1. Introduction
The unique structure and organization of the central nervous system (CNS) makes it particularly vulnerable to changes in its external and internal environment which, if unregulated, can be associated with neurodegenerative disease. The brain and cord, as well as being enveloped and shielded from physical trauma by its meninges and cerebrospinal fluid, is also protected from chemical ionic imbalance and uptake of noxious substances by the blood–brain barrier (BBB) and the blood–cerebrospinal fluid barrier (BCB). It has been suggested that this barrier may be compromised during ageing or adverse environmental conditions that could allow the unregulated entry of metals or other toxicants with potentially damaging consequences. There is supporting evidence that Cu and Fe increase in the brain as it ages and in associated degenerative states.
The hypothesis of metal-based neurodegeneration is now widely accepted [
1,
2,
3] and ageing is a major risk factor. In particular the transition metals, Cu, Fe, and (Mn) are essential metals which have the ability to exist in different ionic states that can provoke the formation of reactive oxygen species (ROS), and are especially incriminated. Protein aggregation in the brain is the putative result of oxidative stress, with the formation of plaques in Alzheimers disease (AD), Lewy bodies in Parkinsons disease (PD), amyotrophic lateral sclerosis (ALS), Huntingtons disease (HD) and the prion diseases (Pr).
The pathobiology of these plaques commands extensive research but less attention has been given to why and how this ionic dyshomeostasis occurs in the brain, given that the blood–brain barrier appears to be tightly regulated. This mini-review addresses this imbalance by presenting an alternative viewpoint from studies of copper uptake in a primitive sheep model, the North Ronaldsay sheep, in which copper regulation in the brain is adapted to serve an evolutionary stratagem.
2. Metal Dyshomeostasis and Oxidative Stress
Both iron (Fe) and copper (Cu) are essential trace elements for human and animal health, and can exist in a variety of oxidation states such as Fe2+, Fe3+ and Cu+, Cu2+. These multiple oxidation states allow the metals to readily participate in oxidation–reduction reactions. Copper is an integral component of various cupro enzymes including cytochrome C oxidase, lysyl oxidase, superoxide dismutase (SOD), tyrosinase, dopamine B-oxidase, and caeruloplasmin. An important function of Fe is to transport oxygen in hemoglobin and myoglobin. Fe is also required for the normal function of cytochrome oxidase, peroxidase, and catalase. Furthermore, Fe is involved in mitochondrial respiration, DNA synthesis, and biosynthesis of neurotransmitters. Conversely, free Fe and Cu ions can readily interact with oxygen to initiate cascades of biochemical reactions, leading to the production of free radicals and oxidative stress. Thus, a stable homeostasis of the redox balance from Cu+/Cu2+ and Fe2+/Fe3+ is necessary for brain function.
The internal milieu of the brain is exceptionally susceptible to oxidative stress by toxic reactive oxygen species (ROS) due to its high metabolic demands, accounting for up to 20% of all oxygen consumption, and its rich concentration of polyunsaturated by fatty acids. (Oxidant stress is regarded as the imbalance between pro-oxidant free radical production and opposing antioxidant defense). The superoxide radical O
2− is produced disproportionally in the brain mitochondria during cellular respiration and undergoes dismutation to hydrogen peroxide (H
2O
2), and thence to water, with catalase. Alternatively, transformation of H
2O
2 in the cytosol may occur in the presence of Cu
+ and Fe
2+ to produce the highly reactive hydroxyl radical OH. Unfortunately the antioxidant defence, chiefly in the form of superoxide dismutase (Cu Zn SOD), catalase, and glutathione peroxidase can, be overwhelmed in the CNS by the high level of oxidizing activity in this organ, and possible occurrence of the unbound transitional metals Cu
+ and Fe
2+. These transition metals are essential for life but also inimical if unregulated. Reactive oxygen species (ROS) are responsible for DNA, cell membrane, and protein damage. Protein folding can also be a casualty of free radical damage, leading to misfolded proteins with aggregation and loss of function. Such plaque formation is a characteristic feature of the degenerative diseases aforementioned. The roles of copper and/or iron in oxidative stress and neurodegenerative disease have been reviewed in depth [
4,
5].
