



Organoruthenium Complexes Containing Phosphinodicarboxamide Ligands

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Results and Discussion
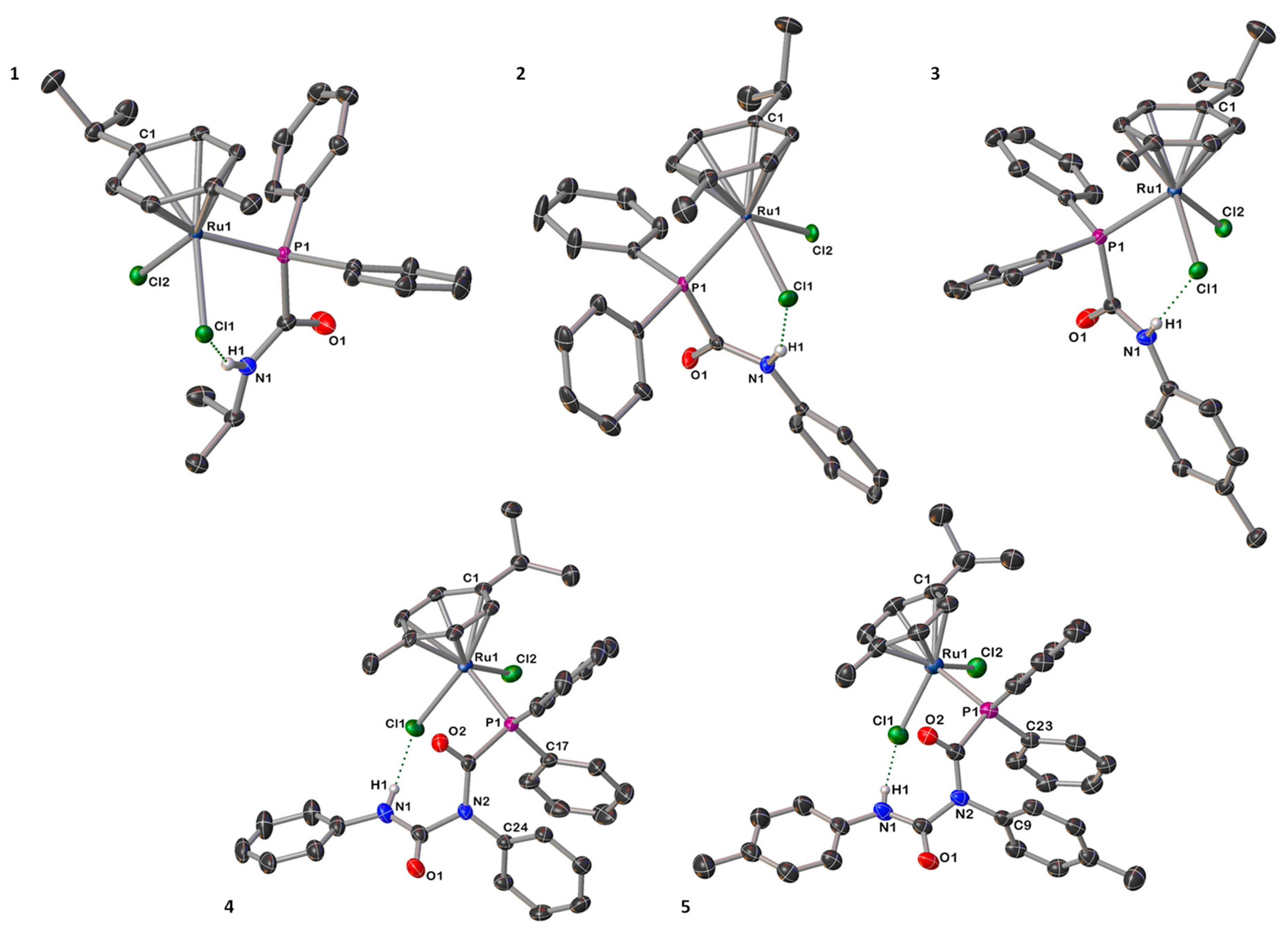
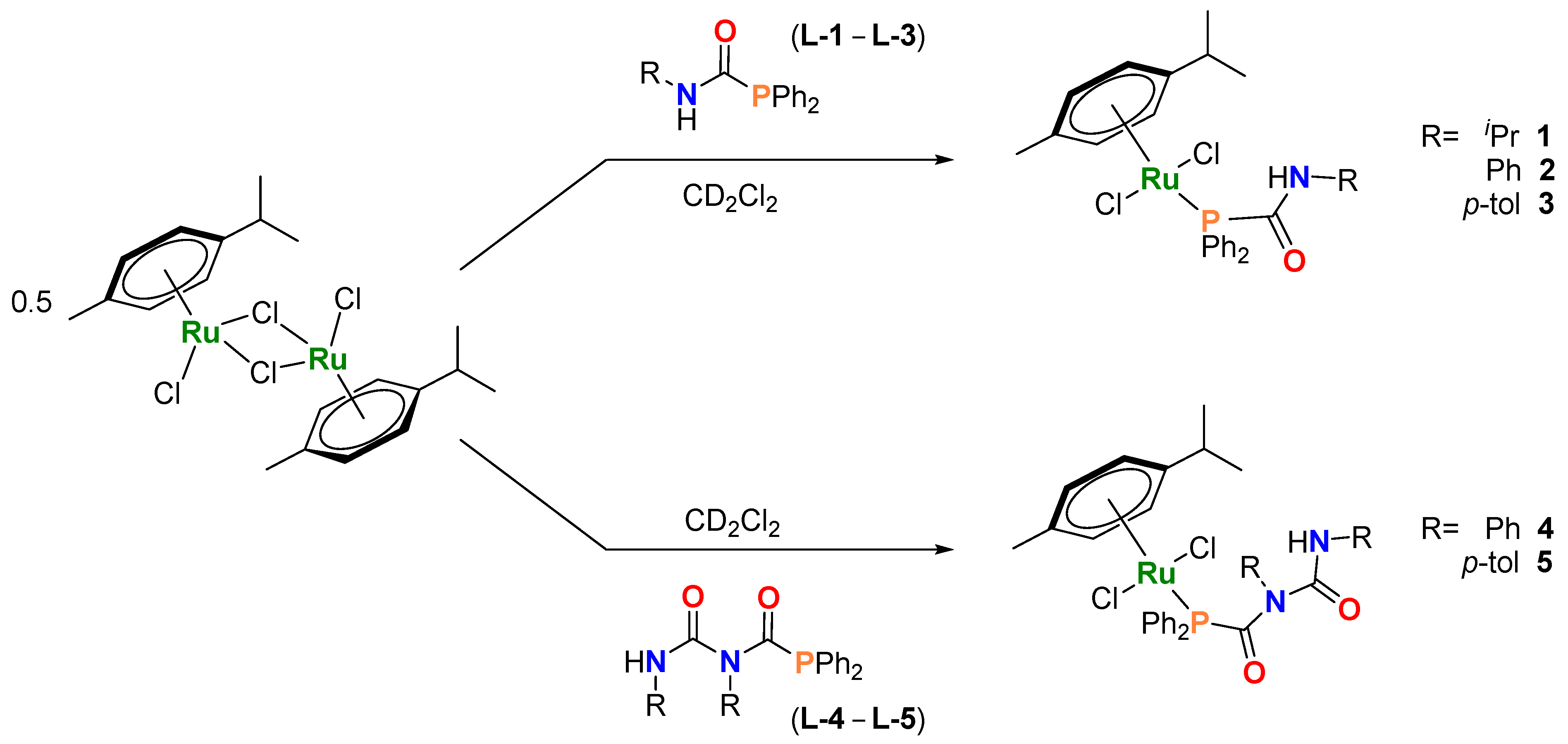
2.1. Synthesis and Characterization of [Ru(p-cym){PPh2C(=O)N(H)R}Cl2] [R = iPr (1), Ph (2), p-tol (3)] and [Ru(p-cym){PPh2C(=O)N(R)C(=O)N(H)R}Cl2] ([R = Ph (4), p-tol (5)]
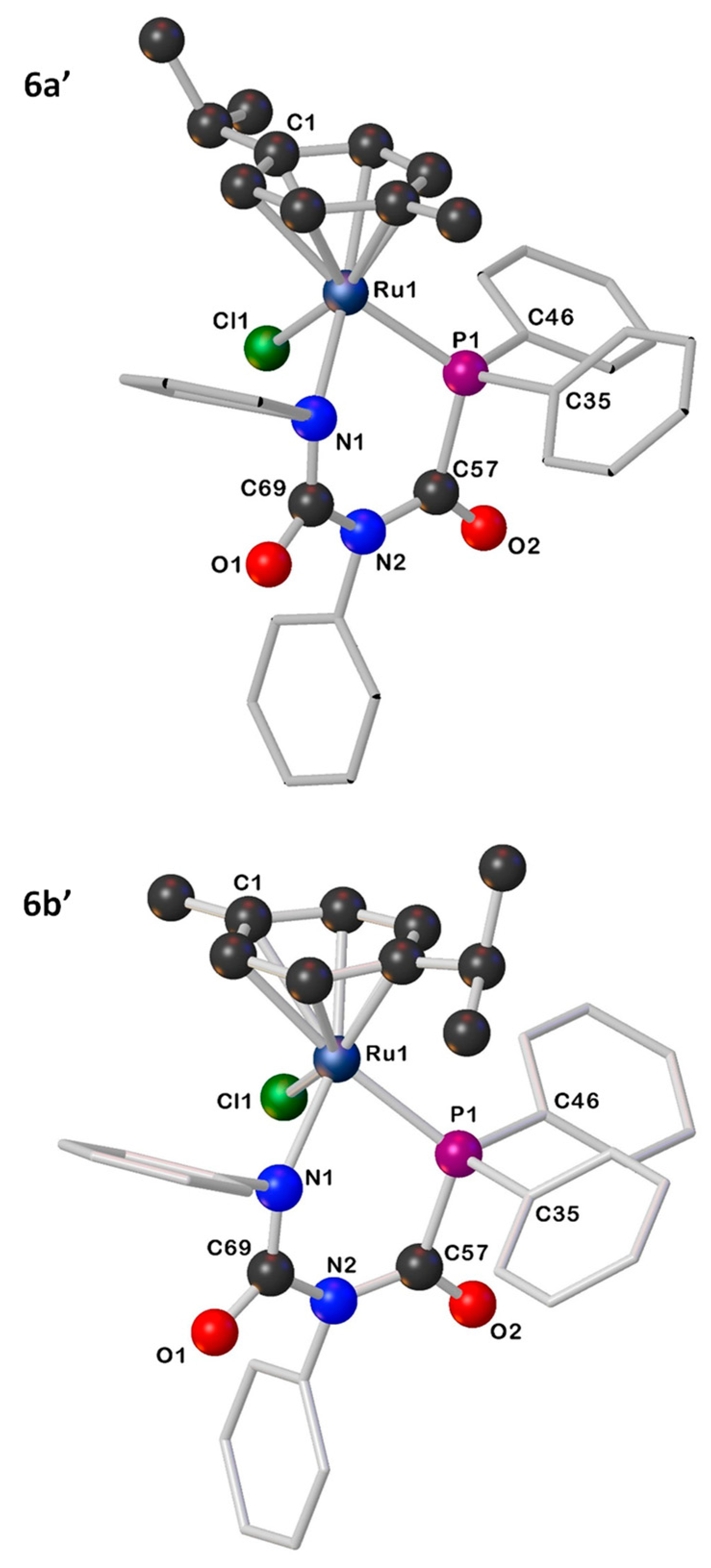
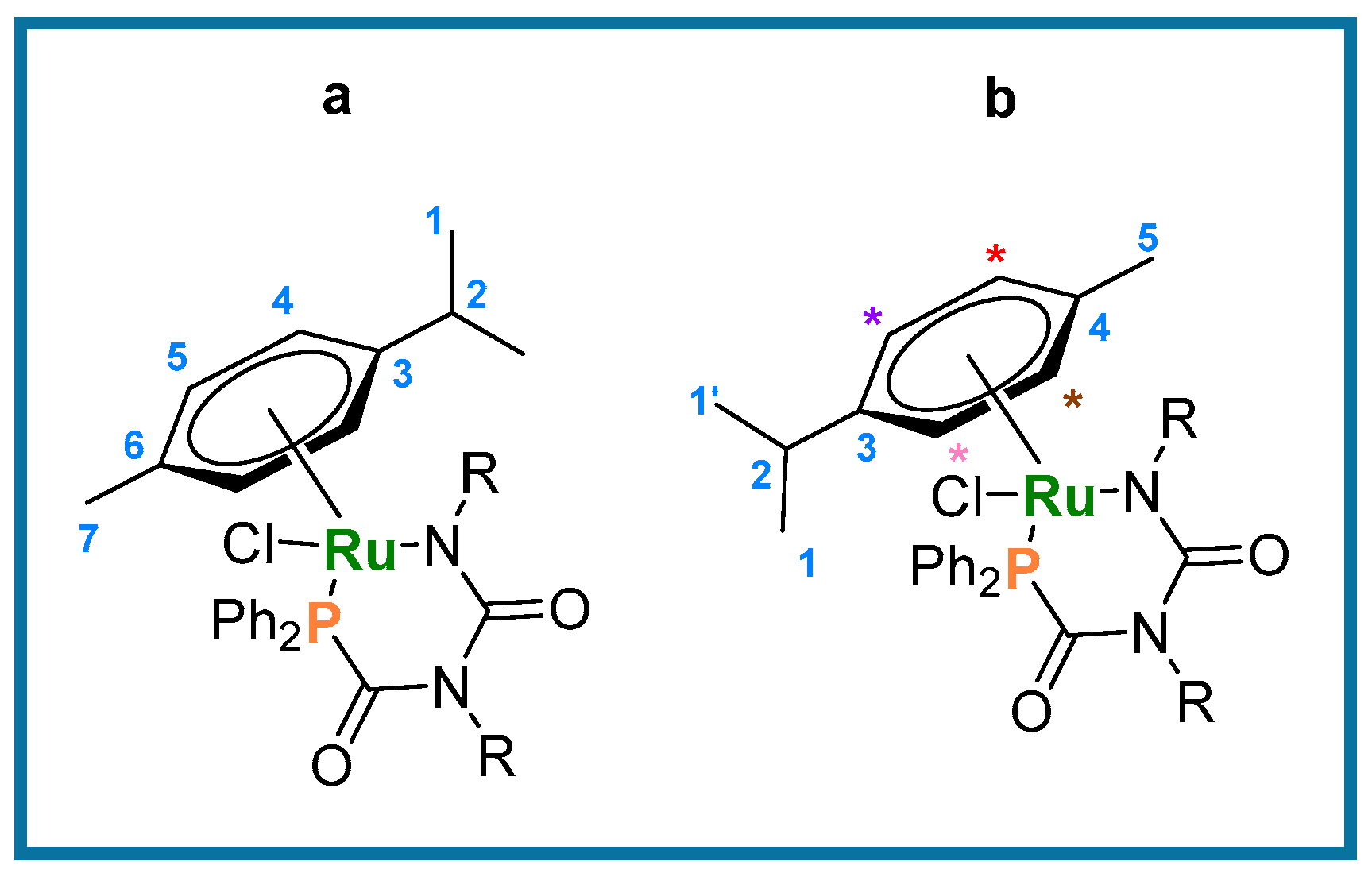
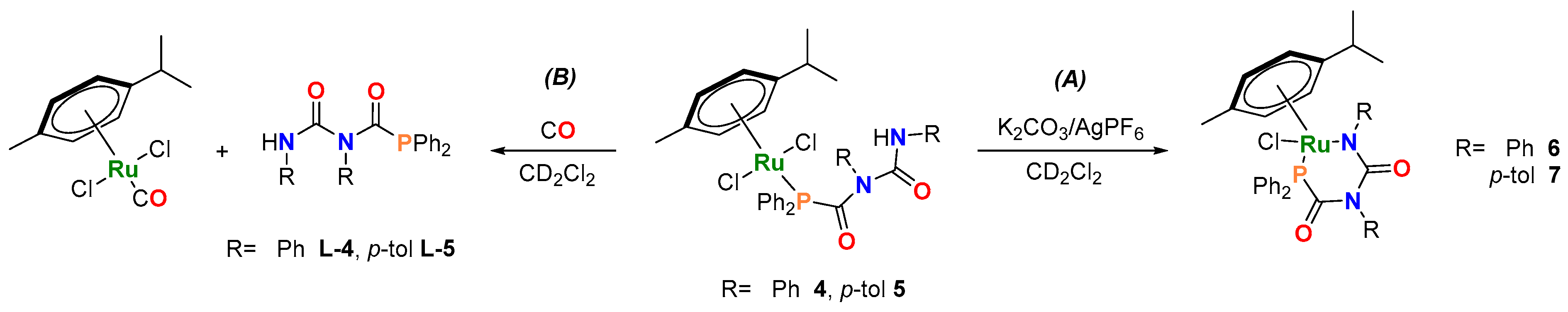
2.2. Synthesis and Characterization of [Ru(p-cym){κ2-P,N-PPh2C(=O)N(R)C(=O)NR}Cl] ([R = Ph (6), p-tol (7)]
2.3. Ligand Displacement Studies
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Štěpnička, P. Phosphino-Carboxamides: The Inconspicuous Gems. Chem. Soc. Rev. 2012, 41, 4273–4305. [Google Scholar] [CrossRef] [PubMed]
- Jupp, A.R.; Trott, G.; Payen De La Garanderie, É.; Holl, J.D.G.; Carmichael, D.; Goicoechea, J.M. Exploiting the Brønsted Acidity of Phosphinecarboxamides for the Synthesis of New Phosphides and Phosphines. Chem. Eur. J. 2015, 21, 8015–8018. [Google Scholar] [CrossRef] [PubMed]
