticle Dynamics for the Advancement of Cancer Nanomedicine. Bioengineering 2020, 7, 56. https://doi.org/10.3390/bioengineering7020056
Bannister A, Dissanayake D, Kowalewski A, Cicon L, Bromma K, Chithrani DB. Modulation of the Microtubule Network for Optimization of Nanoparticle Dynamics for the Advancement of Cancer Nanomedicine. Bioengineering. 2020; 7(2):56. https://doi.org/10.3390/bioengineering7020056
Chicago/Turabian StyleBannister, Aaron, Dushanthi Dissanayake, Antonia Kowalewski, Leah Cicon, Kyle Bromma, and Devika B. Chithrani. 2020. "Modulation of the Microtubule Network for Optimization of Nanoparticle Dynamics for the Advancement of Cancer Nanomedicine" Bioengineering 7, no. 2: 56. https://doi.org/10.3390/bioengineering7020056
APA StyleBannister, A., Dissanayake, D., Kowalewski, A., Cicon, L., Bromma, K., & Chithrani, D. B. (2020). Modulation of the Microtubule Network for Optimization of Nanoparticle Dynamics for the Advancement of Cancer Nanomedicine. Bioengineering, 7(2), 56. https://doi.org/10.3390/bioengineering7020056