Abstract
The release of CO2 into the atmosphere has become a primary issue nowadays. Recently, researchers found Metal-Organic Frameworks M-CPO-27 (M = Mg, Co, Ni, and Zn) to be revolutionary for CO2 adsorption due to the presence of open metal sites enhancing CO2 binding and leading to higher capacity. This study aims to select the best metal center for CPO-27 with the high performance of CO2 adsorption by screening metal centers using simulation as a preliminary selection method. Then, the different metal centers were synthesized using the solvothermal process for validation. The synthesis of MOFs is confirmed through PXRD and FTIR analysis. Subsequently, by using simulation and experimental methods, it is discovered that Ni-CPO-27 gives the best performance compared with magnesium, zinc, and cobalt metal centers. The CO2 adsorption capacity of synthesized Ni-CPO-27 is 5.6 mmol/g, which is almost 20% higher than other MOFs. In conclusion, the prospective outcome of changing the metal from Mg-CPO-27 to Ni-CPO-27 would be advantageous in this investigation owing to its excellent performance in capturing CO2.
1. Introduction
One of the pressing challenges that we face on our planet is carbon dioxide. The uncontrolled release of carbon dioxide (CO2) into the atmosphere leads to a rise in global temperatures and climate change [1,2]. The United Nations Intergovernmental Panel on Climate Change (IPCC) claims that an approximately 50% reduction in present-day CO2 emission is needed to avoid a worldwide average temperature upward thrust of 2–2.5 °C by the year 2050 [3]. In an industrial process, the separation of CO2 from gas mixtures plays an important role, such as the separation of CO2 from coal-gas flues and CO2/CH4 in natural gas, hence reducing the effects of carbon dioxide emission [4,5,6]. Therefore, CO2 capture by selective adsorption is considered one of the most promising methods due to its ease of implementation, lack of potentially harmful ingredients, and reduced energy cost demand [7].
Adsorbents of carbon dioxide must meet certain requirements in order to be used in carbon capture and storage technology. These requirements include the ability to selectively adsorb CO2 over nitrogen and water at low carbon dioxide partial pressure, as well as being resilient in the presence of water and other contaminants [8,9]. The most-used adsorbents are alumina, activated carbon, and zeolites, as they have been reported to satisfy the operation of adsorption. However, these adsorbents result in low carbon dioxide capacities (5.0 wt% at 298 K and 0.15 bar) with difficulties in the regeneration process [10]. A new invention of adsorbents has been found by researchers, which provides a higher adsorption capacity of CO2 with a higher surface area of solid sorbents compared with conventional adsorbents [7,11]. Currently, multiple promising adsorbents, such as Zeolite 5A, MIL-101 (Cr), Mg-Gallate, and Metal-Organic Frameworks, offer higher CO2 adsorption capacities [12,13]. The addition of certain binding sites at the pore surface, such as amines acting as Lewis bases and coordinatively unsaturated metal cations actinig as Lewis acids, can improve high absorption under ambient conditions in porous materials [7]. Therefore, metal-organic frameworks were found to be novel adsorbents due to their ability to obtain a high surface area with good capacity in capturing CO2, large pore volumes, and easily tuneable position [14,15,16]. A type of crystalline material known as Metal-Organic Frameworks (MOFs) is made up of coordination bonds between ions or clusters that are joined by organic ligands [17]. Metal-Organic Frameworks (MOFs) offer CO2 storage due to their ordered structures, high thermal stability, and adjustable chemical functionality [18]. The complexity of the structure needs the continuous interplay of experimental and modeling computational tools for advanced characterization during all stages of structures [19]. Over 5000 MOFs shave been captured in the literature for carbon dioxide adsorption properties. However, only several MOFs give better results, such as Mg/DOBDC (dobdc = 2,5-dioxido-1,4-benzenedicarboxylate) or (CPO-27), which adsorb twice as much carbon dioxide compared with zeolites at 0.1 bar [20,21].
Tremendous studies have been performed for MOFs in various applications in order to replace them with other adsorbents due to their performance in capturing and simulation [3,8,22]. One of the most anticipated study series of MOFs is known as M-CPO-27 (2,5-DOT = 2,5-dioididoterephthalate linker where M = Mg (II), Zn(II), Co(II) and Ni(II) of transition metals in periodic series), also named M-MOF-74 and M2(dobdc) [4,8]. The presence of a metal site in CPO-27 favors CO2 binding mode with a pore diameter of 1.1 Å to 1.2Å (7,14). Among other MOF materials, CPO-27 was reported to have the highest uptake capacity of up to 27.5 wt% for conditions under 298 K and 1 bar [8]. CPO-27 is known as an isostructural structure MOF because it has different metal centers with the same organic ligands. Magnesium metal is reported to be the best metal center due to its lighter molecular weight, which is 23.405 g/mol compared with zinc (65.38 g/mol) and nickel (58.69 g/mol) [23]. Ismail et al. [13] also worked on MOFs using gallic acid as an alternative linker for enhancing the CO2 capacity. The authors found that Mg-gallate has a maximum predicted adsorption capacity of 7.79 mmol/g. This suggests that the most significant interaction takes place between CO2 molecules and the exposed metal Mg2+ sites. However, there is no solid reason that the molecular weight of the metal will affect the adsorption capacity of CO2. In addition, the capacity of CO2 adsorption for Mg/DOBDC was found to decrease and lose stability with the presence of water compared to Co/DOBDC and Ni/DOBDC, which retained 60% and 85% of the initial capacities, respectively [3]. Most research focuses on the magnesium metal center due to its promising result in capturing CO2, but the performance deteriorates severely upon long-term exposure to relevant impurities such as water and oxygen in CO2. Thus, the promising result for CO2 adsorption of other metal centers will be an advantage in this study. However, synthesizing all M-CPO-27 (M = Mg, Co, Zn, and Ni) without promising performance will be costly and time-consuming [1].
