
Process Intensification Strategies for Power-to-X Technologies


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Boundary Conditions for PtX Processes
- (a)
- the thermal instability and transient hot spot formation,
- (b)
- the possibly enhanced catalyst deactivation and degradation due to thermal cycling and load changes,
- (c)
- and the transient changes in product quality or composition and possible undesired side product formation.
- (a)
- no compensation of fluctuating electricity using grid electricity,
- (b)
- the elaborative production of utilities onsite using renewable resources,
- (c)
- the high costs of operation and maintenance,
- (d)
- and the limited available area for construction.
1.2. Objectives of This Work
1.3. Background and Process Intensification Approaches
2. Process Intensification for Power-to-Ammonia Processes
2.1. Background
2.2. Conventional Haber-Bosch Process
2.3. Power-to-Ammonia
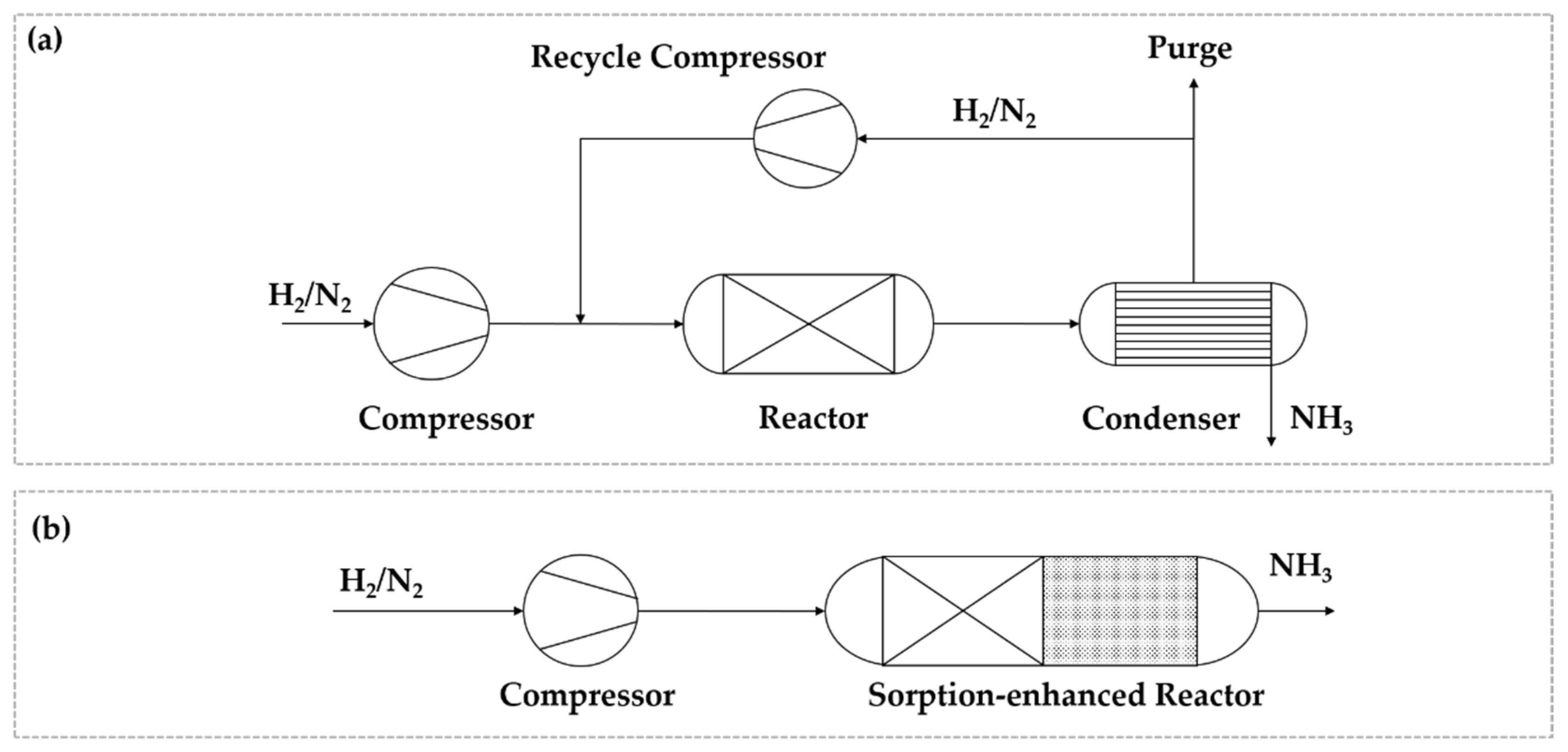
2.4. Process Intensification Methods
3. Process Intensification for Power-to-DME Processes
3.1. Background
3.2. Process Intensification Methods
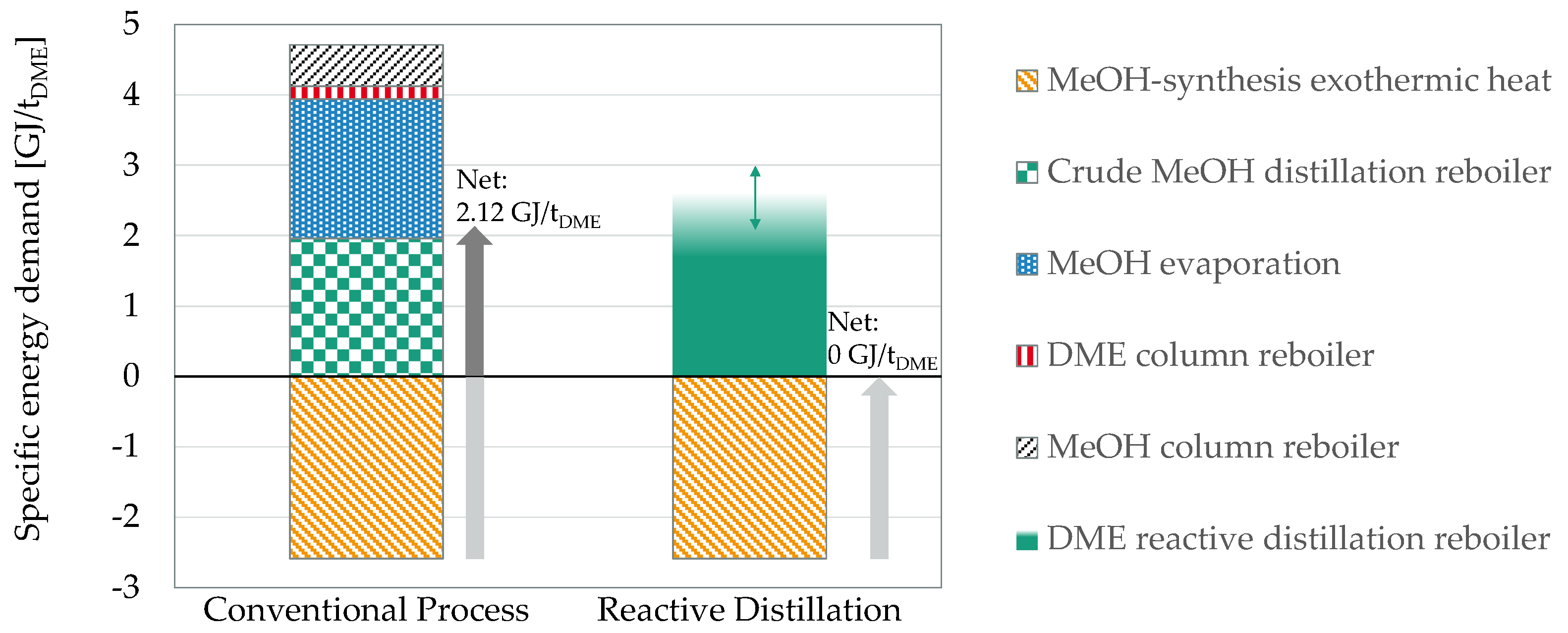
3.3. DME Synthesis by Reactive Distillation
- (a)
- the reaction is limited by chemical equilibrium,
- (b)
- the reaction is exothermic, which allows the utilization of the reaction enthalpy to reduce the reboiler heat demand,
- (c)
- and the components MeOH, DME and water exhibit a high relative volatility, thus allowing a good thermal separation capability.
- (a)
- the feedstock purification (crude MeOH distillation),
- (b)
- the DME synthesis reactor and
- (c)
- the product separation.
4. Process Intensification for Power-to-OME Processes
4.1. Background
4.2. Power-to-OME
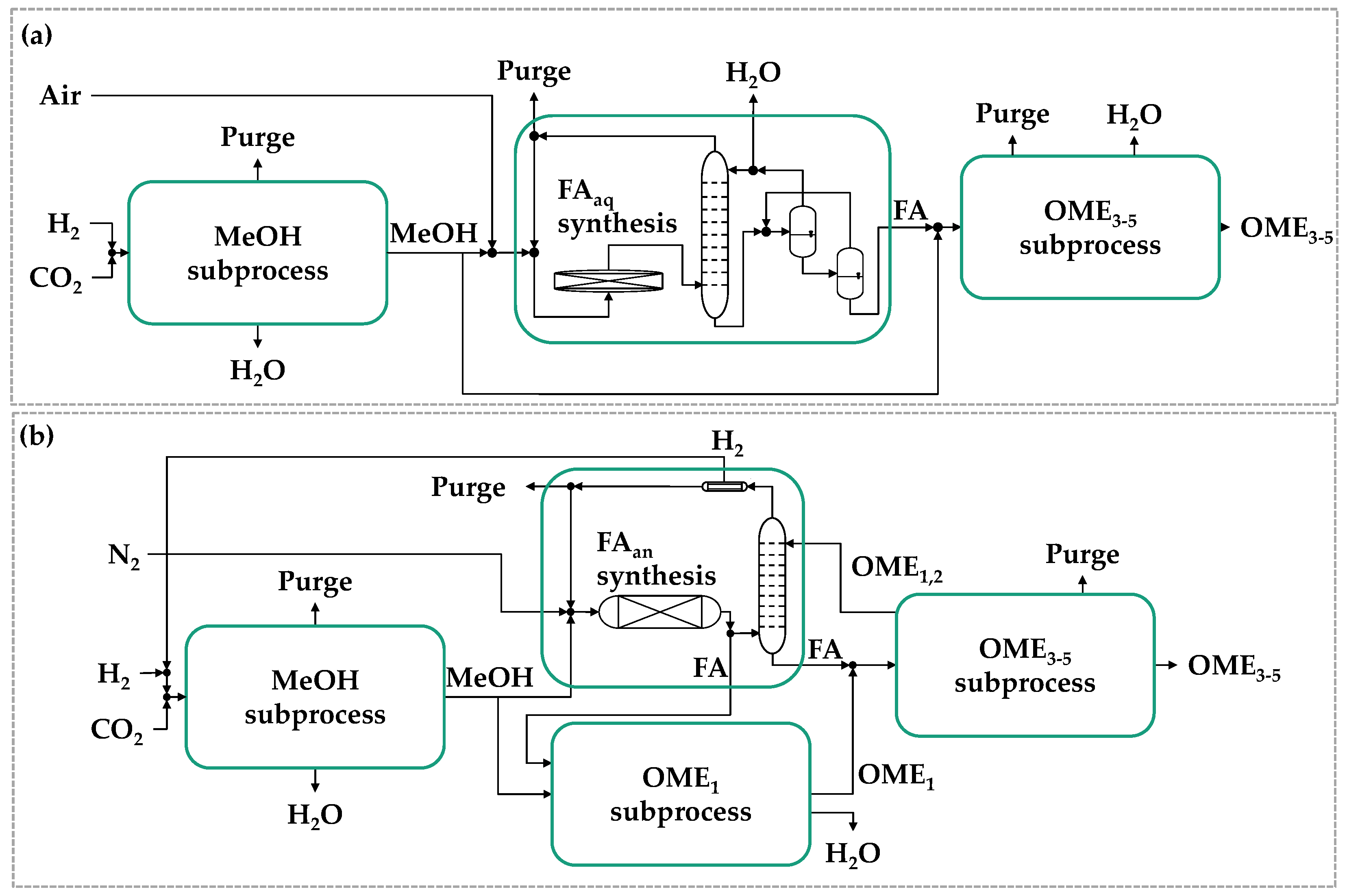