2.1. Copper Homeostasis–Distribution and Dysregulation
The brain has a high requirement for copper, second only to the liver. On account of its oxidation potential, regulation is necessary to ensure only sufficient but not excess copper is present in the brain. Copper concentrations are maintained at a constant level when maturity is achieved; in human beings this is 2.9–10.7 µg/g wet weight, whilst rat brains appear to have a lower copper content of 1–2.3 µg/g wet weight [
6,
7,
8]. There is some ambiguity in the literature concerning the reporting of CNS copper levels at differing time points in the life cycles of individuals in health and disease, from the in utero/neonatal period (Cu increasing), maturity/adult (Cu stable), to ageing +/− dementia (Cu increase). Studies in the literature are consistent in their support of ageing being characterized by elevated plasma copper levels, from which it is inferred that these changes are associated with concomitant changes in the brain copper [
9]. Other studies have shown that copper elevation (in the range 7.8–37.8 µg/L) has been observed in patients with neurological symptoms related to dementia; moreover, in Alzheimers-like dementia, copper levels in the CNS were reported to be twofold higher than in age matched controls [
8]. The metal is not uniformly distributed within the CNS. Studies in adult rats show copper enhancement in the medial geniculate nucleus (visual), superior colliculus (motor functions), periaqueductal grey matter (pain and defensive behaviour); with levels also high in the lateral amygdala (memory and emotional response) and dorsomedial diencephalon (thalamus/hypothalamus) [
10].
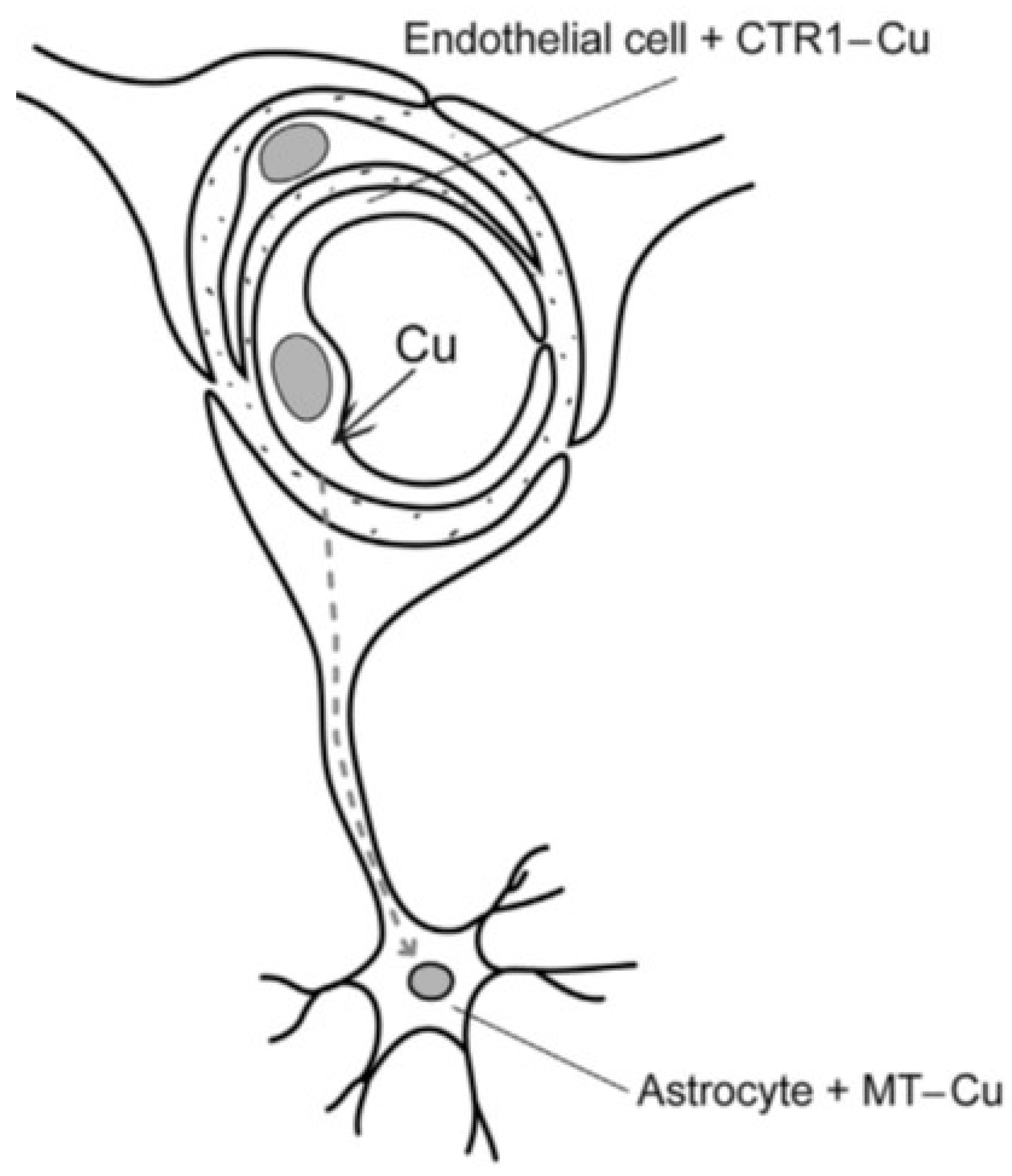
Copper enters the system loosely bound to serum albumin, from which it is released at the BBB and taken up into the luminal endothelium of the blood capillaries (
Figure 1). Distribution is accomplished by a coordinated system of copper transporters and chaperones which convey copper safely across cell membranes to where it is required. Three major groups are recognized: copper uptake transporters, which transfer copper into the cytosol; copper chaperones, which facilitate copper distribution to intracellular target organelles; and copper transporting ATP-ases, which translocate copper into the secretory pathways and small vesicles for delivery to newly synthesized cuproenzymes. In the first category is the recognized high-affinity copper transporter (CTR1) in the brain which has high concentrations in the blood–brain barrier (BBB) and blood–cerebrospinal fluid barrier (BCB) as well as the divalent metal transporter (DMT1) which includes the transfer of Zn
2+, Fe
2+, and Mn
2+, in addition to Cu
2+. In the second category, are the CCS or copper chaperones for the inclusion of the metal in superoxide dismutase—the major antioxidant in cytosol and mitochondria–copper chaperone Atox1, which transfers copper to the binding sites of the ATP-ases, and cytochrome oxidase (CCO) assembly factors which transfer copper (Cu
2+) to the terminal enzyme of the respiratory chain in mitochondria. Finally, the copper transporting ATP-ases, ATP7A and ATP7B, are mutations which are associated with Menkes disease and Wilsons disease, respectively. This subject was reviewed in [
8] and more recently in [
11]. Studies on the distribution of the copper transporters and chaperones are in their infancy, but it has been shown that ATPA and ATP7B show cell specific distribution in the adult cerebellum, and have distinct enzymatic characteristics and are regulated differently during development [
12]. Studies on the human brain have been more limited in deference to its greater size, but one study has identified significant relationships between copper transporter levels, CTR1, Atox1, ATPA, ATPB and brain copper content [
