
High-Capacity Ion Batteries Based on Ti2C MXene and Borophene First Principles Calculations



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
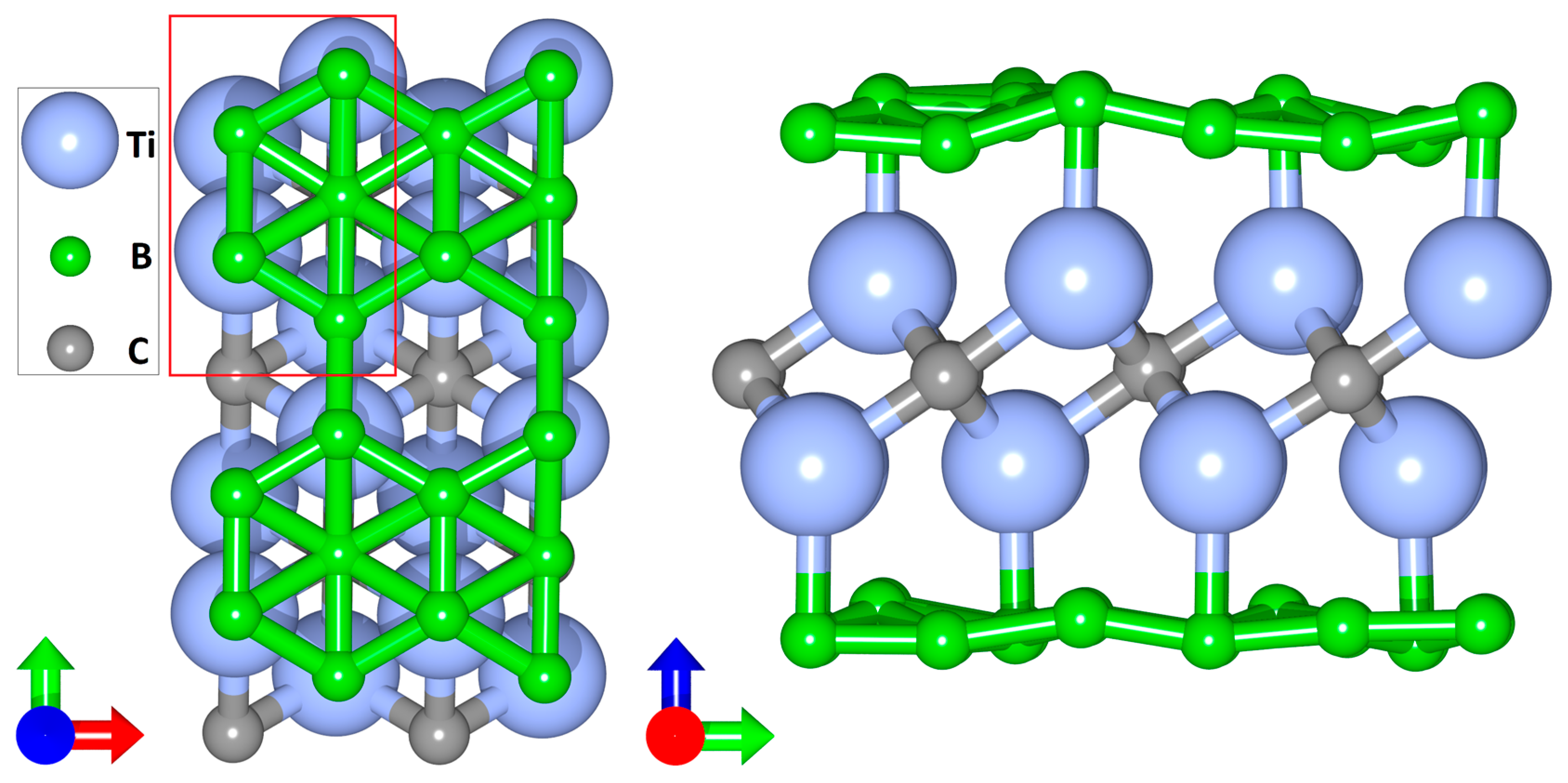
2.1. Atomistic Models
2.2. Electronic and Transport Properties
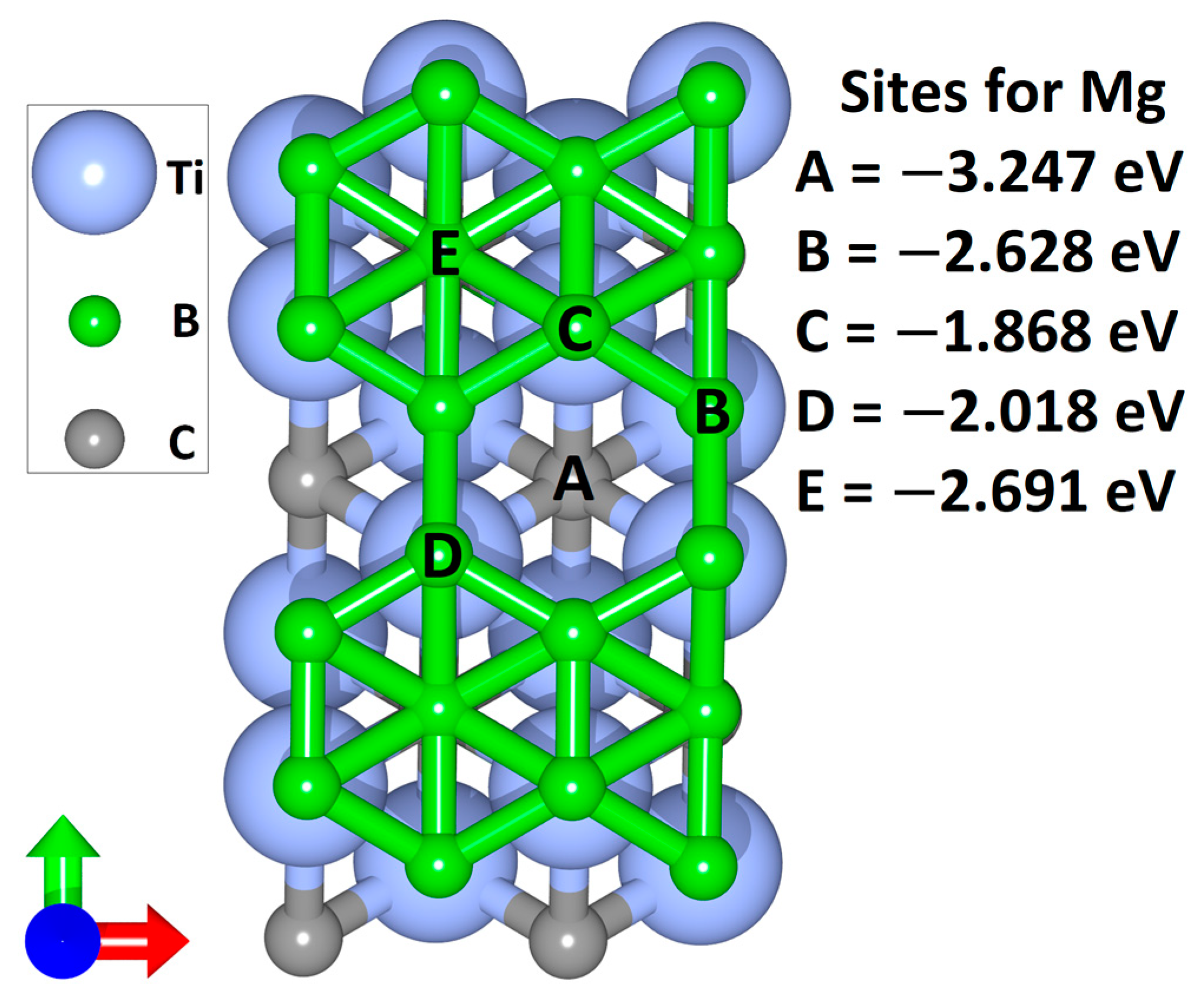
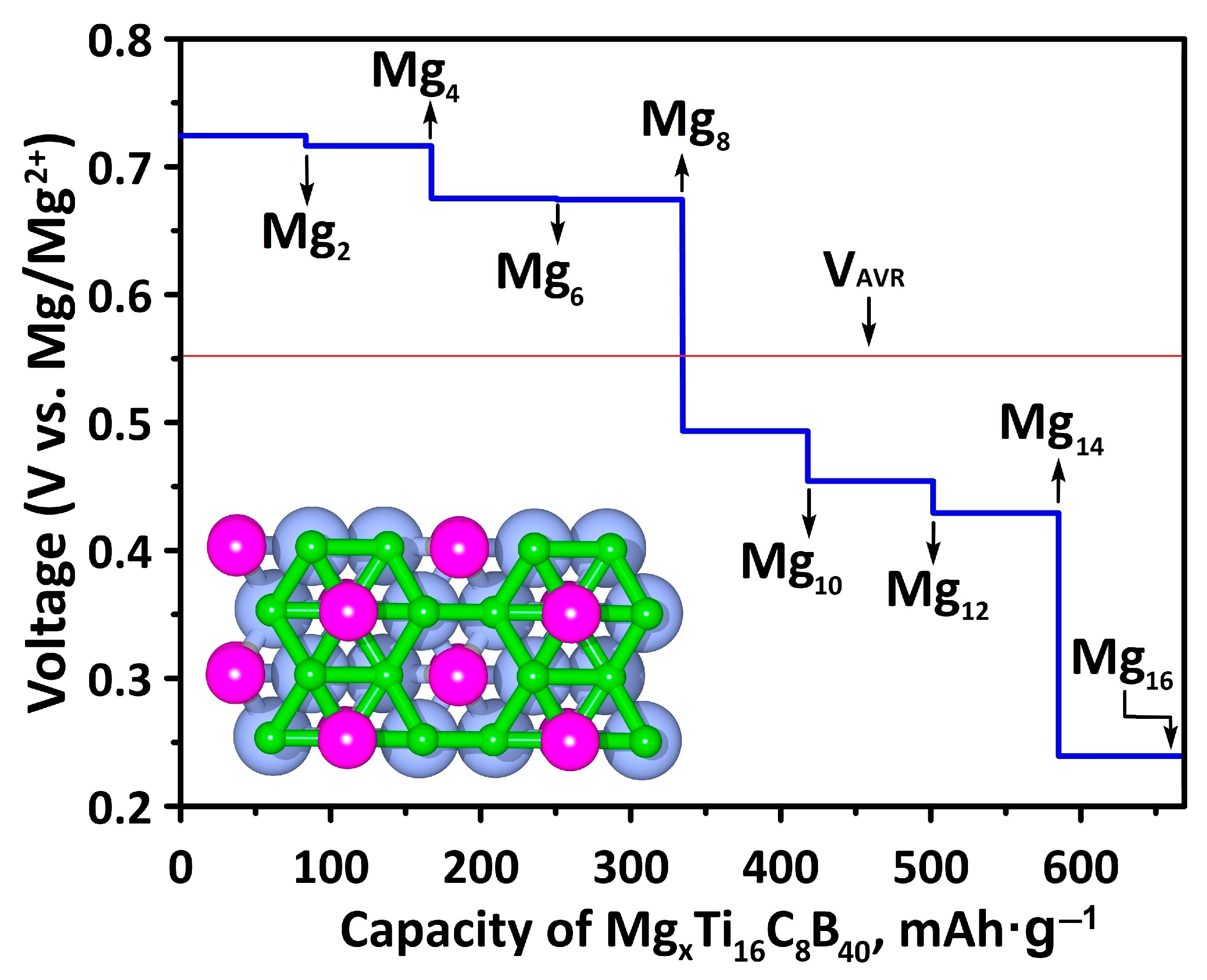
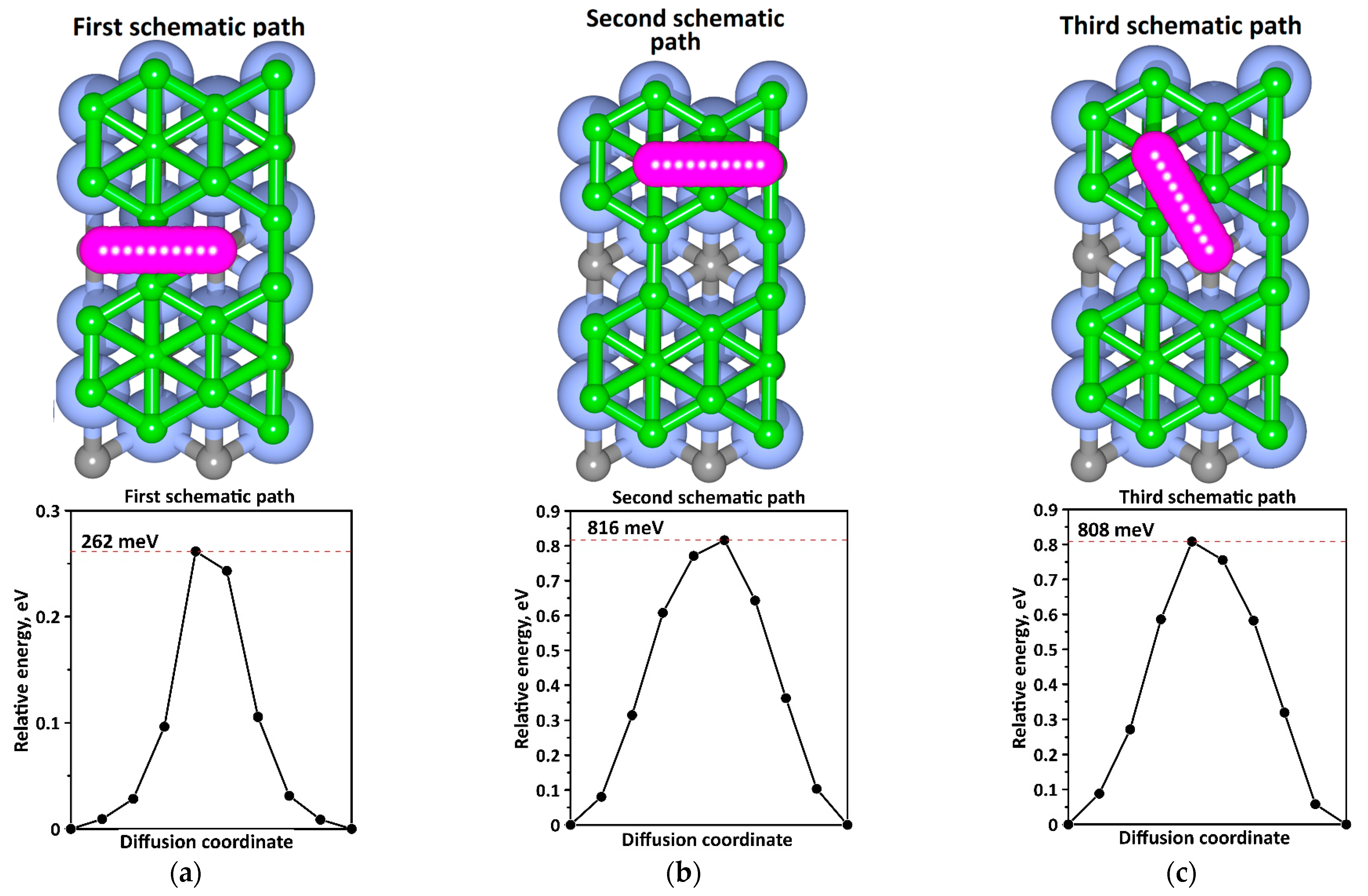
2.3. Adsorption, OCV and Diffusion Barrier
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.-T.; Hao, Y.; Xu, L.-C.; Yang, Z.; Di, M.-Y.; Liu, R.-P.; Li, X.-Y. Insight into the Discharge Products and Mechanism of Room-Temperature Sodium–Sulfur Batteries: A First-Principles Study. J. Phys. Chem. C 2019, 123, 3988–3995. [Google Scholar] [CrossRef]
- Xie, Y.; Dall’Agnese, Y.; Naguib, M.; Gogotsi, Y.; Barsoum, M.W.; Zhuang, H.L.; Kent, P.R. Prediction and characterization of mxene nanosheet anodes for nonlithium-ion batteries. ACS Nano 2014, 8, 9606–9615. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hu, J. Facile synthesis of multi-walled carbon nanotubes/Co9s8 composites with enhanced performances for sodium-ion battery. Mater. Lett. 2017, 195, 26–30. [Google Scholar] [CrossRef]
- Aurbach, D.; Lu, Z.; Schechter, A.; Gofer, Y.; Gizbar, H.; Turgeman, R.; Cohen, Y.; Moshkovich, M.; Levi, E. Prototype systems for rechargeable magnesium batteries. Nature 2000, 407, 724–727. [Google Scholar] [CrossRef]
- Johnson, I.D.; Ingram, B.J.; Cabana, J. The Quest for Functional Oxide Cathodes for Magnesium Batteries: A Critical Perspective. ACS Energy Lett. 2021, 6, 1892–1900. [Google Scholar] [CrossRef]