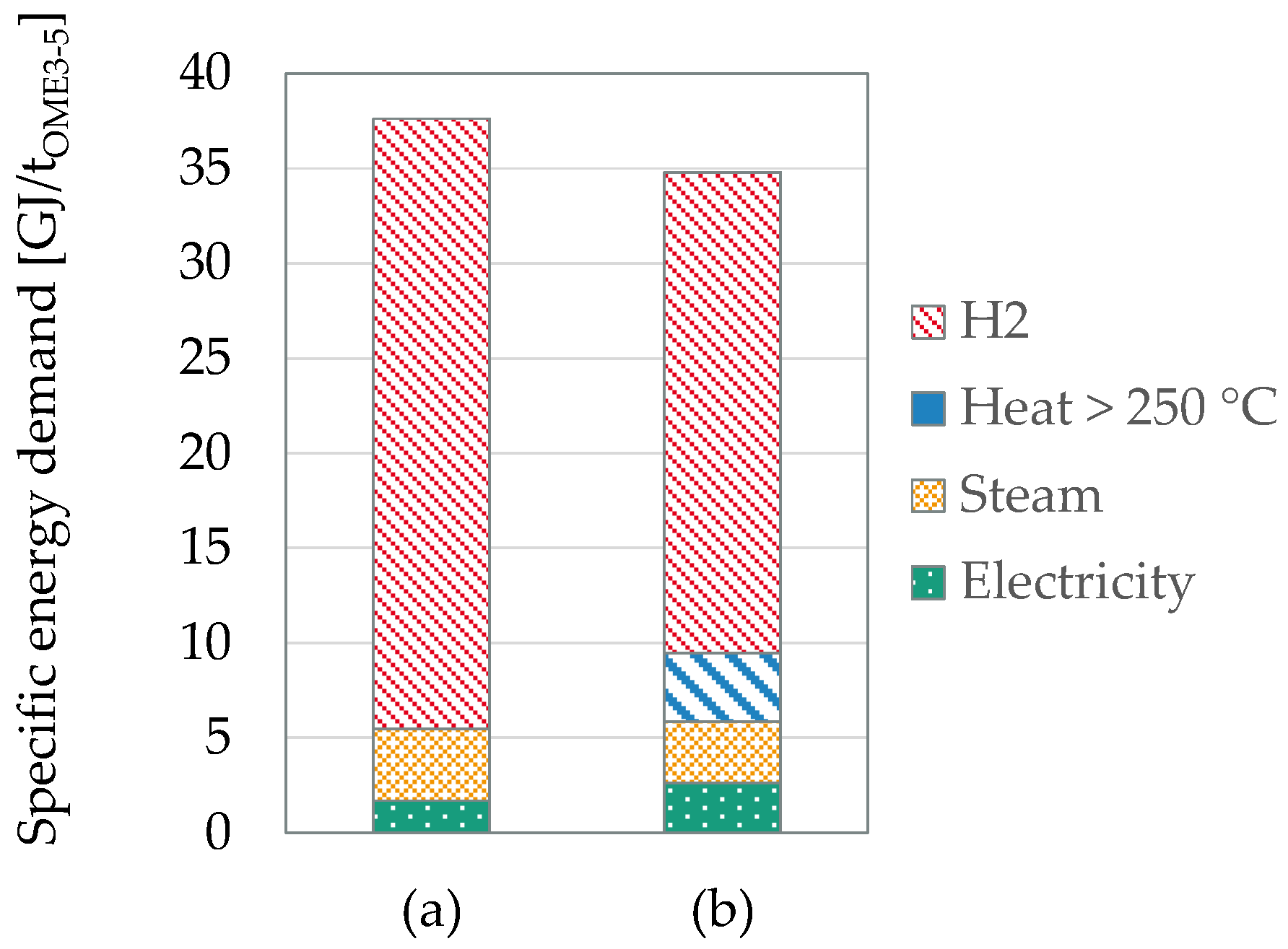
4.3. Process Intensification Methods
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IPCC, 2. Summary for Policymakers. In Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change: The Physical Science Basis; Cambridge University Press: Cambridge, UK, 2021; in press. [Google Scholar]
- Rogelj, J.; Shindell, D.; Jiang, K.; Fifita, S.; Forster, P.; Ginzburg, V.; Handa, C.; Kheshgi, H.; Kobayashi, S.; Kriegler, E.; et al. Mitigation pathways compatible with 1.5 °C in the context of sustainable development. In Global Warming of 1.5 °C; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2018; pp. 93–174. [Google Scholar]
- International Renewable Energy Agency. Green Hydrogen: A Guide to Policy Making. 2020. Available online: https://www.irena.org/-/media/Files/IRENA/Agency/Publication/2020/Nov/IRENA_Green_hydrogen_policy_2020.pdf (accessed on 8 December 2021).