13].
2.2. Iron Homeostasis
Fe (Fe
2+) is first oxidized to (Fe
3+) by Cu-caeruloplasmin on entering the circulation and is bound to B-globulin apotransferrin to form transferrin (Tf), which serves as the major vehicle for Fe transport in the body. Upon arriving at the target cells, transferrin binds with transferrin receptors (TfR) and through endocytosis is carried into the cell where it is utilized in metabolic processes or conjugated with apoferritin to form the storage protein ferritin [
14]. The brain regulates Fe balance via (i) TfR mediated transport or non-mediated transport at brain barriers, (ii) storage of Fe as ferritin, or (iii) efflux of Fe from the BCB back into circulation. Alternatively, DMT1 can transport Fe across the BBB. Fe is primarily transported to the brain parenchyma by the BBB, and the correlation between TfR density and Fe concentration in various brain regions further suggests that a TfR transport mechanism at the BBB is primarily responsible for the rate of Fe entry into the brain parenchyma [
14]. There is evidence that an uneven distribution of TfR receptors in cerebral capillaries may be responsible for the uneven distribution Fe in brain regions, e.g., >Fe in the striatum and hippocampus [
15]. There is some uncertainty regarding the means by which excess Fe exits the brain, but the consensus is that this occurs from the BCB via DMT1 or TfR [
14]. Iron dysregulation (Fe
2+/Fe
3+) has a major role in PD (discussed further in
Section 6.2), and in AD, and is implicated in amyotrophic lateral sclerosis, Huntingtons and the prion diseases. All these diseases are linked by similar mechanisms of oxidative stress, protein aggregation, and mitochondrial dysfunction [
16].
3. The Brain Barrier System and the Regulation of Metal Homeostasis
The overall concentration of copper and iron, and their compartmentalization within the mature brain, must be maintained to ensure optimum activity with minimum oxidative damage and consequent disease. Ageing and the onset of related disease has been associated with an increase in internal copper and iron content, and discompartmentalization [
8,
16]. Probably the most important system for maintaining overall copper and iron homeostasis is the brain barrier, which, it has been suggested, becomes more permeable during ageing due to a variety of developmental and genetic factors The brain barrier is a protective system consisting of the blood–brain barrier (BBB) and the blood–cerebrospinal fluid barrier (BCB), which together regulate these metals entering and leaving the brain [
14].
The sequence of events that takes place in this process is currently better understood with regard to copper than to iron, and the following argument takes account of this accordingly.
The BBB (
Figure 1) exerts a selective discrimination for copper at the level of the cerebral microvascular endothelium. This consists of a non-fenestrated capillary endothelium that, with adhering pericytes and astrocyte end feet on the abluminal surface, constitutes a unique neurovascular unit [
17]. Astrocytes are considered an integral feature of copper homeostasis in the brain, since it has been shown that excess copper is stored in these cells bound to metallothionien [
18,
19,
20]. Furthermore, the strategic localization of astrocytes makes them ideally positioned to regulate the transport of copper from the BBB to neuronal cells [
11]. The BBB is concerned mainly with copper influx mediated by CTR1 [
11,
14,
20], from which it is transferred via ATP7A directly into the brain for neuronal activities [
14], or taken up by astrocyte end feet, stored, and later released into neurons as required [
11].