- Ruiz, R.; Pérez-Vicente, C.; Rubio, S.; Stoyanova, R.; Zuo, W.; Yang, Y.; Ortiz, G.F. A Cubic Mg2MnO4 Cathode for non-aqueous Magnesium Batteries. Energy Storage Mater. 2022, 48, 12–19. [Google Scholar] [CrossRef]
- Guo, W.; Hanaor, D.A.H.; Kober, D.; Wang, J.; Bekheet, M.F.; Gurlo, A. Rechargeable Magnesium Ion Batteries Based on Nanostructured Tungsten Disulfide Cathodes. Batteries 2022, 8, 116. [Google Scholar] [CrossRef]
- Xue, X.; Song, X.; Tao, A.; Yan, W.; Zhang, X.L.; Tie, Z.; Jin, Z. Boosting the cycling stability of rechargeable magnesium batteries by regulating the compatibility between nanostructural metal sulfide cathodes and non-nucleophilic electrolytes. Nano Res. 2022, 16, 2399–2408. [Google Scholar] [CrossRef]
- Xiao, Z.; Li, Z.; Li, P.; Meng, X.; Wang, R. Ultrafine Ti3C2 mxene nanodots-interspersed nanosheet for high-energy-density lithium-sulfur batteries. ACS Nano 2019, 3, 3608–3617. [Google Scholar] [CrossRef]
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional Nanocrystals Produced by Exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248–4253. [Google Scholar] [CrossRef]
- Naguib, M.; Mashtalir, O.; Carle, J.; Presser, V.; Lu, J.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional Transition Metal Carbides. ACS Nano 2012, 6, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Ji, H.; Kim, J.; Kim, H.; Jung, Y. Exploring the possibilities of two-dimensional transition metal carbides as anode materials for sodium batteries. Phys. Chem. Chem. Phys. 2015, 17, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kang, W.; Xue, J. Role of Strain and Concentration on the Li Adsorption and Diffusion Properties on Ti2C Layer. J. Phys. Chem. C 2014, 118, 14983–14990. [Google Scholar] [CrossRef]