- Hank, C.; Sternberg, A.; Köppel, N.; Holst, M.; Smolinka, T.; Schaadt, A.; Hebling, C.; Henning, H.-M. Energy efficiency and economic assessment of imported energy carriers based on renewable electricity. Sustain. Energy Fuels 2020, 4, 2256–2273. [Google Scholar] [CrossRef]
- International Energy Agency. World Energy Outlook 2020, Outlook for Energy Demand. Available online: https://www.iea.org/reports/world-energy-outlook-2020/outlook-for-energy-demand (accessed on 3 August 2021).
- Sterchele, P.; Brandes, J.; Heilig, J.; Wrede, D.; Kost, C.; Schlegl, T.; Bett, A.; Henning, H.-M. Wege zu Einem Klimaneutralen Energiesystem: Die Deutsche Energiewende im Kontext Gesellschaftlicher Verhaltensweisen 2020; Fraunhofer-Institut für Solare Energiesysteme ISE: Freiburg im Breisgau, Germany, 2020. [Google Scholar]
- Nanba, T. Hydrogen Storage and Utilization by Using Carrier Compounds for effective usage of renewable energy. In Proceedings of the World Hydrogen Technologies Convention, Tokyo, Japan, 3 June 2019. [Google Scholar]
- Nazir, H.; Muthuswamy, N.; Louis, C.; Jose, S.; Prakash, J.; Buan, M.E.; Flox, C.; Chavan, S.; Shi, X.; Kauranen, P.; et al. Is the H2 economy realizable in the foreseeable future? Part II: H2 storage, transportation, and distribution. Int. J. Hydrog. Energy 2020, 45, 20693–20708. [Google Scholar] [CrossRef]
- Schemme, S.; Breuer, J.L.; Köller, M.; Meschede, S.; Walman, F.; Samsun, R.C.; Peters, R.; Stolten, D. H2-based synthetic fuels: A techno-economic comparison of alcohol, ether and hydrocarbon production. Int. J. Hydrog. Energy 2020, 45, 5395–5414. [Google Scholar] [CrossRef]
- Jenck, J.F.; Agterberg, F.; Droescher, M. Products and processes for a sustainable chemical industry: A review of achievements and prospects. Green Chem. 2004, 6, 544–556. [Google Scholar] [CrossRef]
- Stankiewicz, A.I.; Moulijn, J.A. Process intensification: Transforming chemical engineering. Chem. Eng. Prog. 2000, 96, 22–34. [Google Scholar]
- Ramshaw, C. Process Intensification and Green Chemistry. Green Chem. 1999, 1, G15–G17. [Google Scholar] [CrossRef]
- Gallucci, F.; van Sint Annaland, M. (Eds.) Process Intensification for Sustainable Energy Conversion; Wiley: Chichester, UK, 2015; ISBN 978-1-118-44935-6. [Google Scholar]
- Hüther, A.; Geißelmann, A.; Hahn, H. Prozessintensivierung—Eine strategische Option für die chemische Industrie. Chem. Ing. Tech. 2005, 77, 1829–1837. [Google Scholar] [CrossRef]
- Sitter, S.; Chen, Q.; Grossmann, I.E. An overview of process intensification methods. Curr. Opin. Chem. Eng. 2019, 25, 87–94. [Google Scholar] [CrossRef]
- Agar, D.W.; Ruppel, W. Multifunktionale Reaktoren für die heterogene Katalyse. Chem. Ing. Tech. 1988, 60, 731–741. [Google Scholar] [CrossRef]
- Bundesministerium für Wirtschaft und Energie (BMWi) (2020): Die Nationale Wasserstoffstrategie. Hg. v. Bundesministerium für Wirtschaft und Energie (BMWi). Berlin. Available online: https://www.bmwi.de/Redaktion/DE/Publikationen/Energie/die-nationale-wasserstoffstrategie.html (accessed on 8 December 2021).
- Yasmina, B.; Andras, P.; Anish, P.; van Chritian, E.S. Power to Ammonia: Rethinking the Role of Ammonia—From a Value Product to a Flexible Energy Carrier (FlexNH3); Hanzehogeschool Groningen: Groningen, The Netherlands, 2016; p. 100. [Google Scholar]
- Appl, M. Ammonia: 2. Production Processes. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; ISBN 9783527306732. [Google Scholar]
- Nayak-Luke, R.; Bañares-Alcántara, R.; Wilkinson, I. “Green” Ammonia: Impact of Renewable Energy Intermittency on Plant Sizing and Levelized Cost of Ammonia. Ind. Eng. Chem. Res. 2018, 57, 14607–14616. [Google Scholar] [CrossRef] [Green Version]
- Afif, A.; Radenahmad, N.; Cheok, Q.; Shams, S.; Kim, J.H.; Azad, A.K. Ammonia-fed fuel cells: A comprehensive review. Renew. Sustain. Energy Rev. 2016, 60, 822–835. [Google Scholar] [CrossRef]
- Klüssmann, J.; Ekknund, L.; Ivarsson, A.; Schramm, J. Ammonia Application in IC Engines. 2020. Available online: https://iea-amf.org/app/webroot/files/file/other%20publications/Ammonia%20Application%20in%20IC%20Engines.pdf (accessed on 8 December 2021).