The BCB is located in the choroid plexus, a polarized and highly vascularized organ in the roof of the brain ventricles. The BCB comprises a single layer of tightly appositional epithelial cells facing the cerebrospinal fluid (CSF), underlying connective tissue, and an inner layer of capillaries lined by fenestrated endothelial cells. The function of the BCB, in addition to secreting CSF, is concerned with the removal of excess copper (Cu
2+) and certain other substances from the CSF [
21].
4. Ontological Development of the Brain Barrier
It is important to recognize that the functional behaviour of the brain barrier is not fixed throughout life. The ontogenesis of the brain barrier is rarely discussed, although it is recognized that the BBB is immature at birth, and for some time thereafter, in several species [
22]. This is often referred to as “leaky”, an unfortunate description since research has shown that functionally developed transport mechanisms are well developed in the embryonic and neonatal brain [
23,
24].
Regulation of the brain barrier (BBB and BCB) is strictly controlled and operates within defined time-related functional domains in the mammalian brain, i.e., fetal/neonatal and postnatal. Each of these domains is structurally and physiologically adapted to the particular life stage of the organism. During the developmental stage (the fetal and neonatal periods) the brain is growing rapidly and there is a high demand for copper and iron, which in the adult is no longer required.
Regulation of divalent metals takes place at the brain barriers which shield the brain from ionic perturbations in the blood. These barriers consist of tight junctions at the blood–brain interface and the blood–cerebrospinal fluid interface, which are present from a very early stage in embryonic development and act as a diffusion restraint. The transfer of essential nutrients into (ingress) and out of (egress) the brain is by means of specific energy-dependent transporters.
The key genes for Cu, Zn, and Fe import in vascular endothelial cells of the fetus/neonate, compared with the adult, are upregulated and expressed as required. Transport mechanisms in the BBB and BCB of the developing brain are more active, with enhanced expression during development than in the adult, reflecting the greater needs of the brain for copper during intrauterine life [
23]. This fetal/neonatal transport is progressively lost with age, and by adulthood is undetectable. The enhanced metal transporter function in the developing BBB comes at a cost since it is recognized that there is a greater vulnerability of these brain barrier mechanisms to adverse circumstances and that damage during development may have serious consequences, both neurological and neuropsychological, in later life such as schizophrenia, Alzheimers disease, and multiple sclerosis [
25]. However, there is much scope for investigation into important changes in influx (efflux) mechanisms including metals following a pathological insult to the developing brain, which should be pursued.
On reaching maturity, the requirement for copper by the adult brain is reduced and the barrier mechanisms appear to enter into a new state. Fetal/neonatal transporter mechanisms appear to be replaced by more robust mechanisms, although information on this actual transformation is sketchy, even non-existent. However it is well established that CTR1 is the main copper transporter in adult life [
26,
27]. A breakdown in this regulatory sequence can result in metal dyshomeostasis with adverse consequences. The North Ronaldsay sheep has proved an excellent example of just such a variation, driven by evolutionary pressure.
5. North Ronaldsay Sheep as an Animal Model of Copper-Related Disease
The North Ronaldsay (NR) sheep have long attracted interest due to their physiological adaptation to a copper deficient environment, but later in-depth studies have shown how this genetic adaptation can have much wider significance in the elucidation of disease. NR sheep are a semi-feral breed of sheep which have adapted to an ecological niche on the island, feeding on seaweed which is low in copper (<5 ppm) (
Figure 2). Conversely, they display an abnormal sensitivity to copper (Cu) and copper toxicosis when transferred to a copper-replete (11 ppm) habitat. Furthermore, NR sheep and their crossbreeds differ from other breeds in absorbing more dietary copper, showing that this trait is heritable [
28,
29]. Comparative trials have shown a tenfold difference between the ability of NR sheep and a domesticated breed to accumulate copper in the liver [
18]. It would appear that in the NR sheep, infantile Cu import mechanisms from the gut may still be operable in adult life, having been selected for a uniquely Cu deficient environment.
Other studies on these sheep have shown that Cu can accumulate in the liver to toxic concentrations, with a pathology reminiscent of non-Wilsonian toxicities of childhood—e.g., Indian childhood cirrhosis, Endemic Tyrolean infantile cirrhosis, and idiopathic copper toxicosis [
30,
31]—for which they have been recognized as a possible model. This relationship to childhood disease has been further strengthened by investigations that have shown NR sheep as being uniquely sensitive to Cu-induced oxidative stress in which mitochondria are incriminated and play a key role, coupled with increased lysosomal activity and hepatic stellate cell activation leading to fibrosis, which further simulates childhood cirrhosis [
32,
33,
34].
Other studies have shown that liver and brain copper concentrations are highly correlated in NR sheep, suggesting a blood–brain barrier permeability. Copper accumulation in the brain is associated with Alzheimer type II astrocytes with histochemical copper localization, the significance of which is debatable since Cu-induced liver malfunction coexists with brain changes [
18]. Plaques have not been identified, and terminal behavioural changes are more likely a consequence of hepatic encephalopathy.