- Jupp, A.R.; Goicoechea, J.M. Phosphinecarboxamide: A Phosphorus-Containing Analogue of Urea and Stable Primary Phosphine. J. Am. Chem. Soc. 2013, 135, 19131–19134. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Correia, J.D.G.; Gano, L.; Santos, I.; Seifert, S.; Syhre, R.; Bergmann, R.; Spies, H. Dramatic Effect of the Tridentate Ligand on the Stability of 99mTc “3 + 1” Oxo Complexes Bearing Arylpiperazine Derivatives. Bioconjug. Chem. 2005, 16, 660–668. [Google Scholar] [CrossRef]
- Meléndez-Alafort, L.; Nadali, A.; Pasut, G.; Zangoni, E.; De Caro, R.; Cariolato, L.; Giron, M.C.; Castagliuolo, I.; Veronese, F.M.; Mazzi, U. Detection of Sites of Infection in Mice Using 99mTc-Labeled PN2S-PEG Conjugated to UBI and 99mTc-UBI: A Comparative Biodistribution Study. Nucl. Med. Biol. 2009, 36, 57–64. [Google Scholar] [CrossRef]
- Léautey, M.; Jubault, P.; Pannecoucke, X.; Quirion, J.C. Synthesis and Evaluation of a Broad Range of New Chiral (Aminoalkyl)Phosphane Ligands for Asymmetric Hydrogen-Transfer Reduction of Prochiral Ketones. Eur. J. Org. Chem. 2003, 19, 3761–3768. [Google Scholar] [CrossRef]
- Sladojevich, F.; Trabocchi, A.; Guarna, A.; Dixon, D.J. A New Family of Cinchona-Derived Amino Phosphine Precatalysts: Application to the Highly Enantio- and Diastereoselective Silver-Catalyzed Isocyanoacetate Aldol Reaction. J. Am. Chem. Soc. 2011, 133, 1710–1713. [Google Scholar] [CrossRef]