The interactions between metal-organic frameworks (MOFs) and adsorbates have been increasingly predicted, and studies employ computer simulations, particularly Grand Canonical Monte Carlo (GCMC), as this method enables comparing the results with experimental results and provides a degree of molecular-level detail that is difficult to obtain in experiments [1]. The prediction of performance for M-CPO-27 (M = Mg, Ni, Co, and Zn) before synthesizing by using molecular simulation is able to reduce time the and cost of the selection process [1,24,25]. Between adsorption processes, there are various types of simulation methods proposed, and the most widely used is the Monte Carlo and Molecular Dynamic method [26,27]. In 1953, Metropolis was the first to use this computer simulation, which was carried out in Los Alamos National configuration generation [26]. Crystalline, ordered, and amorphous porous materials such as zeolites, silica, alumina, and metal-organic frameworks (MOFs) were commonly used in this technique as they have larger surface areas with a pore network that enhances sorption and separation activity [26]. Developing new processes based on M-CPO-27 requires the crucial step of screening and comprehending the underlying structure and functional relationships. The GCMC model utilized in this process considers various interactions, including electrostatic and Lenard–Jones, among the atoms present in the system [28]. As the GCMC model is able to process interactions between the atoms, this molecular modelling is also able to verify the structural properties of the materials such as Lewis’ interaction between the metal and carbon dioxide. Therefore, it is important to screen and identify the material’s behavior before the selection process can be performed.
As discussed, although recently, Mg-CPO-27 has been able to capture higher CO2, there are drawbacks that are faced when using magnesium as a metal center due to its inability to withstand stability in the presence of water [3,29]. Therefore, the findings of other metal centers that provide higher CO2 than previously reported will be an advantage. This study is proposed to screen open metal site MOFs, namely, M-CPO-27 (M = Co, Ni, Mg, and Zn), as the preliminary selection method and perform experimental synthesis for validation. This is due to the presence of open metal sites in CPO-27 in favor of CO2 binding in the adsorption that was investigated. This research will be focused on the grand canonical ensemble because of its ability to allow fluctuations in the number of molecules and becomes the most suitable ensemble technique for adsorption simulation [27]. This validation study will not only provide information for different metal centers in M-CPO-27 but also provide an alternative to Mg-CPO-27 adsorbents in capturing CO2.
2. Materials and Methods
2.1. Screening of M-CPO-27
This study used the Material Studio tools license from Biovia Dassault Systemes with a sorption module to perform the adsorption of CO2 in CPO-27. The structure of M-CPO-27 (M = Mg, Co, Zn, and Mg) was obtained from the Cambridge Structural Database (CSD). The impurities in the structures were cleaned in order to obtain the hexagonal structure of the R3 space group [30]. CO2 is drawn to be set as the adsorbate for CPO-27 with an angle of 180° and a C=O double bond length of 1.16 Å. Figure 1a,b show the structure of Ni-CPO-27 and CO2, respectively, before performing adsorption analysis. In the CPO-27 structure, simulation boxes were constructed with periodic boundary conditions in order to obtain an optimum adsorption system [31].
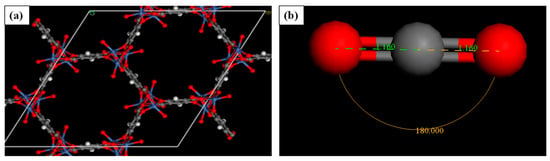
Figure 1.
{(a): Molecular structure of Ni-CPO-27; (b) CO2 Structure} (Blue: Nickel atoms, Red: Oxygen atoms, White: Hydrogen atoms, and Grey: Carbon atoms).
Forcite is used to identify van der Waals attraction between the adsorbent and adsorbate. It is operated depending on the forcefield applied. It is crucial to establish a forcefield for this adsorption that is appropriate for open metal sites. The universal forcefield is renowned for its rough and aggressive technique [32]. This forcefield is error-resistant due to the many parameters for the universal forcefield (UFF) that are accessible and hard programmed. For each CPO-74 metal selection, the COMPASS II forcefield is chosen in accordance with the instructions, lowering the inaccuracy achieved [4].
The Grand Canonical Monte Carlo (GCMC) calculation method was determined in the Material Studio by the sorption module. GCMC is applied to calculate the adsorption isotherm for a homogenous solid surface and metal-organic frameworks (MOFs) [31]. GCMC calculation works in the translation, insertion, rotation, and deletion of CO2 in accordance with adsorption into CPO-27 [31]. Monte Carlo moved randomly on the new configuration in which the lower energy was accepted or otherwise rejected [31]. The adsorption Isotherm task is selected for the adsorption of carbon dioxide. CPO-27 was reported to have a decent performance at a pressure of 1 bar and an ambient temperature [8]. Thus, this simulation was set for sorbate components (CO2 and M-CPO-27) at a constant temperature (298 K) and elevated pressure (0 to 1 bar).
2.2. Synthesis and Characterization of M-CPO-27
The M-CPO-27 material (where M = Mg, Ni, Co, and Co) utilized in this study was synthesized through a solvothermal reaction of magnesium (II) nitrate (+99%, Sigma-Aldrich, Darmstadt, Germany) with DOT (2,5-dihydroxytherephthalic acid) in a mixture of N,N-dimethylformamide (DMF), absolute ethanol, and deionized water. The synthesis method employed was consistent with a previously published procedure [21]. The same step was applied for the different metal centers by replacing magnesium (II) nitrate with nickel, zinc, and cobalt (II) nitrate. Mg(NO3)2·6H2O and DOT weights of 0.712 g and 0.167 g, respectively, were dissolved under sonication in a ratio of 15:1:1 (v/v/v) for the mixture of DMF, ethanol, and deionized water (67.5 mL, 4.5 mL, and 4.5 mL, respectively). The homogeneous solution was placed into a Teflon-lined stainless-steel autoclave and carefully sealed before being warmed in an oven to under 125 °C. The samples were taken out of the oven and allowed to cool at ambient temperature after being under autogenous pressure for the reaction for 26 h. Methanol was used to replace the mother liquor after it was thoroughly decanted. DMF was replaced with methanol and rinsed six times over the course of three days. Filtration was used to separate the yellowish microcrystalline precipitate, which was then extensively eroded with methanol. Dark-yellow crystal was formed for Co-CPO-27, whereas a red crystal was obtained by removing the guest molecules under a dynamic vacuum at 150 °C for 15 h.