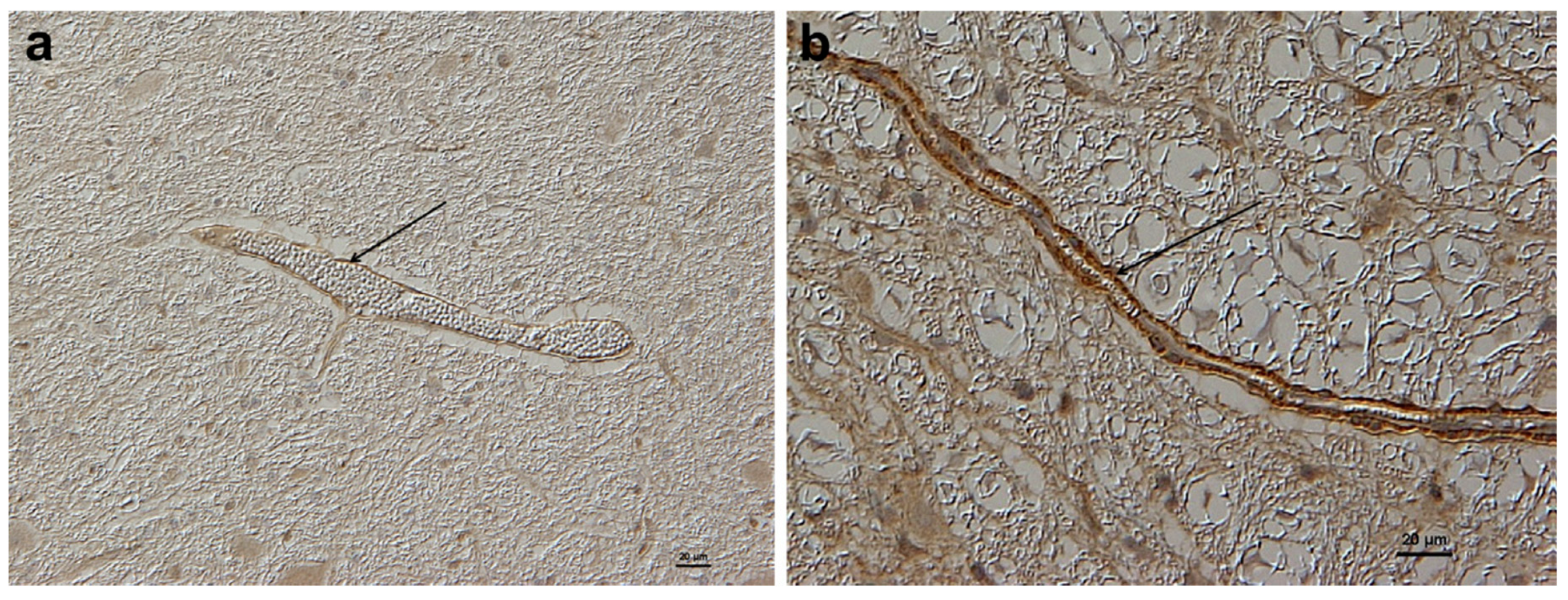
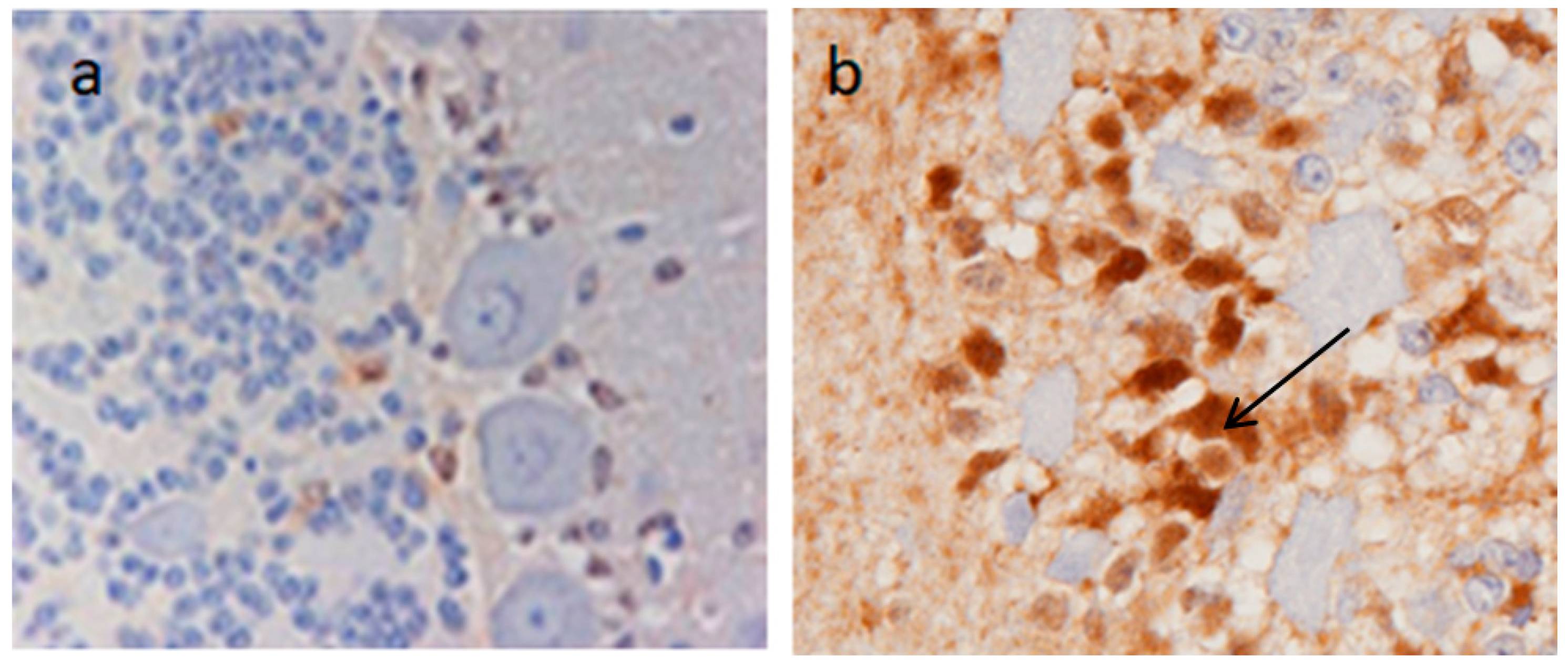
Enhanced blood barrier permeability has been confirmed as being due to the persistent upregulation of CTR1 [
20]. This later study compared the expression of brain CTR1 in adult NR sheep with sheep from a domesticated breed, and found that in the latter CTR1 was only minimally expressed whereas in the NR sheep there was a persistent upregulation of CTR1 (
Figure 3) associated with Cu import and sequestration as MT in astrocytes (
Figure 4) [
20]. It would appear that a genetic mutation has taken place in these NR sheep which allows a continuation of the fetal/neonatal setting regarding systemic copper uptake extending into the brain. This has been selected for by a Darwinian stratagem to allow NR sheep to occupy a unique copper-deficient ecological niche.
This finding in NR sheep [
20] contradicts other findings [
23], in that we have shown that regulatory control of metal transporters into the brain from the BBB is not necessarily time dependent. We surmised further, that the elevated copper content of the ageing (human) brain may derive from a dysregulation of CTR1 at the brain barrier with age and a return to the default (fetal/neonatal) setting, with implications for neurodegenerative disease. The reasons for this must currently remain speculative but may involve changes in the genetic control which may be innate or possibly due to environmentally-induced mutation. Certainly, a study into the control of brain–barrier regulation is well overdue.
6. Neurodegenerative Disorders
Neurological disorders of cognitive and sometimes motor processes are characterized by the presence of misfolded proteins, causing neuronal damage. Such aberrant proteins have a typical tendency to form solid deposits or aggregates, such as the plaques in Alzheimers disease (AD), the Lewy bodies of Parkinsons disease (PD), the Bunini bodies of familial amyotrophic lateral sclerosis (ALS), the inclusions of Huntingtons disease (HD), and lastly, the plaques of the prion diseases. All these disorders have a common endpoint usually involving metals and are collectively known as neurodegenerative diseases.