- Seidenkranz, D.T.; McGrath, J.M.; Zakharov, L.N.; Pluth, M.D. Supramolecular Bidentate Phosphine Ligand Scaffolds from Deconstructed Hamilton Receptors. Chem. Commun. 2017, 53, 561–564. [Google Scholar] [CrossRef]
- Sharpe, H.R.; Geer, A.M.; Lewis, W.; Blake, A.J.; Kays, D.L. Iron(II)-Catalyzed Hydrophosphination of Isocyanates. Angew. Chem. Int. Ed. 2017, 56, 4845–4848. [Google Scholar] [CrossRef]
- Braunstein, P.; Naud, F. Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems. Angew. Chem. Int. Ed. 2001, 40, 680–699. [Google Scholar] [CrossRef]
- Geeson, M.B.; Jupp, A.R.; McGrady, J.E.; Goicoechea, J.M. On the Coordination Chemistry of Phosphinecarboxamide: Assessing Ligand Basicity. Chem. Commun. 2014, 50, 12281–12284. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, Z.; Wang, X.; Li, X.; Weng, L.; Zhou, X. Isocyanate Diinsertion into the N-H Bond of the 2-Pyridylamino Ligand of Organolanthanides. Dalton Trans. 2010, 39, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Iseki, M.; Itazaki, M.; Nakazawa, H. Tetrahedral Cage Complex with Planar Vertices: Selective Synthesis of Pt4L6 Cage Complexes Involving Hydrogen Bonds Driven by Halide Binding. Chem. Commun. 