Abstract
A class of drugs called coxibs (COX-2 inhibitors) were created to help relieve pain and inflammation of osteoarthritis and rheumatoid arthritis with the lowest amount of side effects possible. The presented paper describes a new developed, optimized and validated thin layer chromatographic (TLC)-densitometric procedure for the simultaneous assay of five coxibs: celecoxib, etoricoxib, firecoxib, rofecoxib and cimicoxib. Chromatographic separation was conducted on HPTLC F254 silica gel chromatographic plates as a stationary phase using chloroform–acetone–toluene (12:5:2, v/v/v) as a mobile phase. Densitometric detection was carried out at two wavelengths of 254 and 290 nm. The method was tested according to ICH guidelines for linearity, recovery and specificity. The presented method was linear in a wide range of concentrations for all analyzed compounds, with correlation coefficients greater than 0.99. The method is specific, precise (%RSD < 1) and accurate (more than 95%, %RSD < 2). Low-cost, simple and rapid, it can be used in laboratories for drug monitoring and quality control.
1. Introduction
The search for new non-steroidal anti-inflammatory drugs (NSAIDs), since the synthesis of salicylic acid, has been carried out simultaneously in two main directions, obtaining compounds with higher anti-inflammatory effects, and compounds with weaker side effects. Over time, the availability of an increasing number of new, strong representatives of this group of drugs increased, but the problem of their toxic effects, especially on the gastrointestinal tract and kidneys, remained unresolved. A breakthrough in the search led to the discovery of several isoforms of cyclooxygenase (COX) [1]. The goal of the creation of NSAIDs was to stop prostaglandin production as the main cause of inflammation, pain and fever. The mechanism of their action is the inhibition of the cyclooxygenase enzyme responsible for the transformation of arachidonic acid, and the formation of signal molecules—prostaglandins. However, as it turned out in later studies, there are two isoforms of this enzyme, COX-1 (along with the COX-3 subtype) and COX-2, and prostaglandins performs many varied and physiologically important functions in the body. The elementary description of the differences between the two isoforms is that COX-1 occurs mainly physiologically in the human body, while COX-2 is a constitutive isoform arising primarily in places of tissue damage and dysfunction. COX-1 occurs naturally in many important organs, and regulates their work, e.g., regulation of renal blood flow, formation and functioning of platelets and the presence of blood vessels in the endothelium. The COX-2 isoform is mainly recognized in pathological conditions, but it has been proven that it can also be constitutively present in small amounts in organs, e.g., kidneys, where both types complement each other [2,3,4,5]. The distribution of both enzyme isoforms is subject to fairly high species variability in mammals and even individual variation. The most known and commonly used NSAIDs inhibit both enzyme types (e.g., ibuprofen, diclofenac). In this case, serious side effects associated with COX1 inhibition (gastric mucosal damage, kidney damage) may occur. In order to minimize the adverse effects, more selective blocking of the COX-2 isoform than COX-1 (e.g., meloxicam) and the completely selective blocking of COX-2 (group of “coxibes”) was sought.
The aforementioned group of selective COX-2 inhibitors has a much weaker gastrotoxic effect compared to other NSAIDs, and their therapeutic effectiveness is compared to the action of reference drugs, i.e., ibuprofen or naproxen [6,7]. Coxibs are used primarily to treat inflammation and pain in osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. They are also commonly used for the short-term treatment of acute pain conditions [8]. Conducted research suggests that they may affect cartilage metabolism by inhibiting cartilage matrix formation (e.g., naproxen or indomethacin), or by stimulating it (e.g., aceclofenac) or without affecting its synthesis (e.g., piroxicam) [9]. Unfortunately, some drugs, also from the group of COX-2 inhibitors, may increase the risk of cardiovascular incidents (e.g., rofecoxib) [10].
Celecoxib was the first coxib introduced to human medicine. The development of this group of drugs was stopped when increased cardiotoxicity was found in randomized clinical trials. This led to the withdrawal from the market, among others—rofecoxib and valdecoxib. The use of coxibs in humans has since been significantly reduced [11,12,13,14]. At the same time, many representatives of NSAIDs used in human pharmacotherapy have found use in veterinary medicine. Unfortunately, a large group of these drugs cannot be used in veterinary treatment because of their high toxicity to animals. Deracoxib, firocoxib, mavacoxib, robenacoxib, and cimicoxib have been registered since the 2000s. Osteoarthritis is the main indication for the use of this group of drugs in veterinary medicine. In addition, they are used in postoperative pain and bone fractures [15,16,17,18]. Firocoxib and deracoxib are active ingredients in drugs indicated for use in dogs [19]. In one field trial, firocoxib showed improved efficacy in some measurement over the less COX-1 sparing NSAID carprofen [20]. In the other field trial, dogs treated with firocoxib showed significantly greater improvement in osteoarthritis-related pain and lameness, and a significantly reduced incidence of diarrhea relative to dogs treated with etodolac [21]. Currently, there are more and more studies exploring the possibility of using selective COX2 inhibitors in oncological treatment in both animals and humans [22,23,24,25].