The sample was then characterized by powder XRD using K alpha radiation of a Bench Top X-Ray Diffractometer to confirm its structure and porosity. To obtain high-resolution patterns, step scanning with an increment of 0.02° in 2 and a scan rate of 0.2°/min were used [19]. N2 adsorption and desorption isotherms were used to determine the pore characteristics for the Langmuir region and Brunauer–Emmet–Teller (BET) assessed by Belsorp Mini, Japan for the surface area, pore volume, and pore size distribution. When the gas pressure was raised to 1 bar and the temperature was 298 K, the CO2 adsorption isotherms were measured. The desired temperature was achieved using a Dewar and a circulating jacket that was connected to a thermostatic water bath. Gas adsorption studies were conducted using approximately 0.1 g of adsorbents. To initiate the process, the sample was degassed under a vacuum at a temperature of 150 °C for a duration of 15 h.
The PXRD data for simulation was obtained from Material Studio software starting from the magnesium structure, nickel, zinc, and cobalt by using reflex tools [33,34]. Material Studio reflex tools were used to stimulate X-ray, neutron, and electron powder diffraction upon models of crystalline material [35]. This tool helps to determine the crystal of the structure, assist in the interpretation of diffraction data, and validate the result with experiments and computation. This study will be used to identify the X-Ray Diffraction (XRD) of the molecules. The peak scale and validation of theoretical XRD will be identified using DS BIOVIA Material Studio 2020 software and compared in this study.
3. Results
3.1. Screening of M-CPO-27 (M = Mg, Ni, Co, and Zn)
CPO-27 is known for its pore structure with high surface area and ability to adsorb a high CO2 capacity [30]. The chemical environment in the adsorbent molecule (Mg-CPO-27) can be tuned by exchanging the metal ion incorporated in the structure. This study shows the effect of open metal sites in CPO-27 on the adsorption behavior, with the possibility of increasing the affinity of CO2 molecules [36]. It is reported that CPO-27 predicts the highest uptake due to its large number of open metal sites facing the hexagonal pores, which provide higher sensitivity of the CO2 adsorption prediction on the metal center charge [37]. In recent studies by Jia Dent et.al., 2023, the capacity of CO2 adsorption for CPO-27 using molecular simulation was reported, with Mg-CPO-27 having the highest adsorption followed by other metals [38]. Figure 2 indicates the trend of CPO-27 for different metals using the molecular simulation approach.
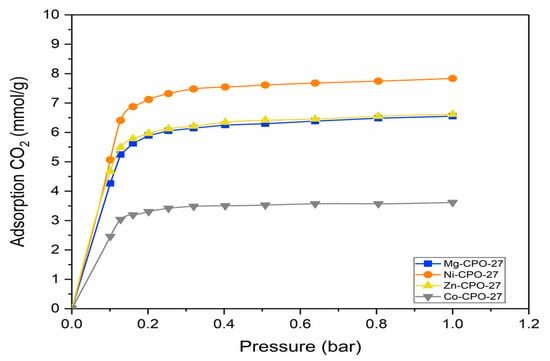
Figure 2.
Adsorption isotherm of CO2 in M-CPO-27 (M = Mg, Ni, Co, and Zn).
The molecular simulation approach is used for the preliminary selection of different metals in CPO-27 materials. Figure 2 shows the trend of different metals achieved in this study. As a result, Ni-CPO-27 shows the highest adsorption capacity followed by zinc, magnesium, and cobalt metal centers. The higher ionic interaction between the OCO2 atom and Mg2+ ions increase the adsorption capacity, and most previous studies selected Mg-CPO-27 for its promising performance in CO2 adsorption [30,31,39]. However, the inconsistencies found due to variations in the synthetic procedure, sample activation, or handling make it difficult to assess the accuracy of the reported result [40]. In addition, the stability of materials in the presence of water must be taken into account due to H2O acting as a strong competitor to CO2 molecules in hydrophilic materials such as Mg-CPO-27 [41]. The study found that Mg-CPO-27 was only able to achieve 16% adsorption capacity recovery compared with Ni-CPO-27 and Co-MOF-74 where it can attain up to 85% and 60% recovery, respectively [42]. However, the promising result of changing the metal from Mg-CPO-27 to other metal centers will be an advantage in this study, where Ni-CPO-27 is able to achieve higher CO2 adsorption compared with Mg-CPO-27. The steep increment in the initial pressure of CO2 adsorption shown in Figure 2 may be due to the strong polarization of adsorption sites that can always be found in the presence of coordinatively unsaturated sites of the metal center [40]. In low-pressure regions, it is reported that open metal sites dominate the adsorption process of CO2, thus, different trends are achieved by different metal centers of isostructural MOFs [40]. Co-CPO-27 found lower adsorption capacity due to the exceptionally ordered structure of gases, with metal centers causing the weak interaction to occur [43]. In addition, the tendency of Co2+ to oxidize to Co3+ at moderate temperatures resulted in low attraction to CO2 [44]. It is also reported that the charge of the metal plays an important role in adsorption capacity, where the isostructural MOFs yield the same isotherm trends [36]. For zinc and nickel metal centers, the trend shows higher magnesium (different from the expected result) attributable to the additional contributions from secondary adsorption sites, where there is an attraction to organic linkers [41]. Three sites of adsorption of CO2 were identified, where site I (primary) was located at the open metal site and showed an attraction of M-OCO2 and site II (secondary) focused on the framework channel, where the attractions of Oframework-CCO2 bonding were identified. Lastly, the tertiary site was found to be disordered at the center of the framework channel [40]. CO2 adsorption was reported to initially attract the metal center and further adsorb to organic linker sites after all metal sites were fully occupied to complete adsorption in M-CPO-27 [21].
The distance of CCO2-Oframework and OCO2-Mframework can be determined by the Lennard Jones potential equation, where van der Waals interactions between molecules can be described [30,45]. The Van der Waals interaction is dominant at 0.1 bar, where the electrostatic interaction between MOFs and CO2 is negligible at a pressure of 0.1 to 1.0 bar [36]. For M-MOF-74, M-O attraction is known for exothermically chemisorbing CO2 and forming MCO3. However, the GCMC technique was applicable for physisorption simulation where the M-O bond in this method was imprecisely determined [30]. Upon increasing the pressure (>0.1 bar), the sites proximal to metal atoms are saturated, and physisorption characteristic results can be accepted for M-CPO-27. Thus, this screening method will then be validated with an experimental technique to find the best metal center selection.