2016, 52, 7205–7208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ding, F.; Sun, Y.; Verpoort, F. O,N-Bidentate Ruthenium Azo Complexes as Catalysts for Olefin Isomerization Reactions. Eur. J. Inorg. Chem. 2010, 10, 1536–1543. [Google Scholar] [CrossRef]
- Rath, R.K.; Nethaji, M.; Chakravarty, A.R. Synthesis, Crystal Structure and Catalytic Properties of (p-cymene)Ruthenium(II) Azophenol Complexes: Azophenyl to Azophenol Conversion by Oxygen Insertion to a Ruthenium-Carbon Bond. J. Organomet. Chem. 2001, 633, 79–84. [Google Scholar] [CrossRef]
- Rath, R.K.; Nethaji, M.; Chakravarty, A.R. Transfer Hydrogenation of Acetophenone Promoted by (Arene)Ruthenium(II) Reduced Schiff Base Complexes: An X-Ray Structure of [(η6-p-cymene)RuCl(OC6H4-2-CH2NHC6H4-p-Me)]. Polyhedron 2001, 20, 2735–2739. [Google Scholar] [CrossRef]
- Bozec, H.; Le Touchard, D.; Dixneuf, P.H. Organometallic Chemistry of Arene Ruthenium and Osmium Complexes. Adv. Organomet. Chem. 1989, 29, 163–247. [Google Scholar]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Naota, T.; Takaya, H.; Murahashi, S.I. Ruthenium-Catalyzed Reactions for Organic Synthesis. Chem. Rev. 1998, 98, 2599–2660. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, C.; Dixneuf, P.H. Metal Vinylidenes in Catalysis. Acc. Chem. Res. 1999, 32, 311–323. [Google Scholar] [CrossRef]