Proteins are the major component of cells and are involved in nearly every biological process taking place. Their functionality depends on their ability to fold into 3D structures, with intermediate stages in the folding process playing significant roles in the translocation of substances and trafficking to specific cellular locations. Whereas increased Cu and Fe have been associated with the diseases of ageing such as AD, ALS, PD, and HD, certain other diseases, such as the prion diseases, occur over a wider age range in man and animals. All of these disorders are associated with cognitive and in some cases motor disturbances, and have one thing in common in that they have, in the main, intracellular aggregates which are the outcome of the deposition of misfolded protein. It is currently realized that oxidative stress is the chief cause of protein aggregation in the ageing brain and that this is the outcome of impaired copper and iron (and possibly other metals) homeostasis. There are two generic reactions of relevance to neurodegenerative disease: (1) a metal–protein association leading to aggregation, and (2) a metal catalyzed protein–oxidation leading to protein damage and denaturation. This subject is reviewed in [
4,
5]. Ageing and some disease states are associated with elevated brain copper (Cu) and other metals, notably iron (Fe). The brain barrier, likewise, and other internal barrier systems, become more permeable, allegedly due to ageing, genetic, or environmental influences affecting metal compartmentalization. AD, PD, ALS, and HD have all been associated with raised metals, chiefly Cu (Zn) and Mn, Fe. The prion diseases, which include Creutzfeldt–Jakob disease in man, and in animals bovine spongiform encephalopathy and scrapie in sheep are all associated with brain copper dyshomeostasis, and in the latter, possible displacement of Mn. Despite the likely involvement of the brain barriers in the internal displacement of metals, there is no reported study or evidence of deep changes in the brain barriers in any of the neurodegenerative diseases.