- Christensen, C.H.; Johannessen, T.; Sørensen, R.Z.; Nørskov, J.K. Towards an ammonia-mediated hydrogen economy? Catal. Today 2006, 111, 140–144. [Google Scholar] [CrossRef]
- Brown, T. Ammonia Production Causes 1% of Total Global GHG Emissions. Available online: https://ammoniaindustry.com/ammonia-production-causes-1-percent-of-total-global-ghg-emissions/ (accessed on 19 November 2019).
- Schlögl, R. Katalytische Ammoniaksynthese—Eine “unendliche Geschichte”? Angew. Chem. 2003, 115, 2050–2055. [Google Scholar] [CrossRef]
- Twigg, M.V.; Spencer, M.S.; Jennings, J.R. Catalytic Ammonia Synthesis; Springer: Boston, MA, USA, 1991; ISBN 978-1-4757-9594-3. [Google Scholar]
- Liu, H. Ammonia synthesis catalyst 100 years: Practice, enlightenment and challenge. Chin. J. Catal. 2014, 35, 1619–1640. [Google Scholar] [CrossRef]
- Schlögl, R. Catalytic Synthesis of Ammonia—A “Never-Ending Story”? Angew. Chem. Int. Ed. 2003, 42, 2004–2008. [Google Scholar] [CrossRef]
- Fujimura, Y.; Kai, M.; Fujimoto, T.; Fujimoto, S.; Atsumi, R.; Nishi, M.; Mochizuki, T.; Nanba, T. Demonstration and optimization of Green Ammonia Production Operation Responding to Fluctuating Hydrogen Production from Renewable Energy. In Proceedings of the AIChE Annual Meeting, Orlando, FL, USA, 10–15 November 2019. [Google Scholar]
- Ayvalı, T.; Tsang, S.C.E.; Van Vrijaldenhoven, T. The Position of Ammonia in Decarbonising Maritime Industry: An Overview and Perspectives: Part I: Technological advantages and the momentum towards ammonia-propelled shipping. Johns. Matthey Technol. Rev. 2021, 65, 275–290. [Google Scholar] [CrossRef]
- Rouwenhorst, K.H.R.; Krzywda, P.M.; Benes, N.E.; Mul, G.; Lefferts, L. Ammonia, 4. Green Ammonia Production. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 1–20. ISBN 9783527306732. [Google Scholar]
- Aika, K.; Hori, H.; Ozaki, A. Activation of nitrogen by alkali metal promoted transition metal I. Ammonia synthesis over ruthenium promoted by alkali metal. J. Catal. 1972, 27, 424–431. [Google Scholar] [CrossRef]
- Nishi, M.; Chen, S.-Y.; Takagi, H. A Mesoporous Carbon-Supported and Cs-promoted Ru Catalyst with Enhanced Activity and Stability for Sustainable Ammonia Synthesis. ChemCatChem 2018, 10, 3411–3414. [Google Scholar] [CrossRef]
- Nishi, M.; Chen, S.-Y.; Takagi, H. Mild Ammonia Synthesis over Ba-Promoted Ru/MPC Catalysts: Effects of the Ba/Ru Ratio and the Mesoporous Structure. Catalysts 2019, 9, 480. [Google Scholar] [CrossRef] [Green Version]
- Javaid, R.; Matsumoto, H.; Nanba, T. Influence of Reaction Conditions and Promoting Role of Ammonia Produced at Higher Temperature Conditions in Its Synthesis Process over Cs-Ru/MgO Catalyst. ChemistrySelect 2019, 4, 2218–2224. [Google Scholar] [CrossRef]
- Rouwenhorst, K.; Krzywda, P.M.; Benes, N.E.; Mul, G.; Lefferts, L. Ammonia Production Technologies. In Techno-Economic Challenges of Green Ammonia as an Energy Vector; Elsevier: Amsterdam, The Netherlands, 2021; pp. 41–83. ISBN 9780128205600. [Google Scholar]
- Liu, H. Ammonia Synthesis Catalysts: Innovation and Practice. World Scientific Pub. Co: Singapore; Hackensack, NJ, USA; Beijing, China, 2013; ISBN 978-981-4355-77-3. [Google Scholar]
- Nikačević, N.; Jovanović, M.; Petkovska, M. Enhanced ammonia synthesis in multifunctional reactor with in situ adsorption. Chem. Eng. Res. Des. 2011, 89, 398–404. [Google Scholar] [CrossRef]
- Liu, C.Y.; Aika, K.-I. Ammonia Adsorption on Ion Exchanged Y-zeolites as Ammonia Storage Material. J. Jpn. Pet. Inst. 2003, 46, 301–307. [Google Scholar] [CrossRef]
- Matito-Martos, I.; García-Reyes, J.; Martin-Calvo, A.; Dubbeldam, D.; Calero, S. Improving Ammonia Production Using Zeolites. J. Phys. Chem. 2019, 123, 18475–18481. [Google Scholar] [CrossRef]
- Smith, C.; Torrente-Murciano, L. Exceeding Single-Pass Equilibrium with Integrated Absorption Separation for Ammonia Synthesis Using Renewable Energy—Redefining the Haber-Bosch Loop. Adv. Energy Mater. 2021, 11, 2003845. [Google Scholar] [CrossRef]
- Smith, C.; McCormick, A.V.; Cussler, E.L. Optimizing the Conditions for Ammonia Production Using Absorption. ACS Sustain. Chem. Eng. 2019, 7, 4019–4029. [Google Scholar] [CrossRef]
- Malmali, M.; Wei, Y.; McCormick, A.; Cussler, E.L. Ammonia Synthesis at Reduced Pressure via Reactive Separation. Ind. Eng. Chem. Res. 2016, 55, 8922–8932. [Google Scholar] [CrossRef]