- Cadierno, V.; Gamasa, M.P.; Gimeno, J. Recent Developments in the Reactivity of Allenylidene and Cumulenylidene Complexes. Eur. J. Inorg. Chem. 2001, 3, 571–591. [Google Scholar] [CrossRef]
- Bruce, M.I. Transition Metal Complexes Containing Allenylidene, Cumulenylidene, and Related Ligands. Chem. Rev. 1998, 98, 2797–2858. [Google Scholar] [CrossRef] [PubMed]
- Touchard, D.; Dixneuf, P.H. A New Class of Carbon-Rich Organometallics. The C3, C4 and C5 Metallacumulenes Ru=(C=)nCR2. Coord. Chem. Rev. 1998, 178, 409–429. [Google Scholar] [CrossRef]
- Berenguer, J.R.; Bernechea, M.; Forniés, J.; García, A.; Lalinde, E. (p-cymene) Ruthenium (II) (Diphenylphosphino) Alkyne Complexes: Preparation of (μ-Cl) (μ-PPh2C≡CR)-Bridged Ru/Pt Heterobimetallic Complexes. Organometallics 2004, 23, 4288–4300. [Google Scholar] [CrossRef]
- Çalık, H.S.; Ispir, E.; Karabuga, Ş.; Aslantas, M. Ruthenium (II) Complexes of NO Ligands: Synthesis, Characterization and Application in Transfer Hydrogenation of Carbonyl Compounds. J. Organomet. Chem. 2016, 801, 122–129. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van Der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Cabrero-Antonino, J.R.; Alberico, E.; Drexler, H.J.; Baumann, W.; Junge, K.; Junge, H.; Beller, M. Efficient Base-Free Hydrogenation of Amides to Alcohols and Amines Catalyzed by Well-Defined Pincer Imidazolyl-Ruthenium Complexes. ACS Catal. 