- Tang, X.; Guo, X.; Wu, W.; Wang, G. 2D Metal Carbides and Nitrides (MXenes) as High-Performance Electrode Materials for Lithium-Based Batteries. Adv. Energy Mater. 2018, 8, 1801897. [Google Scholar] [CrossRef]
- Simonenko, N.P.; Glukhova, O.E.; Plugin, I.A.; Kolosov, D.A.; Nagornov, I.A.; Simonenko, T.L.; Varezhnikov, A.S.; Simonenko, E.P.; Sysoev, V.V.; Kuznetsov, N.T. The Ti0.2V1.8C MXene Ink-Prepared Chemiresistor: From Theory to Tests with Humidity versus VOCs. Chemosensors 2023, 11, 7. [Google Scholar] [CrossRef]
- Yao, C.; Li, W.; Duan, K.; Zhu, C.; Li, J.; Ren, Q.; Bai, G. Properties of S-Functionalized Nitrogen-Based MXene (Ti2NS2) as a Hosting Material for Lithium-Sulfur Batteries. Nanomaterials 2021, 11, 2478. [Google Scholar] [CrossRef]
- Pazniak, H.; Varezhnikov, A.S.; Kolosov, D.A.; Plugin, I.A.; Di Vito, A.; Glukhova, O.E.; Sheverdyaeva, P.M.; Spasova, M.; Kaikov, I.; Kolesnikov, E.A.; et al. Molybdenum Carbide MXenes for Enhanced Selective Detection of Humidity in Air. Adv. Mater. 2021, 33, 2104878. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, M.; Xu, L.-C.; Zhao, W.; Li, R.; Yang, Z.; Liu, R.; Li, X. Achieving superior high-capacity batteries with the lightest Ti2C MXene anode by first-principles calculations: Overarching role of S-functionate (Ti 2CS2) and multivalent cations carrier. J. Power Sources 2020, 451, 227791. [Google Scholar] [CrossRef]
- Hu, J.; Xu, B.; Ouyang, C.; Yang, S.A.; Yao, Y. Investigations on V2C and V2CX2 (X = F, OH) monolayer as a promising anode material for li ion batteries from firstprinciples calculations. J. Phys. Chem. C 2014, 118, 24274–24281. [Google Scholar] [CrossRef]