- Malmali, M.; Le, G.; Hendrickson, J.; Prince, J.; McCormick, A.V.; Cussler, E.L. Better Absorbents for Ammonia Separation. ACS Sustain. Chem. Eng. 2018, 6, 6536–6546. [Google Scholar] [CrossRef]
- Liu, C.Y.; Aika, K.-I. Ammonia Absorption into Alkaline Earth Metal Halide Mixtures as an Ammonia Storage Material. Ind. Eng. Chem. Res. 2004, 43, 7484–7491. [Google Scholar] [CrossRef]
- Klerke, A.; Christensen, C.H.; Nørskov, J.K.; Vegge, T. Ammonia for hydrogen storage: Challenges and opportunities. J. Mater. Chem. 2008, 18, 2304–2310. [Google Scholar] [CrossRef]
- Cussler, E.; McCormick, A.; Reese, M.; Malmali, M. Ammonia Synthesis at Low Pressure. J. Vis. Exp. 2017, 112, e55691. [Google Scholar] [CrossRef]
- Christensen, C.H.; Sørensen, R.Z.; Johannessen, T.; Quaade, U.J.; Honkala, K.; Elmøe, T.D.; Køhler, R.; Nørskov, J.K. Metal ammine complexes for hydrogen storage. J. Mater. Chem. 2005, 15, 4106–4108. [Google Scholar] [CrossRef]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Liu, H.; Han, W.; Huo, C.; Cen, Y. Development and application of wüstite-based ammonia synthesis catalysts. Catal. Today 2019, 355, 110–127. [Google Scholar] [CrossRef]
- Palys, M.J.; McCormick, A.; Cussler, E.L.; Daoutidis, P. Modeling and Optimal Design of Absorbent Enhanced Ammonia Synthesis. Processes 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Fleisch, T.; Basu, A.; Sills, R. Introduction and advancement of a new clean global fuel: The status of DME developments in China and beyond. J. Nat. Gas Sci. Eng. 2012, 9, 94–107. [Google Scholar] [CrossRef]
- Semmel, M.; Ali, R.E.; Ouda, M.; Schaadt, A.; Sauer, J.; Hebling, C. Power-to-DME: A cornerstone towards a sustainable energy system. In Power to Fuel: How to Speed Up a Hydrogen Economy; Spazzafumo, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 123–151. ISBN 9780128228135. [Google Scholar]
- Nestler, F.; Krüger, M.; Full, J.; Hadrich, M.J.; White, R.J.; Schaadt, A. Methanol Synthesis—Industrial Challenges within a Changing Raw Material Landscape. Chem. Ing. Tech. 2018, 90, 1409–1418. [Google Scholar] [CrossRef]
- Pontzen, F.; Liebner, W.; Gronemann, V.; Rothaemel, M.; Ahlers, B. CO2-based methanol and DME—Efficient technologies for industrial scale production. Catal. Today 2011, 171, 242–250. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. Process Intensif. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Van Kampen, J.; Boon, J.; van Berkel, F.; Vente, J.; Annaland, M.V.S. Steam separation enhanced reactions: Review and outlook. Chem. Eng. J. 2019, 374, 1286–1303. [Google Scholar] [CrossRef]
- Van Kampen, J.; Boon, J.; Vente, J.; Annaland, M.V.S. Sorption enhanced dimethyl ether synthesis for high efficiency carbon conversion: Modelling and cycle design. J. CO2 Util. 2020, 37, 295–308. [Google Scholar] [CrossRef]
- Diban, N.; Urtiaga, A.M.; Ortiz, I.; Ereña, J.; Bilbao, J.; Aguayo, A.T. Influence of the membrane properties on the catalytic production of dimethyl ether with in situ water removal for the successful capture of co2. Chem. Eng. J. 2013, 234, 140–148. [Google Scholar] [CrossRef]
- Diban, N.; Aguayo, A.T.; Bilbao, J.; Urtiaga, A.; Ortiz, I. Membrane Reactors for in Situ Water Removal: A Review of Applications. Ind. Eng. Chem. Res. 2013, 52, 10342–10354. [Google Scholar] [CrossRef]
- Bîldea, C.S.; Győrgy, R.; Brunchi, C.C.; Kiss, A.A. Optimal design of intensified processes for DME synthesis. Comput. Chem. Eng. 2017, 105, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Zou, Z.; Dai, C.; Li, Q.; Chen, B. Synthesis of dimethyl ether (DME) by catalytic distillation. Chem. Eng. Sci. 2011, 66, 3195–3203. [Google Scholar] [CrossRef]
- An, W.; Chuang, K.T.; Sanger, A.R. Dehydration of Methanol to Dimethyl Ether by Catalytic Distillation. Can. J. Chem. Eng. 2008, 82, 948–955. [Google Scholar] [CrossRef]
- Zhenova, A.; Pellis, A.; Milescu, R.A.; McElroy, C.R.; White, R.J.; Clark, J.H. Solvent Applications of Short-Chain Oxymethylene Dimethyl Ether Oligomers. ACS Sustain. Chem. Eng. 2019, 7, 14834–14840. [Google Scholar] [CrossRef]
- Schappals, M.; Breug-Nissen, T.; Langenbach, K.; Burger, J.; Hasse, H. Solubility of Carbon Dioxide in Poly (oxymethylene) Dimethyl Ethers. J. Chem. Eng. Data 2017, 62, 4027–4031. [Google Scholar] [CrossRef]