2016, 6, 47–54. [Google Scholar] [CrossRef]
- Adam, R.; Alberico, E.; Baumann, W.; Drexler, H.J.; Jackstell, R.; Junge, H.; Beller, M. NNP-Type Pincer Imidazolylphosphine Ruthenium Complexes: Efficient Base-Free Hydrogenation of Aromatic and Aliphatic Nitriles under Mild Conditions. Chem. Eur. J. 2016, 22, 4991–5002. [Google Scholar] [CrossRef]
- Lee, C.C.; Huang, H.C.; Liu, S.T.; Liu, Y.H.; Peng, S.M.; Chen, J.T. Coordination Chemistry and Catalytic Activity of Ruthenium(II) Complexes Containing a Phospha-Macrocyclic Ligand. Polyhedron 2013, 52, 1024–1029. [Google Scholar] [CrossRef]
- García de la Arada, I.; Díez, J.; Gamasa, P.; Lastra, E. Isomerization Processes on Organoruthenium Complexes Bearing κ2(P,C)- Bidentate Ligands Generated Through Nucleophilic Addition To Coordinated Alkenyl Phosphanes. Eur. J. Inorg. Chem. 2018, 45, 4875–4886. [Google Scholar] [CrossRef]
- Hierso, J.C. Indirect Nonbonded Nuclear Spin-Spin Coupling: A Guide for the Recognition and Understanding of “through-Space” NMR J Constants in Small Organic, Organometallic, and Coordination Compounds. Chem. Rev. 2014, 114, 4838–4867. [Google Scholar] [CrossRef] [PubMed]