- Wang, Y.T.; Shen, J.L.; Xu, L.-C.; Yang, Z.; Li, R.; Liu, R.P.; Li, X.Y. Sulfurfunctionalized vanadium carbide mxene (V2CS2) as a promising anchoring material for lithium-sulfur batteries. Phys. Chem. Chem. Phys. 2019, 21, 18559–18568. [Google Scholar] [CrossRef]
- Berdiyorov, G.R.; Madjet, M.E.; Mahmoud, K.A. First-Principles Density Functional Theory Calculations of Bilayer Membranes Heterostructures of Ti3C2T2 (MXene)/Graphene and AgNPs. Membranes 2021, 11, 543. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.R.; Lu, Z.; Wu, M.C.; Ciucci, F.; Zhao, T.S. Borophene: A promising anode material offering high specific capacity and high rate capability for lithium-ion batteries. Nano Energy 2016, 23, 97–104. [Google Scholar] [CrossRef]
- Mortazavi, B.; Rahaman, O.; Ahzi, S.; Rabczuk, T. Flat borophene films as anode materials for Mg, Na or Li-ion batteries with ultra high capacities: A first-principles study. Appl. Mater. Today 2017, 8, 60–67. [Google Scholar] [CrossRef]
- Kaneti, Y.V.; Benu, D.P.; Xu, X.; Yuliarto, B.; Yamauchi, Y.; Golberg, D. Borophene: Two-dimensional Boron Monolayer: Synthesis, Properties, and Potential Applications. Chem. Rev. 2022, 122, 1000–1051. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, M.; Yang, M.; Yang, Q.; Zhang, Z.; Zhang, Y. High-Performance Borophene/Graphene Heterostructure Anode of Lithium-Ion Batteries Achieved via Controlled Interlayer Spacing. ACS Appl. Energy Mater. 2020, 3, 11699–11705. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, M.; Yang, M.; Zhang, Y.; Xu, B.; Li, X.; Tao, H. Pristine and Defective 2D Borophene/Graphene Heterostructure as the Potential Anode of Lithium-Ion Batteries. Adv. Mater. Interfaces 2022, 9, 2102088. [Google Scholar] [CrossRef]