There are a number of spectrophotometric and liquid chromatographic (with various detectors) methods reported in the literature for the assays of the selected five representatives of selective COX-2 inhibitors (celecoxib, etoricoxib, cimicoxib, firocoxib and robenacoxib) in various materials, biological samples and pharmaceuticals [26,27,28,29,30,31,32,33,34,35]. There were also publications in which these drugs were determined in animal and environmental samples [16,36,37,38]. However, no method for the simultaneous determination of those active substances in dosage forms has been studied so far. On this basis, it became apparent to develop and validate a new, simple, sensitive and accurate methodology for the simultaneous estimation of these drugs (Figure 1) in pharmaceutical preparations (human and veterinary), using the thin layer chromatographic (TLC) technique. TLC with densitometric detection is a rapid and non-complicated analytical technique for the separation, identification and quantification of medicinal substances. It is simple and low cost, and the minimal need for cleaning of the sample allows for the conduction of various types of analyses in different areas of science.
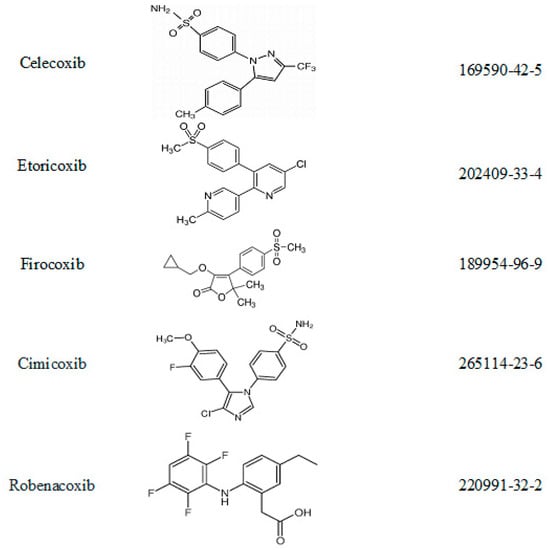
Figure 1.
Chemical structure and CAS numbers of analyzed compounds.
2. Materials and Methods
2.1. Materials and Methods
2.1.1. Chemicals and Apparatus
Methanol, chloroform, acetone, toluene, ethyl acetate, ammonia, glacial acetic acid, and other organic solvents were purchased from Merck (Darmstadt, Germany). All chemicals were analytical grade. HCl, NaOH and H2O2 solutions were purchased from POCh, Gliwice, Poland. Buffer solutions (pH 2—citrate buffer, pH 7—phosphate buffer, pH 8—borate buffer) were purchased from Witko (Łódź, Poland). Densitometer TLC Scanner 3 with Cat4 software (CAMAG, Muttenz, Switzerland), Linomat 4 (CAMAG, Switzerland), analytical balance XA 52/Y (Radwag, Poland) were used. Chromatographic plates TLC Silica gel 60F254 (no 1.05554), HPTLC Silica gel 60F254 (no 1.05548), TLC Kieselgel 60WF254s (no 16484), TLC Cellulose F (no 1.05574) were all purchased from Merck (Darmstadt, Germany).
2.1.2. Standard Substances and Solutions
Celecoxib (no PHR1683), etoricoxib (no 32097) and firocoxib (no 32236) were purchased from Sigma-Aldrich (Laramie, WY, USA). Due to the lack of available standard substances, pure compounds of cimicoxib and robenacoxib were obtained from their preparations. Extraction from their preparations was carried out using diethyl ether (POCh, Gliwice, Poland). The weight of the preparation was shaken successively with three portions of ether, the filtrate was collected, and then the solvent was evaporated until a dry residue was obtained. The obtained compounds were subjected to infrared spectrophotometric analysis. Methanol solutions (about 1% (w/v)) were also made, and subjected to TLC analysis under the conditions described below (analysis of RF values and absorption spectra). No additional peaks were received on the obtained chromatograms.
The identities of the obtained substances were confirmed by infrared spectrophotometry. Finally, pure substances were obtained, and they were used in later studies.
Pharmaceutical preparations: Celebrex (Pfizer Europe, UK) containing 200 mg of celecoxib, Arcoxia (MSD, Warszawa, Poland) containing 120 mg of etoricoxib, Previcox (Boehringer Ingelheim Vetmedica GmbH, Ingelheim am Rhein, Germany) containing 227 mg of firocoxib, Cimalgex (Vétoquinol SA, France) containing 80 mg of cimicoxib and Onsior (Elanco GmbH, Cuxhaven, Germany) containing 40 mg of robenacoxib, were used. All preparations were purchased from the local pharmacy or veterinary office. Preparations had an expiry of no less than one year at the time of study.