- Hodson, E.; Simpson, S.J. Synthesis and Characterisation of [(η6-cymene)Ru(L)X2] Compounds: Single Crystal X-ray Structure of [(η6- cymene)Ru(P{OPh}3)Cl2] at 203 K. Polyhedron 2004, 23, 2695–2707. [Google Scholar] [CrossRef]
- Dulebohn, J.I.; Berglund, K.A.; Haefner, S.C.; Dunbar, K.R. Reversible Carbon Monoxide Addition to Sol-Gel Derived Composite Films Containing a Cationic Rhodium(I) Complex: Towards the Development of a New Class of Molecule-Basedcarbon Monoxide Sensors. Chem. Mater. 1992, 4, 506–508. [Google Scholar] [CrossRef]
- Dunbar, K.R. New Applications of Weak Donor Atoms to Coordination, Organometallic and Materials Chemistry. Comments Inorg. Chem. 1992, 13, 313–357. [Google Scholar] [CrossRef]
- Cosier, J.; Glazer, A.M. A nitrogen-gas-stream cryostat for general X-ray diffraction studies. J. Appl. Crystallogr. 1986, 19, 105–107. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Blake, A.J.; Champness, N.R.; Schroder, M. OLEX: New software for visualization and analysis of extended crystal structures. J. Appl. Crystallogr. 2003, 36, 1283–1284. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Available online: http://checkcif.iucr.org (accessed on 15 September 2023).