- Vigier, F.; Coutanceau, C.; Léger, J.; Dubois, J. Polyoxymethylenedimethylether (CH3O(CH2O)nCH3) oxidation on Pt and Pt/Ru supported catalysts. J. Power Sources 2008, 175, 82–90. [Google Scholar] [CrossRef]
- Devaux, D.; Yano, H.; Uchida, H.; Dubois, J.-L.; Watanabe, M. Electro-oxidation of hydrolysed poly-oxymethylene-dimethylether on PtRu supported catalysts. Electrochim. Acta 2011, 56, 1460–1465. [Google Scholar] [CrossRef]
- Baranton, S.; Uchida, H.; Tryk, D.A.; Dubois, J.L.; Watanabe, M. Hydrolyzed polyoxymethylenedimethylethers as liquid fuels for direct oxidation fuel cells. Electrochim. Acta 2013, 108, 350–355. [Google Scholar] [CrossRef]
- Kakinuma, K.; Kim, I.-T.; Senoo, Y.; Yano, H.; Watanabe, M.; Uchida, M. Electrochemical Oxidation of Hydrolyzed Poly Oxymethylene-dimethyl Ether by PtRu Catalysts on Nb-Doped SnO2−δ Supports for Direct Oxidation Fuel Cells. ACS Appl. Mater. Interfaces 2014, 6, 22138–22145. [Google Scholar] [CrossRef] [PubMed]
- Sakaida, S.; Sugiyama, M.; Nagayama, R.; Tanaka, K.; Konno, M. Potential of hydrolyzed oxymethylene dimethyl ether for the suppression of fuel crossover in polymer electrolyte fuel cells. Int. J. Hydrog. Energy 2021, 46, 10892–10902. [Google Scholar] [CrossRef]
- Wolfgang, M.; Jacob, E.; Härtl, M.; Wachtmeister, G. (Eds.) Synthetic Fuels—OME 1: A Potentially Sustainable. In Proceedings of the Wiener Motorensymposium, Vienna, Austria, 8–9 May 2014. Diesel Fuel. [Google Scholar]
- Härtl, M.; Gaukel, K.; Pélerin, D.; Wachtmeister, G. Oxymethylene Ether as Potentially CO2-neutral Fuel for Clean Diesel Engines Part 1: Engine Testing. MTZ Worldw. 2017, 78, 52–59. [Google Scholar] [CrossRef]
- Jacob, E.; Maus, W. Oxymethylene Ether as Potentially Carbon-neutral Fuel for Clean Diesel Engines Part 2: Compliance with the Sustainability Requirement. MTZ Worldw. 2017, 78, 52–57. [Google Scholar] [CrossRef]
- Münz, M.; Mokros, A.; Töpfer, D.; Beidl, C. OME—Assessment of Particle Emissions in Real Driving Conditions. MTZ Worldw. 2018, 79, 16–21. [Google Scholar] [CrossRef]
- Lumpp, B.; Rothe, D.; Pastötter, C.; Lämmermann, R.; Jacob, E. Oxymethylene Ethers as Diesel Fuel Additives of the Future. Mtz Worldw. 2011, 72, 34–38. [Google Scholar] [CrossRef]
- Schmitz, N.; Ströfer, E.; Burger, J.; Hasse, H. Conceptual Design of a Novel Process for the Production of Poly (oxymethylene) Dimethyl Ethers from Formaldehyde and Methanol. Ind. Eng. Chem. Res. 2017, 56, 11519–11530. [Google Scholar] [CrossRef]
- Burger, J. A novel process for the production of diesel fuel additives by hierarchical design. Ph.D. Thesis, University of Kaiserslautern, Kaiserslautern, Germany, 2012. [Google Scholar]
- Li, X.; Cao, J.; Nawaz, M.A.; Hu, Y.; Liu, D. Experimental and Correlated Liquid–Liquid Equilibrium Data for Ternary Systems (Water + Poly (oxymethylene) Dimethyl Ethers + Toluene) at T = 293.15 and 303.15 K and p = 101.3 kPa. J. Chem. Eng. Data 2019, 64, 5548–5557. [Google Scholar] [CrossRef]
- Li, X.; Tian, H.; Zhang, W.; Liu, D. Production Process for Diesel Fuel Components Polyoxymethylene Dimethyl Ethers from Methanol and Formaldehyde Solution. Int. J. Chem. Mol. Eng. 2018, 12, 536–541. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, S.; Li, P.; Li, P.; Li, Q.; Yu, Y. Measurement and Thermodynamic Models for Ternary Liquid–Liquid Equilibrium Systems {Water + Polyoxymethylene Dimethyl Ethers + 4-Methyl-2-pentanol} at Different Temperatures. J. Chem. Eng. Data 2018, 63, 3074–3082. [Google Scholar] [CrossRef]
- Shi, M.; Yu, X.; He, G.; Li, Q. Liquid–Liquid equilibrium for the ternary systems water + DMM3 + (p-xylene, toluene, and n-heptane) at different temperatures. Can. J. Chem. Eng. 2017, 96, 968–977. [Google Scholar] [CrossRef]
- Shi, M.; He, G.; Gan, F.; Yu, X.; Li, Q. Extraction of Low Concentration Aqueous Solution of Methylal: Liquid–Liquid Equilibrium in Water + Methylal + (Cyclohexane and n-Heptane) Ternary Systems. J. Chem. Eng. Data 2017, 62, 2183–2190. [Google Scholar] [CrossRef]
- Zhuang, Z.; Zhang, J.; Liu, D. Liquid-Liquid equilibria for ternary systems polyoxymethylene dimethyl ethers + water + n-hexane. CIESC J. 2016, 67, 3545–3551. [Google Scholar] [CrossRef]