- Zhou, D.; Wang, Z.; Cheng, J.; Pu, C. Metallic B2C3P Monolayer as Li-Ion Battery Materials: A First-Principles Study. Processes 2022, 10, 1809. [Google Scholar] [CrossRef]
- Cui, W.; Hu, Z.-Y.; Unocic, R.R.; Van Tendeloo, G.; Sang, X. Atomic defects, functional groups and properties in MXenes. Chin. Chem. Lett. 2021, 32, 339–344. [Google Scholar] [CrossRef]
- Kolosov, D.A.; Levitsky, S.G.; Glukhova, O.E. Adhesion and Electron Properties of Quasi-2D Mo2C, Ti2C, and V2C MXene Flakes after Van Der Waals Adsorption of Alcohol Molecules: Influence of Humidity. Lubricants 2022, 10, 159. [Google Scholar] [CrossRef]
- Dihrab., S.S.; Sopian, K.; Zaharim, A. Membrane and bipolar plates materials for regenerative fuel cells. In Proceedings of the 8th WSEAS International Conference on Simulation, Modeling and Optimization (SMO’08), Santander, Spain, 23–25 September 2008; pp. 183–188. [Google Scholar]
- Tantardini, C.; Oganov, A.R. Thermochemical electronegativities of the elements. Nat. Commun. 2021, 12, 2087. [Google Scholar] [CrossRef]
- Wu, P.; Li, P.; Huang, M. Potential Application of Graphene/Antimonene Herterostructure as an Anode for Li-Ion Batteries: A First-Principles Study. Nanomaterials 2019, 9, 1430. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Rao, D.; Zhang, L.; Meng, Z.; Zhang, X.; Wang, Y.; Qiao, G.; Shen, X.; Xia, H.; Liu, J.; Lu, R. Ultrahigh energy storage and ultrafast ion diffusion in borophene-based anodes for rechargeable metal ion batteries. J. Mater. Chem. A 2017, 5, 2328–2338. [Google Scholar] [CrossRef]