3.2. Physical Properties of M-CPO-27 (M = Co, Mg, Ni, and Zn)
3.2.1. X-ray Diffraction
To achieve the objective of preparing metal-based CPO-27, we compared the experimental and simulation results of XRD with previously reported studies [45,46]. The research objective is not to report and discuss the failed conditions that lead to a non-pure CPO-27 group but rather to point out the key point of obtaining CPO-27 materials. The use of a nitrate source (e.g., Mg (NO3)2), which is commonly used in synthesizing CPO-27, led to the presence of impurities [34]. Figure 3 shows the X-ray Diffraction (XRD) pattern obtained for the Ni-CPO-27 sample prepared according to previous studies [21,34]. Similarities of the pattern (the presence of crystals at the same peaks) are evident, and our sample crystallized from the system has a CPO-27 topology [34].
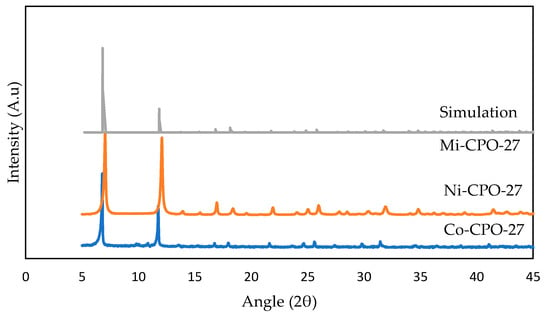
Figure 3.
XRD Analysis of M-CPO-27 for this study.
XRD patterns prepared for M-CPO-27 (M = Co and Ni) from the solvothermal method are shown in Figure 3 for experimental and simulation results, and the same trends were achieved by Mg-CPO-27 and Zn-CPO-27. The main peak is identified and compared with the theoretical pattern from a previous study [47]. As displayed, there are fewer peaks showing broadening found in the XRD pattern, which explains the presence of the crystalline nature in CPO-27 [35]. In the simulation study, we can perceive that the structure has less crystallinity compared with the experimental results as the intensity (second peak onwards) is lower. A further improvement in structure optimization should be performed to achieve a high-crystal structure. High-quality crystals could not be achieved easily based on single-crystal X-ray methods, but this PXRD pattern helps us to estimate unit cell parameters based on the trigonal spatial group (R-3) corresponding to CPO-27 materials [34,48,49]. The agreement between the experiment and other literature data was quite good and proves CPO-27 materials have been achieved.
The simulation structure is somehow questionable as the lesser crystal peak is estimated in the unit cell. Due to the nature of CPO-27 being isostructural, the XRD cannot distinguish the presence of different metals in the CPO-27 group [48]. Therefore, all metals in CPO-27 share the same space group (R-3) and topology, and the primary difference is the identity of cations at the open metal sites, and the variance can be identified through the lattice parameter shown in Table 1, here a = b ≠ c [48].

Table 1.
Lattice constant and M-O bond length of M-CPO-27.
The simulated lattices obtained are consistent with the literature values of M-CPO-27 (where M = Mg, Ni, Co, and Zn). The mean absolute errors for lattice parameters (a = b) and c are within the range of (0.1–0.7%) and (1.9–3.9%), respectively. The discrepancy between the values of a and b could be linked to the size of the metal center, which is likely to correspond to the size of the micro-pore diameter. The substitution of metal does not affect the marked discrepancy in the c dimension. However, this dimension is anticipated to be either less sensitive or insensitive to the size of the metal ion in cases of isomorphic substitution within 1-D dimensionality and a hexagonal space group (R3), which corresponds to the similar behavior observed among metal centers in CPO-27 with a trigonal space group [34,50]. The decent agreement between the lattice parameter behavior from the simulation with the literature data has proven that the structure tested is the CPO-27 group, and any difference in achieving adsorption capacity may be due to the bonding length and the attraction of the adsorption sites.
3.2.2. Fourier Transform IR (FTIR) Spectroscopy
Similar to powder X-ray Diffraction (XRD), the IR spectroscopy region of the organic ligand in CPO-27 is between 650 and 1800 cm−1, which was commonly taken as fingerprints for this material. XRD is sensitive to the crystal structure of the material, while that region in spectroscopy is more sensitive to the conformational or local environment of organic molecules (short-range information) [34]. Figure 4 shows the FTIR spectra for the CPO-27 group, and similarities between the IR region and the experimental result can confirm (dobdc = 2,5-dioxido-1,4-benzenedicarboxylate) that the organic ligand is present, as strongly indicated by M-based CPO-27 groups.
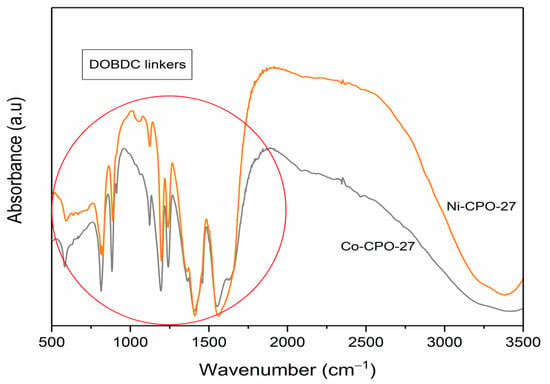
Figure 4.
FTIR Spectroscopy of M-CPO-27 (M = Ni (II) and Co (II)).
FTIR analysis of this study has proven that the structure of M-CPO-27 (M = Co, Mg, Ni, and Co) has been achieved. Mg-CPO-27 and Zn-CPO-27 (not shown in the figure) follow a common pattern due to the isostructural nature of CPO-27.
3.2.3. Adsorption CO2 of Synthesis M-CPO-27 (M = Co, Mg, Ni and Zn)
It is important to validate the computational method using the experimental approach as the structure in the simulation was assumed in bulk and pure conditions without any defects [36,51]. Meanwhile, in real conditions, it is very difficult to synthesize defect-free crystals [36]. Therefore, an experimental study is needed to validate the simulation method. Figure 5 shows the low pressure of CO2 adsorption isotherms collected for M-CPO-27 (M = Co, Mg, Ni, and Zn) using the solvothermal method at 298 K.