6.1. Alzheimers Disease
Alzheimers disease (AD) is associated initially with memory loss, a decline in cognitive function, and ultimately a common endpoint involving metal dyshomeostasis. Extracellular senile plaques of amyloid-AB polypeptide and intracellular neurofibrillary tangles are diagnostic hallmarks [
5,
11]. The AB peptides are generated from the amyloid precursor protein (APP) a membrane protein widely distributed in the brain.
Whilst the plaques have attracted most attention, increasingly they are seen as secondary to metal dyscompartmentalization. The plaques are copper-enriched (to the overall detriment of the brain generally) and this has given rise to the Metal Hypothesis [
2,
16]. Indeed, the role of copper dyshomeostasis in AD has been further emphasized in that it has been postulated that an “epidemic of AD” has been seen in developed countries which have nearly universal copper piping, aided by copper supplements and a high fat component in the diet [
35]. More recently, AD metal dyshomeostasis has extended to include Fe (Fe
2+), which has been observed surrounding the plaques, and it has been suggested that it is the combination of redox metals (Fe and Cu) in the vicinity of plaques which is responsible for the neurotoxicity of the AD plaques [
16].
6.2. Parkinsons Disease (PD)
Parkinsons disease (PD) is the second most prevalent neurodegenerative disease world-wide and is characterized by motor disturbances tremor, rigidity, and bradykinesia. PD, like AD, is generally a disease of advanced age. The pathological changes in PD include degeneration and death of dopaminergic neurons within the substantia nigra pars compacta (SN) and the accumulation in neurons of aggregated α-synuclein protein in Lewy bodies. Most cases of PD are sporadic (90%) but 10% are familial, linked mainly to mutations in synuclein [
16]. The selective molecular basis of PD is not entirely clear but a scientific consensus suggests multiple factors are involved in the degenerative process which include oxidative stress, inflammation, and protein degradation with the aggregation of synuclein. The role of Fe has been recognized for some time, with the involvement of Fe transporters DMT1 and ferroportin, but also with strong and growing evidence for Cu and Zn miscompartmentalization [
5]. However the central role of Fe in PD has become increasingly emphasized. Fe increases in the brain during normal ageing and disproportionately so in the substantia nigra (SN). Dopamine is initially chelated by neuromelanin in the SN but increasingly is overwhelmed by accumulating Fe which initiates the pathophysiology of PD [
36] that leads to neuronal cell death of dopinergic neurones and the accumulation of intracellular inclusions, designated as Lewy bodies [
5,
16].
There is also recognition of a link between the occurrence of PD and industrialization, with the further implication of heavy metal involvement, and in particular Mn, known as manganism, although this has some differences in presentation from the sporadic age-related entity [
37]. However, Mn is now being more widely incriminated in both developmental and neurodegenerative disorders [
38,
39].
Therapeutic intervention using iron chelation is currently a useful strategy, using Deferiprone, a recognized treatment for Fe overload disorder [
16]. Proactively, in the long run it would seem that with all neurodegenerative disorders an understanding of the genetic and environmental influences which determine the movement of metals into and out of the brain would be a most useful approach.
6.3. Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease clinically manifested by weakness and wasting of affected muscles mediated by the loss of motor neurons of the anterior horns of the spinal cord. ALS is, in the main, sporadic (90%) but does possess a familial genetic (autosomal dominant) component (FALS) in another 10% of cases. Both sporadic ALS and FALS, are characterized by the malfunction of Cu–Zn SOD, with loss of antioxidant function and misfolding of the protein resulting in cytoplasmic aggregates, Bunini bodies, and Lewy body-like inclusions. Superoxide dismutases are the major antioxidant scavenging enzymes with a Cu ion as the catalytic function and a Zn ion that maintains structure; see the review [
5] on the association between FALS and the SOD1 mutation. Furthermore, the diminished antioxidant capacity of the motor cell is further aggregated by the SOD gain of toxic function, in which Cu bound to the protein plays a central role. Additionally, the involvement of SOD in regulating Fe proteins plays an important role in Fe accumulation complicating oxidative stress [
5].
6.4. Huntingtons Disease
Huntingtons disease is an autosomal dominant neurodegenerative disorder characterized by progressive motor, cognitive, and psychiatric deterioration with neuronal loss widespread. Disruption in Fe homeostasis, free radical generation, and protein precipitation are all features with, once more, increasing evidence of an as yet unexplored disturbance of copper metabolism [
5,
11].