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- te Velde, G.; Bickelhaupt, F.M.; van Gisbergen, S.J.A.; Fonseca Guerra, C.; Baerends, E.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 31P Free PCA/PDCA Ligand a | 31P PCA/PDCA Complex b | 13C{1H}C=O PCA/PDCA Complex b | 1HNH PCA/PDCA Complex b |
|---|---|---|---|---|
| 1 | −4.0 | 29.8 a | 167.4 a | 8.6 a |
| 2 | −0.2 | 37.2 | 167.6 | 10.1 |
| 3 | −0.9 | 36.6 | 167.5 | 10.0 |
| 4 | 8.3 | 33.5 | 178.2/177.7 c | 9.2 |
| 5 | 8.0 | 33.8 | 177.9/177.5 c | 9.2 |
| 6 | - | 52.6 | - d | - |
| 7 | - | 52.3 | 170.9/162.1c | - |
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| C1plane–Ru1 | 1.7010 (8) | 1.7007 (10) | 1.6989 (7) | 1.7010 (11) | 1.702 (2) |
| Ru1–Cl1 | 2.4183 (5) | 2.4142 (6) | 2.4171 (4) | 2.3941 (7) | 2.4302 (11) |
| Ru1–Cl2 | 2.4115 (5) | 2.4151 (6) | 2.4078 (4) | 2.4296 (9) | 2.3974 (10) |
| Ru1–P1 | 2.3476 (5) | 2.3448 (6) | 2.3517 (4) | 2.3678 (7) | 2.3635 (10) |
| H1⋯Cl1 | 2.54 (3) a | 2.3455 (6) | 2.28 (2) | 2.3723 (8) | 2.44 (5) |
| Cl1–Ru1–P1 | 86.96 (2) | 85.17 (2) | 90.44 (2) | 87.45 (3) | 86.49 (4) |
| Cl2–Ru1–P1 | 87.35 (2) | 86.87 (2) | 83.29 (2) | 89.33 (2) | 89.70 (4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolla-Saltiel, R.; Geer, A.M.; Sharpe, H.R.; Huke, C.D.; Taylor, L.J.; Linford-Wood, T.G.; James, A.; Allen, J.; Lewis, W.; Blake, A.J.; et al. Organoruthenium Complexes Containing Phosphinodicarboxamide Ligands. Inorganics 2023, 11, 372. https://doi.org/10.3390/inorganics11090372
Nolla-Saltiel R, Geer AM, Sharpe HR, Huke CD, Taylor LJ, Linford-Wood TG, James A, Allen J, Lewis W, Blake AJ, et al. Organoruthenium Complexes Containing Phosphinodicarboxamide Ligands. Inorganics. 2023; 11(9):372. https://doi.org/10.3390/inorganics11090372
Chicago/Turabian StyleNolla-Saltiel, Roberto, Ana M. Geer, Helen R. Sharpe, Cameron D. Huke, Laurence J. Taylor, Thomas G. Linford-Wood, Ashleigh James, Jamie Allen, William Lewis, Alexander J. Blake, and et al. 2023. "Organoruthenium Complexes Containing Phosphinodicarboxamide Ligands" Inorganics 11, no. 9: 372. https://doi.org/10.3390/inorganics11090372
APA StyleNolla-Saltiel, R., Geer, A. M., Sharpe, H. R., Huke, C. D., Taylor, L. J., Linford-Wood, T. G., James, A., Allen, J., Lewis, W., Blake, A. J., McMaster, J., & Kays, D. L. (2023). Organoruthenium Complexes Containing Phosphinodicarboxamide Ligands. Inorganics, 11(9), 372. https://doi.org/10.3390/inorganics11090372