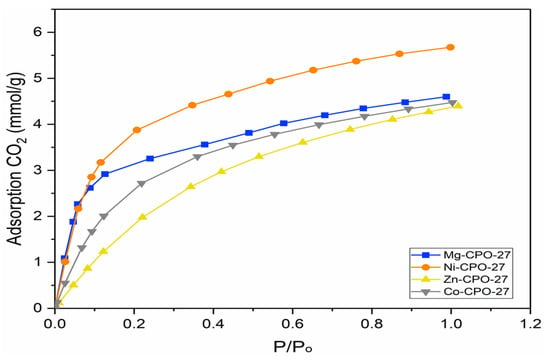
Figure 5.
CO2 adsorption on M-CPO-27.
Adsorption is reported to occur when molecules of gases such as CO2 stick to the surface of solid material, and in this case, the CPO-27 group indicates that adsorption is a phenomenon that occurs at the surface of materials [52,53]. In Figure 5, it is shown that Ni-CPO-27 achieves a higher adsorption capacity compared with Mg, Co, and Zn metals. At a pressure of 1 bar, the nickel-metal center provides a value of 5.6 mmol/g of CO2 adsorption followed by magnesium, cobalt, and zinc, which have 4.6 mmol/g, 4.5 mmol/g, and 4.4 mmol/g, respectively. Thus, it provides the trend of Ni > Mg > Co > Zn compared with the screening method of Ni > Zn > Mg > Co for metal center performance on CO2 adsorption. However, both methods agree that nickel is the best metal center. The difference in trends for the zinc metal center in the experiment is due to defects caused by the synthesis procedure. In addition, it is reported that the charge of the metal tendency of Zn2+ oxidizes and forms Zn3+, which could be the result of crystalline defects and provides lower adsorption capacity [34,36,42]. The same condition occurs for cobalt metal centers as described in Section 3.1. This study shows agreement that the nickel-metal center should be selected due to the promising result in achieving high CO2 adsorption compared with other metals. The promising robustness in water for Ni-CPO-27 compared with other metal centers provides added value for future works [3,39,54].
4. Discussion
In experimental works, metal centers in an organic ligand are expected, when not fully activated, to attract CO2 molecules compared with simulation, resulting in a difference in the adsorption trends achieved. The function of loading achieved in the experiment is ~0.8 CO2 per metal center, while in the simulation, every metal center was activated to attract CO2 molecules (one CO2 loading per metal in the simulation) [35,50]. Adsorption is significantly influenced by open metal sites, where the strong Lewis acid–base interaction between the metal ions and the oxygen atom of CO2 at the primary site, as well as the carbon atom of CO2 with the oxygen atom in the organic linker at the secondary site, results in high CO2 adsorption [28]. At a low pressure of 0.1 bar, as seen in Figure 5, it is shown that magnesium and nickel metal centers achieve almost the same adsorption capacity compared with cobalt and zinc, which may be due to the low interaction that occurs for its metal attraction to CO2. As increasing pressure is applied, the interaction of Ni-CPO-27 with CO2 leads to the trend of providing higher adsorption capacity, which may be due to the structure purity, as in XRD, Ni-CPO-27 is able to achieve higher peak crystallinity compared with other metals. The primary adsorption is expected to vary throughout the series, but as the pressure increase, the saturation of CO2 atoms at the metal center leads to the secondary site’s interaction occurring and is expected to resemble one another due to the nature of the framework being isostructural [34,36]. Indeed, magnesium, cobalt, and zinc metals have very similar CO2 adsorption at 1.0 bar, which may cause the materials to already approach the saturation capacity of primary adsorption [36]. Certain reasons may cause the difference in trends achieved compared with other literature, such as structural changes in the framework that are assumed to occur, the presence of impurities, remaining ligand/solvent blocking the pores or open metal sites, or crystalline defects that can block CO2 access to the channels [36]. Therefore, the aid of a simulation study can help to support the result achieved in this study.
Due to the higher adsorption capacity achieved in this work, the Ni-MOF-74 molecule’s behavior and attraction were further studied in order to identify its applicability. Table 2 indicates the adsorption capacity in this work compared with the previous study.
Table 2.
Adsorption capacity of CO2 for this experimental study.
Table 2 specifies that this study is able to achieve a higher capacity of CO2 for the Ni-CPO-27 metal center of 5.56 mmol/g, compared to other studies [3,21]. It is shown that both previous studies have a magnesium metal center with higher adsorption capacity, where the trend of the metal center for Caskey et al. and Andirova et al. showed Mg > Co > Ni > Zn and Mg > Ni > Zn > Co, respectively [3,21]. The difference in the value achieved may be due to the factor of crystalline defects or the blockage of pores [36]. However, this study provides a satisfying value that can be compared with other literature data, and Ni-CPO-27 can be selected as promising metal centers for further study.
The attraction of OCO2-Mframework in Table 1 can explain why the adsorption of Ni-CPO-27 provides the highest value compared with other metals for this work. CO2 shows a closer distance from the nickel metal center (2.04 Å) compared with magnesium (2.61 Å), hence the consequences of better performance in CO2 uptake. The guest–host interaction becomes less important in the limit of high CO2 uptake where the accessible volumes for CO2 are dominant [22]. Further improvement should be performed in simulation and experimental work to identify the strength of metal ions with CO2 attraction, such as including a polarization technique and using the Density Functional Theory (DFT) calculation with Hubbard U analysis with certain metal centers (e.g., cobalt) due to the presence of the semi-d orbital, which cannot properly describe the strong electronic interaction [16,22]. In addition, the molecular structure and metal center binding energy plays a certain role in the determination of adsorption, requiring a detailed study of this factor.