All the evidence indicates disturbances of metal functioning (Cu and Fe) with metal transporters and altered compartmentalization implicated. Mechanisms of damage elicited by Cu and Fe common to AD, PD ALS, and HD include free radical production, protein aggregation, and metal transport alteration. All this suggests miscompartmentalization of metals, which in turn may implicate genetic regulatory regulation.
6.5. Prion Disease
A final major category of metal-associated neurodegenerative diseases is the Prion disorders, which have crossed the species boundary and are recognized as Creutzfeldt–Jakob disease (CJD) in man, with more recently, a variant form (vCJD) in young people. In animals, the prion disorders are given the generic name of transmissible spongiform encephalopathies (TSEs) and are recognized in cattle as bovine spongiform encephalopathy (BSE), as scrapie in sheep (PrP
sc), and recognized latterly as transmissible mink encephalopathy (TME), chronic wasting disease in mule deer (CWD), and in reports of TSEs in zoo animals and felines [
40,
41]. They have similarities with AD and the other neurodegenerative disorders in that they all show protein aggregation or plaques as a dominant part of their pathology, which consist of an abnormal protease isoform of the prion protein PrP
sc. However, they differ from the above in that they may occur in a variety of ages and also that they are associated with a so called “infectious” agent, the prion protein.
Several decades ago, prion disease was limited to sporadic CJD in humans and the exotic rarity Kuru seen in cannibalistic tribes in New Guinea. Now, certain point genetic mutations in the prion protein gene are recognized in the familial forms of the disease, Gerstmann–Straussler–Scheinker syndrome, fatal familial insomnia, and inherited CJD. In animals, scrapie has long been known to be endemic in the UK for more than 250 years. In 1986, bovine spongiform encephalopathy (BSE) was identified in UK cattle. Transmissible mink encephalopathy (TME), chronic wasting in mule deer (CWD), and identification of TSEs in zoo animals and felines were also reported at that time. The national press was alerted when a variant form of CJD, vCJD was identified in young people. The supposition at the time was that BSE had crossed the species barrier to infect young people through consumption of beef products, though this was never proven. All these diseases are progressively fatal degenerative disorders of the CNS and have a pathology consisting of spongiform changes in the neurons together with the deposition of large amyloid plaques. [
40,
41,
42,
43]. The cause is the prion protein, PrP, which is a normal component of the nervous system found in the cell wall, and is a copper-containing protein with alleged SOD-like activity [
40]. The infective form, PrP
sc, has been shown to be a Mn-substituted isoform which is protease resistant and lacks antioxidant capacity. It is not known how the change occurs and it is thought possible that Mn from enriched soils is responsible for the metal substitution in herbivores, and that a concentration of the isoform provokes self-replication in that it recruits normal PrP in a hijacking of the synthetic machinery, which then operates in an uncontrollable fashion. A unique distinguishing feature of the prion disorders, or TSEs, is that despite lacking nucleic acid they can spread to another host by lateral spread, ingestion of contaminated tissues, and possibly maternal transmission [
40].
A recent report that incriminates Mn in neurodegeneration which may be of relevance to prion disease, is that citrate binds to Mn at the BBB and facilitates the uptake of the metal into the brain several fold [
38]. This may be a path whereby the prion diseases, particularly diseases of herbivores (BSE) on postulated Mn-enriched soils, convert normal PrP
Cu to PrP
sc, the infective prion protein. A final intriguing piece of information is that the conversion of PrP
Cu to PrP
Mn may occur in the astrocyte component of the BBB, all of which links the defective brain barrier into the genesis of the metal-induced neuropathies described [
44].
The mechanisms linking metal-induced dyshomeostasis and neuronal disturbance are still unproven but in general these diseases, even the prion disorders, represent a group of neuropathies characterized by a dysregulation of metalloprotein chemistry deriving from defective metal compartmentalization with protein misfolding, the likely outcome of disordered barrier regulation.
7. Conclusions
It is recognized that dyshomeostasis and miscompartmentalization of the transition metals Cu and Fe (Mn) can take place during brain ageing, with associated oxidative stress and neurodegenerative disease. The integrity of the brain barrier is integral to the transportation and distribution of these metals. The increased need of the developing brain for Cu and Fe is reflected in upregulated brain–barrier transport at this time, which is later downregulated in post-natal life.
Studies on North Ronaldsay sheep have shown that a neonatal setting for high Cu import CTR1 persists into adulthood. Should a similar genetic variation of metal transport take place as a default setting in the brain–barrier of the ageing brain, it would allow the import of metals to upset the delicate equilibrium promoting deleterious oxidative stress.
It is suggested that it may be a productive line of enquiry to study the lifelong ontogeny of the brain barrier and its influence on metal import into the brain and, furthermore, how this can be modulated.

{kind=link}
{kind=link}
{kind=link}
{kind=link}