- Guo, G.; Wang, D.; Wei, X.; Zhang, Q.; Liu, H.; Lau, W.; Liu, L. First-Principles Study of Phosphorene and Graphene Hetero-structure as Anode Materials for Rechargeable Li Batteries. J. Phys. Chem. Lett. 2015, 6, 5002–5008. [Google Scholar] [CrossRef] [PubMed]
- Thinius, S.; Islam, M.M.; Heitjans, P.; Bredow, T. Theoretical Study of Li Migration in Lithium–Graphite Intercalation Com-pounds with Dispersion-Corrected DFT Methods. J. Phys. Chem. C 2014, 118, 2273–2280. [Google Scholar] [CrossRef]
- Zhang, H.; Xia, Y.; Bu, H.; Wang, X.; Zhang, M.; Luo, Y.; Zhao, M. Graphdiyne: A promising anode material for lithium ion batteries with high capacity and rate capability. J. Appl. Phys. 2013, 113, 044309. [Google Scholar] [CrossRef]
- Zhao, B.; Ran, R.; Liu, M.; Shao, Z. A comprehensive review of Li4Ti5O12-based electrodes for lithium-ion batteries: The latest advancements and future perspectives. Mater. Sci. Eng. R Rep. 2015, 98, 1–71. [Google Scholar] [CrossRef]
- Han, X.; Liu, C.; Sun, J.; Sendek, A.D.; Yang, W. Density functional theory calculations for evaluation of phosphorene as a potential anode material for magnesium batteries. RSC Adv. 2018, 8, 7196–7204. [Google Scholar] [CrossRef]
- Ordejón, P.; Artacho, E.; Soler, J.M. Self-consistent order-N density-functional calculations for very large systems. Phys. Rev. B 1996, 53, R10441. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Johnson, D.D. Modified Broyden’s method for accelerating convergence in self-consistent calculations. Phys. Rev. B 1988, 38, 12807. [Google Scholar] [CrossRef]