5. Conclusions
In this preliminary investigation, the screening of different metal centers for CPO-27 groups (M = Mg, Ni, Co, and Zn) allowed us to discover the trend of metal centers in the adsorption of CO2, where the initial trend gives Ni > Zn > Mg > Co. This simulation study uses GCMC calculation for its ability to identify the adsorption capacity of CO2 with insertion, deletion, translation, and rotation movement. It is found that adsorption may occur at three different sites (where site I focuses on the metal center, site II on the attraction at organic linkers, and site III on the disordered center of the framework). However, this study focuses on lower pressure, where the adsorption of CO2 at site I becomes dominant. The selection of the metal center procedure further continues with experimental work, where the simulation study may vary with the real situation due to the absence of impurities and structure decomposition applied; thus, the aid of experimental studies is needed for the selection of the metal center. M-CPO-27 was further synthesized using the solvothermal method at ambient temperature and pressure of up to 1 bar. This study found that Ni-CPO-27 provides the highest adsorption capacity of 5.6 mmol/g of CO2 compared with other metal centers. In the experimental result, the trend was found to be Ni > Mg > Zn > Co, where there is agreement that nickel is the best metal for selection. The different positions for the zinc metal center in the screening and experiment are due to the metal ion charge, where it oxidizes and become Zn3+. Additional information regarding XRD analysis determined that the crystalline structure of CPO-27 has been achieved in this study. Higher adsorption of Ni-CPO-27 achieved in this work will be an advantage for future work as it is reported to be robust in the presence of water. Further study should be performed on the crystallographic position of CPO-27 to identify the isosteric heat of attraction, the binding energy for the metal center, structure stability applications, and the selectivity of CO2 over other gases.
Author Contributions
Conceptualization, M.A.B.; methodology, N.E.F.K., K.K. and M.I.; software, N.E.F.K.; validation, N.E.F.K., M.A.B., K.K. and M.I.; formal analysis, N.E.F.K., M.A.B., M.I. and K.K.; resources, N.E.F.K., M.I. and K.K.; data curation, M.I. and K.K.; writing—original draft preparation, N.E.F.K. and A.A.; writing—review and editing, N.E.F.K., A.A. and M.A.B.; visualization, N.E.F.K. and M.A.B.; supervision, M.A.B. and T.L.C.; project administration, N.E.F.K., M.A.B., K.K. and T.L.C.; funding acquisition, M.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research work is supported by Yayasan Universiti Teknologi Petronas (YUTP)-PRG 2022 grant number 015PBC-014.
Data Availability Statement
As the data is currently part of an ongoing study, it is not possible to share the raw and processed data required to replicate these findings.
Acknowledgments
We would like to acknowledge Universiti Teknologi Petronas for providing all the facilities to accomplish this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, S.; Zhu, M.; Tang, Y.; Fu, Y.; Li, W.; Xiao, B. Molecular simulation and experimental investigation of CO2 capture in a polymetallic cation-exchanged 13X zeolite. J. Mater. Chem. A 2018, 6, 19570–19583. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, M.; Majeed, Z.; Xie, Y.; Zheng, K.; Luo, Z.; Li, C.; Zhao, C.J.S. Application of Hdrogen-Bonded Organic Frameworks in Environmental Remediation: Recent Advances and Future Trends. Separations 2023, 10, 196. [Google Scholar] [CrossRef]
- Andirova, D.; Cogswell, C.F.; Lei, Y.; Choi, S. Effect of the structural constituents of metal organic frameworks on carbon dioxide capture. Microporous Mesoporous Mater. 2016, 219, 276–305. [Google Scholar] [CrossRef]
- Mercado, R.; Vlaisavljevich, B.; Lin, L.-C.; Lee, K.; Lee, L.; Mason, J.A.; Xiao, D.J.; Gonzalez, M.I.; Kapelewski, M.T.; Neaton, B.; et al. Force Field Development from Periodic Density Functional Theory Calculations for Gas Separation Applications Using Metal–Organic Frameworks. J. Phys. Chem. 2016, 23, 12590–12604. [Google Scholar] [CrossRef]
- Hassan, R.Z. Adsorption of Gases (CO2, CH4) Using Novel Porous Materials (MOFs). Ph.D. Thesis, Curtin University, Bentley, WA, Australia, 2016. [Google Scholar]
- Xiang, S.; He, Y.; Zhang, Z.; Wu, H.; Zhou, W.; Krishna, R.; Chen, B. Microporous metal-organic framework with potential for carbon dioxide capture at ambient conditions. Nat. Commun. 2012, 3, 954. [Google Scholar] [CrossRef]
- Bolotov, V.A.; Kovalenko, K.A.; Samsonenko, D.G.; Han, X.; Zhang, X.; Smith, G.L.; McCormick, L.J.; Teat, S.J.; Yang, S.; Lennox, M.J.; et al. Enhancement of CO2 Uptake and Selectivity in a Metal-Organic Framework by the Incorporation of Thiophene Functionality. Inorg. Chem. 2018, 57, 5074–5082. [Google Scholar] [CrossRef] [PubMed]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Gutiérrez-Serpa, A.; Fernandez, I.P.; Pasan, J.; Pino, V.J. Metal-Organic Frameworks as Key Materials for soild-Phase Microextraction Devices-A Review. Separations 2019, 6, 47. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, S.-J. A review on solid adsorbents for carbon dioxide capture. J. Ind. Eng. Chem. 2015, 23, 1–11. [Google Scholar] [CrossRef]