- Zhuang, Z.; Zhang, J.; Liu, X.; Liu, D. Liquid–Liquid equilibria for ternary systems polyoxymethylene dimethyl ethers + para-xylene + water. J. Chem. Thermodyn. 2016, 101, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Oestreich, D.; Lautenschütz, L.; Arnold, U.; Sauer, J. Production of oxymethylene dimethyl ether (OME)-hydrocarbon fuel blends in a one-step synthesis/extraction procedure. Fuel 2018, 214, 39–44. [Google Scholar] [CrossRef]
- Ferre, A.; Burger, J. Coadsorption Equilibria on Molecular Sieves 3A and Densities of Liquid Mixtures Containing Formaldehyde, Methanol, and Water at 295.15 and 313.15 K. Ind. Eng. Chem. Res. 2021, 60, 15256–15263. [Google Scholar] [CrossRef]
- Schmitz, N.; Breitkreuz, C.F.; Ströfer, E.; Burger, J.; Hasse, H. Separation of water from mixtures containing formaldehyde, water, methanol, methylal, and poly(oxymethylene) dimethyl ethers by pervaporation. J. Membr. Sci. 2018, 564, 806–812. [Google Scholar] [CrossRef]
- Held, M.; Tönges, Y.; Pélerin, D.; Härtl, M.; Wachtmeister, G.; Burger, J. On the energetic efficiency of producing polyoxymethylene dimethyl ethers from CO2 using electrical energy. Energy Environ. Sci. 2019, 12, 1019–1034. [Google Scholar] [CrossRef]
- Ullmann’s Encyclopedia of Industrial Chemistry: Formaldehyde; Franz, A.W.; Kronemayer, H.; Pfeiffer, D.; Pilz, R.D.; Reuss, G.; Disteldorf, W.; Gamer, A.O.; Hilt, A. (Eds.) Wiley-VCH Verlag GmbH & Co. KG: Weinheim, Germany, 2016. [Google Scholar]
- Mantei, F.; Ali, R.E.; Baensch, C.; Voelker, S.; Haltenort, P.; Burger, J.; Dietrich, R.-U.; von der Assen, N.; Schaadt, A.; Sauer, J.; et al. Techno-economic assessment and carbon footprint of processes for the large-scale production of oxymethylene dimethyl ethers from carbon dioxide and hydrogen. Sustain. Energy Fuels 2021, 6, 528–549. [Google Scholar] [CrossRef]
- FReSMe Project—From Residual Steel Gases to Methanol. Available online: https://www.carbonrecycling.is/news-media/fresme-project-reaches-final-milestone-from-blast-furnace-waste-emissions-to-ferry-fuel (accessed on 8 December 2021).
- Hackbarth, K.; Haltenort, P.; Arnold, U.; Sauer, J. Recent Progress in the Production, Application and Evaluation of Oxymethylene Ethers. Chem. Ing. Tech. 2018, 90, 1520–1528. [Google Scholar] [CrossRef]
- Dolton Jiangsu Dolton Chemical Technology Co., Ltd. DMMn Plant. Available online: https://www.aldehydeepc.com/dmmn-plant-15609099648755723.html (accessed on 8 December 2021).
- Breitkreuz, C.F.; Schmitz, N.; Ströfer, E.; Burger, J.; Hasse, H. Design of a Production Process for Poly (oxymethylene) Dimethyl Ethers from Dimethyl Ether and Trioxane. Chem. Ing. Tech. 2018, 90, 1489–1496. [Google Scholar] [CrossRef]
- Haltenort, P.; Hackbarth, K.; Oestreich, D.; Lautenschütz, L.; Arnold, U.; Sauer, J. Heterogeneously catalyzed synthesis of oxymethylene dimethyl ethers (OME) from dimethyl ether and trioxane. Catal. Commun. 2018, 109, 80–84. [Google Scholar] [CrossRef]
- Su, S.; Zaza, D.-C.-I.P.; Renken, A. Catalytic dehydrogenation of methanol to water-free formaldehyde. Chem. Eng. Technol. 1994, 17, 34–40. [Google Scholar] [CrossRef]
- Sauer, I.J.; Emig, G. The catalyzed dehydrogenation of methanol to formaldehyde at high temperatures: New insights by modelling of transport phenomena and reaction. Chem. Eng. Technol. 1995, 18, 284–291. [Google Scholar] [CrossRef]
- Ouda, M.; Mantei, F.; Hesterwerth, K.; Eleonora Bargiacchi, E.; Klein, H.; White, R.J. A hybrid description and evaluation of oxymethylene dimethyl ethers synthesis based on the endothermic dehydrogenation of methanol. React. Chem. Eng. 2018, 3, 676–695. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cholewa, T.; Semmel, M.; Mantei, F.; Güttel, R.; Salem, O. Process Intensification Strategies for Power-to-X Technologies. ChemEngineering 2022, 6, 13. https://doi.org/10.3390/chemengineering6010013
Cholewa T, Semmel M, Mantei F, Güttel R, Salem O. Process Intensification Strategies for Power-to-X Technologies. ChemEngineering. 2022; 6(1):13. https://doi.org/10.3390/chemengineering6010013
Chicago/Turabian StyleCholewa, Thomas, Malte Semmel, Franz Mantei, Robert Güttel, and Ouda Salem. 2022. "Process Intensification Strategies for Power-to-X Technologies" ChemEngineering 6, no. 1: 13. https://doi.org/10.3390/chemengineering6010013
APA StyleCholewa, T., Semmel, M., Mantei, F., Güttel, R., & Salem, O. (2022). Process Intensification Strategies for Power-to-X Technologies. ChemEngineering, 6(1), 13. https://doi.org/10.3390/chemengineering6010013