- Brandbyge, M.; Mozos, J.-L.; Ordejon, P.; Taylor, J.; Stokbro, K. Density-functional method for nonequilibrium electron transport. Phys. Rev. B 2002, 65, 165401. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Ferrante, F.; Prestianni, A.; Bertini, M.; Duca, D. H2 Transformations on Graphene Supported Palladium Cluster: DFT-MD Simulations and NEB Calculations. Catalysts 2020, 10, 1306. [Google Scholar] [CrossRef]
- Mortazavi, B.; Dianat, A.; Rahaman, O.; Cuniberti, G.; Rabczuk, T. Borophene as an anode material for Ca, Mg, Na or Li ion storage: A first-principle study. J. Power Sources 2016, 329, 456–461. [Google Scholar] [CrossRef]
- Kavalsky, L.; Mukherjee, S.; Singh, C.V. Phosphorene as a Catalyst for Highly Efficient Nonaqueous Li–Air Batteries. ACS Appl. Mater. Interfaces 2019, 11, 499–510. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolosov, D.A.; Glukhova, O.E. High-Capacity Ion Batteries Based on Ti2C MXene and Borophene First Principles Calculations. Inorganics 2023, 11, 95. https://doi.org/10.3390/inorganics11030095
Kolosov DA, Glukhova OE. High-Capacity Ion Batteries Based on Ti2C MXene and Borophene First Principles Calculations. Inorganics. 2023; 11(3):95. https://doi.org/10.3390/inorganics11030095
Chicago/Turabian StyleKolosov, Dmitry A., and Olga E. Glukhova. 2023. "High-Capacity Ion Batteries Based on Ti2C MXene and Borophene First Principles Calculations" Inorganics 11, no. 3: 95. https://doi.org/10.3390/inorganics11030095
APA StyleKolosov, D. A., & Glukhova, O. E. (2023). High-Capacity Ion Batteries Based on Ti2C MXene and Borophene First Principles Calculations. Inorganics, 11(3), 95. https://doi.org/10.3390/inorganics11030095