- Ullah, S.; Bustam, M.A.; Assiri, M.A.; Al-Sehemi, A.G.; Sagir, M.; Kareem, F.A.A.; Elkhalifah, A.E.; Mukhtar, A.; Gonfa, G. Synthesis, and characterization of metal-organic frameworks-177 for static and dynamic adsorption behavior of CO2 and CH4. Microporous Mesoporous Mater. 2019, 288, 109569. [Google Scholar] [CrossRef]
- Mahdipoor, H.R.; Halladj, R.; Babakhani, E.G.; Amjad-Iranagh, S.; Ahari, J.S. Adsorption of CO2, N2 and CH4 on a Fe-based metal organic framework, MIL-101 (Fe)-NH2. Colloids Surf. A Physicochem. Eng. Asp. 2021, 619, 126554. [Google Scholar] [CrossRef]
- Ismail, M.; Bustam, M.A.; Kari, N.E.F. Screening of Gallate-Based Metal-Organic Frameworks for Single-Component CO2 and CH4 Gas. In E3S Web of Conferences; EDP Sciences: Les Ulis, France, 2021; p. 02005. [Google Scholar]
- Krishna, R. Screening metal–organic frameworks for mixture separations in fixed-bed adsorbers using a combined selectivity/capacity metric. RSC Adv. 2017, 7, 35724–35737. [Google Scholar] [CrossRef]
- Ullah, S.; Bustam, M.A.; Assiri, M.A.; Al-Sehemi, A.G.; Gonfa, G.; Mukhtar, A.; Kareem, F.A.A.; Ayoub, M.; Saqib, S.; Mellon, N.B. Synthesis and characterization of mesoporous MOF UMCM-1 for CO2/CH4 adsorption; an experimental, isotherm modeling and thermodynamic study. Microporous Mesoporous Mater. 2020, 294, 109844. [Google Scholar] [CrossRef]
- Sunder, N.; Fong, Y.-Y.; Bustam, M.A.; Lau, W.-J. Study on the Performance of Cellulose Triacetate Hollow Fiber Mixed Matrix Membrane Incorporated with Amine-Functionalized NH2-MIL-125(Ti) for CO2 and CH4 Separation. Separations 2023, 10, 41. [Google Scholar] [CrossRef]
- Alonso, G.; Bahamon, D.; Keshavarz, F.; Giménez, X.; Gamallo, P.; Sayós, R. Density Functional Therory-Based Adsorption Isotherm for Pure and Flue Gas Mixtures om Mg-MOF-74. Application in CO2 Capture Swing Adsorption Processes. J. Phys. Chem. C 2018, 122, 3945. [Google Scholar] [CrossRef]
- de Oliveira, A.; de Lima, G.F.; De Abreu, H.A. Structural and electronic properties of M-MOF-74 (M=Mg, Co or Mn). Chem. Phys. Lett. 2018, 691, 283. [Google Scholar]
- Maurin, G. Role of molecular simulations in the structure exoplaration of Metal Organic Frameworks Illustrations through recent advances in the field. CR Chim. 2016, 19, 207. [Google Scholar] [CrossRef]
- Wilmer, C.E.; Leaf, M.; Lee, C.Y.; Farha, O.; Hauser, B.; Hupp, J.T.; Snurr, R.Q. Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 2011, 171, 83–89. [Google Scholar]
- Bao, Z.; Yu, L.; Ren, Q.; Lu, X.; Deng, S. Adsorption of CO2 and CH4 on a magnesium-based metal organic framework. J. Colloid. Interface. Sci. 2011, 353, 549–556. [Google Scholar] [CrossRef]
- Bautista, P.R.; Mancera, I.T.; Pasan, J.; Pino, V. Metal-Organic Frameworks in Green Analytical Chemistry. Separations 2019, 6, 33. [Google Scholar] [CrossRef]
- Caskey, S.R.; Adam, J. Dramatic Tuning of Carbon Dioxide Uptake via Metal Substitution in a Coordination Polymer with Cylindrical Pores. J. Am. Chem. Soc. 2008, 130, 10870–10871. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.M.; Lin, L.C.; Dubbeldam, D.; Vlugt, T.J. Polarizable Force Field for CO2 in M-MOF-74 Derived from Quantum Mechanics. J. Phys Chem. C 2018, 122, 24488. [Google Scholar] [CrossRef]
- Wang, X.; Ramirez-Hinestrosa, S.; Dobnikar, J.; Frenkel, D. The Lennard-Jones potential: When (not) to use it. Phys. Chem. Chem. Phys. 2020, 22, 10624–10633. [Google Scholar] [CrossRef] [PubMed]
- Konstantakou, M.; Gotzias, A.; Kainourgiakis, M.; Stubos, A.K.; Steriotis, T.A. GCMC Simulation of Gas Adsorption in Carbon Pore Structure. In Applications of Monte Carlo Method in Science and Engineering; Intechopen: London, UK, 2011. [Google Scholar]
- Fairen, D.F.; Seaton, N.A.; Düren, T. Unusual Adsorption Behavior on Metal-Organic Frameworks. Langmuir 2010, 26, 14694–14699. [Google Scholar] [CrossRef] [PubMed]
- Yazaydın, A.O.; Snurr, R.Q.; Park, T.H.; Koh, K.; Liu, J.; LeVan, M.D.; Benin, A.I.; Jakubczak, P.; Lanuza, M.; Galloway, D.B.; et al. Screening of Metal-Organic Frameworks for Carbon Dioxide from Fuel Gas Using a Combined Experimental and Modeling Approcah. J. Am. Chem. Soc. 2009, 131, 18189–18199. [Google Scholar] [CrossRef] [PubMed]
- Remy, T.P.; Sunil, A.; Perre, A.; Valvekens, P.; Vos, P.; Baron, G.; Joeri, J. Selective Dynamic CO2 Seperation on Mg-MOF-74 at Low Pressures: A Detailed Comparison with 13X. J. Phys. Chem. C 2013, 117, 9301–9310. [Google Scholar] [CrossRef]
- Xin, J.H.; He, P.; Li, H.; Wang, X. Understanding the Adsorption of C2H2, CO2, and CH4 in Isostructural Metal-Organic Frameworks with Cooridinatively Unsaturated Metal Sites. J. Phys. Chem. C 2013, 117, 2824–2834. [Google Scholar]
- Keskin, S.; Altintas, C. Computational screening of MOFs for C2H6/C2H4 and C2H6/CH4 separations. Chem Eng. Sci. 2016, 139, 49–60. [Google Scholar]
- Tim, M.B.; Heinen, J.; Dubbeldam, D.; Lin, L.; Vlugt, T.H. Polarized Force Field for CO2 and CH4 Adsorption in M-MOF-74. J. Phys. Chem. C 2017, 121, 4659–4673. [Google Scholar]
- Jung, D.H.; Kim, D.; Lee, T.B.; Choi, S.B.; Yoon, J.H.; Kim, J.; Choi, K.; Choi, S.-H. Grand Canonical Monte Carlo Simulation Study on the Catenation Effect on Hydrogen Adsorption onto the Interpenetrating Metal−Organic Frameworks. J. Phys. Chem. B 2006, 100, 22987–22990. [Google Scholar] [CrossRef]
- García, M.D.; Sanchez, M. Synthesis and characterization of a new Cd-based metal-organic framework isostructural with MOF-74/CPO-27 material. Micropor. Mesopor. Mat. 2014, 190, 248–254. [Google Scholar] [CrossRef]
- Biovia, D.S. Modules Tutorials Materials Studio; Dassault Systèmes: San Diego, CA, USA, 2017; Volume 470, pp. 226–233. [Google Scholar]
- Agrawal, A.; Agrawal, M.; Suh, D.; Fei, S.; Alizadeh, A.; Ma, Y.; Matsuda, R.; Hsu, W.; Daiguji, H. Augmenting the Carbon Dioxide Uptake and Selectivity of Metal-Organic Frameworks by Metal Substitution: Molecular Simulations of LMOF-202. ACS Omega 2020, 5, 17193–17198. [Google Scholar] [CrossRef]
- Sladekova, K.; Campbell, C.; Grant, C.; Fletcher, A.J.; Gomes, J.R.; Jorge, M. The Effect of Atomic Point Charges on Adsorption Isotherms of CO2 and Water in Metal Organic Frameworks. Adsorption 2020, 26, 663–685. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, G.; Zhang, L.; Ma, H.; Rong, Y. CO2 Adsorption and Separation Properties of M-MOF-74 Materials Determined by Molecular Simulation. Capillarity 2023, 6, 13–18. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, J.; Ma, X.; Liu, J.; Lin, Y. Mechanism of CO2 adsorption on Mg/DOBDC with elevated CO2 loading. Fuel 2016, 181, 340–346. [Google Scholar] [CrossRef]
- Queen, W.L.; Hudson, M.R.; Bloch, E.D.; Mason, J.A.; Gonzalez, M.I.; Lee, J.S.; Gygi, D.; Howe, J.D.; Lee, K.; Darwish, T.A.; et al. Comprehensive study of carbon dioxide adsorption in the metal–organic frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn). Chem. Sci. 2014, 5, 4569. [Google Scholar] [CrossRef]
- Canivet, J.; Fateeva, A.; Coasne, B.; Farrusseng, D. Water adsorption in MOFs: Fundamentals and applications. Chem. Soc. Rev. 2014, 43, 5594. [Google Scholar] [CrossRef]
- Liu, J.; Thallapally, P.K.; McGrail, B.P. Selective CO2 Capture from Flue Gas Using Metal Organic Frameworks—A Fixed Bed Study. J. Phys. Chem. C 2012, 116, 9575–9581. [Google Scholar] [CrossRef]
- Gonzalez, M.I.; Mason, J.A.; Bloch, E.D.; Teat, S.J.; Gagnon, K.J.; Morrison, G.Y.; Queen, W.L.; Long, J.R. Structural characterization of framework–gas interactions in the metal–organic framework Co2(dobdc) by in situ single-crystal X.-ray diffraction. R. Soc. Chem. 2017, 9, 4387–4398. [Google Scholar] [CrossRef] [PubMed]
- Botas, J.A.; Calleja, G.; Sánchez-Sánchez, M.; Orcajo, M.G. Effect of Zn/Co ratio in MOF-74 type materials containing exposed metal sites on their hydrogen adsorption behaviour and on their band gap energy. Int. J. Hydrogen Energy 2011, 36, 10834–10844. [Google Scholar] [CrossRef]
- Naeem, R. Lennard Jones Potential; UC Davis Chem Wiki: Davis, CA, USA, 2015. [Google Scholar]
- Zhao, Z.; Zuhra, Z.; Qin, L.; Zhou, Y.; Zhang, L.; Tang, F.; Mu, C. Confinement of microporous MOF-74(Ni) within mesoporous γ-Al2O3 beads for excellent ultra-deep and selective adsorptive desulfurization performance. Fuel Process. Technol. 2018, 176, 276–282. [Google Scholar] [CrossRef]
- Thakkar, H.; Eastman, S.; Al-Naddaf, Q.; Rownaghi, A.A.; Rezaei, F. 3D-Printed Metal–Organic Framework Monoliths for Gas Adsorption Processes. ACS Appl. Mater. Interfaces 2017, 9, 35908–35916. [Google Scholar] [CrossRef]
- Lee, K.; Howe, J.D.; Lin, L.C.; Smit, B.; Neaton, J.B. Small-Molecule Adsorption in Open-Site Metal−Organic Frameworks: A Systematic Density Functional Theory Study for Rational Design Chemistry of Materials. Chem. Mater. 2015, 3, 668–678. [Google Scholar] [CrossRef]
- Koh, H.S.; Rana, M.K.; Wong-Foy, A.G.; Siegel, D.J. Predicting Methane Storage in Open-Metal-Site Metal–Organic Frameworks. J. Phys. Chem. C 2015, 119, 13451–13458. [Google Scholar] [CrossRef]
- Concepcih, P.; Mifsud, A. Preparation and Characterization of Mg-Containing AFI and Chabazite-Type Material. Zeolites 1996, 16, 56–64. [Google Scholar] [CrossRef]
- Valenzano, L.; Civalleri, B.; Chavan, S.; Palomino, G.T.; Areán, C.O.; Bordiga, S. Computational and Experimental Studies on the Adsorption of CO, N2, and CO2 on Mg-MOF-74. J. Phys. Chem. C 2010, 114, 11185–11191. [Google Scholar] [CrossRef]
- Ullah, S.; Bustam, M.A.; Assiri, M.A.; Al-Sehemi, A.G.; Sagir, M.; Kareem, F.A.A.; Elkhalifah, E.I.; Mukhtar, A.; Gonfa, G. Influence of post-synthetic graphene oxide (GO) functionalization on the selective CO2/CH4 adsorption behavior of MOF-200 at different temperatures; an experimental and adsorption isotherms study. Microporous Mesoporous Mater. 2019, 296, 110002. [Google Scholar] [CrossRef]
- Kizzie, A.C.; Wong-Foy, A.G.; Matzger, A.J. Effect of Humidity on the Performance of Microporous Coordination Polymers as Adsorbents for CO2 Capture. Langmuir 2011, 27, 6368–6373. [Google Scholar] [CrossRef]
- Lopez, M.G.; Canepa, P.; Thonhauser, T. NMR study of small molecule adsorption in MOF-74-Mg. J. Chem. Phys. 2013, 138, 154704. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).