Standard solutions were prepared individually by dissolving an accurately-weighed 10.0 mg of each standard in 10.0 mL of methanol, and stored in the dark at 4 °C for up to three days.
2.1.3. Samples Solutions
Samples solutions were prepared by powdering five tablets or capsules. Next, each tablet powder was weighed to an accuracy of 0.1 mg, and a quantity of equivalent to 10.0 m. Each drug was weighed and transferred separately into a 10.0 mL volumetric flask containing approximately 5.0 mL methanol. Each samples were shaken on a laboratory shaker (Lab Dancer V Vario, Imlab) for 5 min and the volume was made up to the mark with methanol. Prepared solutions were filtered through filter paper and all filtrates were used for the analysis.
2.1.4. Chromatographic Conditions
Chromatography was performed on 10 × 10 cm aluminum sheets precoated with 200 µm layers of silica gel 60 F254 (E. Merck, Darmstadt, Germany). Samples were applied to the plates as bands that were 5 mm wide and 10 mm apart by means of a Linomat V (CAMAG, Switzerland) sample applicator equipped with a 100 L syringe (Hamilton, Switzerland). The first application was 10 mm from the bottom and 10 mm from the left edge of the plate. The application rate was 600 nL/s. Samples were developed in different mobile phases and various separation lengths. Plates were taken into a chromatographic chamber (18 × 16 × 8 cm; Sigma-Aldrich, Laramie, WY, USA) previously saturated with mobile phase vapor for 20 min in room temperature. Good separation and well-developed peaks were obtained with mobile phase containing with chloroform–acetone–toluene (12:5:2, v/v/v). The development distance was 10 cm during 20 min. After development, plates were dried in room temperature for about 20 min. Densitometric scanning was performed using a CAMAG TLC Scanner 3 with winCats 4 software (Muttenz, Switzerland). The source of radiation was the deuterium lamp emitting a continuous UV spectrum between 200 to 400 nm. Scanning speed was 20 mm/s and slit dimensions were 4.00 × 0.45 mm. Plates were scanned over the range of 200–400 nm and the spectra were overlaid. Based on the obtained spectra, analytical wavelengths were chosen for detection all analyzed coxibs.
2.2. Validation of the Method
The validation procedure of the proposed method was performed with respect to specificity, linearity and range, limits of detection and quantification, precision, and robustness, according to ICH guidelines [39].
2.2.1. Selectivity
The separation factor α, calculated by equation α = [(1/RF1) − 1]/[(1/RF2) − 1] (where: RF1 and RF2—the retardation factor for 1 and 2 substance), is a chromatographic parameter that determines the selectivity of separation of two adjacent compounds in a given chromatographic system. Its value should be higher than 1.0.
To ensure the accuracy of quantitative analysis, it is also important to determine the value of the resolution factor R, calculated by the equation R = [2 (Z2 − Z1)]/(W1 + W2) (where: Z1 and Z2—the separation distance of two adjacent peaks 1 and 2, W1 and W2—the width of two adjacent peaks 1 and 2). It determines the possibility of resolution analysis for two adjacent peaks, and it should be no less than 1.
2.2.2. Range and Linearity
The range and linearity of the method were verified by analyzing of a set of ten calibration mixtures in the concentration ranges of 0.10–10 mg/mL for celecoxib, 0.02–10 mg/mL for etoricoxib, 0.40–10 mg/mL for firocoxib, 0.05–7 mg/mL for cimicoxib and 0.05–10 mg/mL for robenacoxib. To develop the calibration curves, peak area and drug concentration data were treated by linear least-square regression analysis.
The dependence of the substance concentration on the densitogram peak area is rectilinear only within a certain range. Regression parameters were determined, such as slope (a), intercept (b), standard deviation of a slope (Sa), standard deviation of an intercept (Sb), estimation error (Se), correlation coefficient (r). Based on the obtained results for each regression equation, the analysis of residuals was also completed.
2.2.3. Limit of Detection (LOD) and Limit of Quantification (LOQ)
The LOD is the smallest amount (concentration) of the tested substance in the sample that can be detected but not necessarily determined with appropriate accuracy. The smallest amount of substance that can be determined with a certain precision and accuracy is named the LOQ. Both parameters are very important and needed when using the method for determining a drug substance in pharmaceutical preparations. The LOD and LOQ were calculated based on obtained calibration curve values, respectively as: LOD = (3.3 · Sb)/a and LOQ = (10 · Sb)/a, where: Sb—the standard deviation of the intercept, a—the slope of the calibration curve.
2.2.4. Precision
Precision is defined as checking the extent in which the results of analyses carried out on the same sample will be compatible with each other. The precision of analytical methods was assessed regarding repeatability (intra-day precision) and intermediate precision (inter-day precision). The percentage of relative standard deviation (%RSD) of the obtained peak areas was taken as the measure of precision. To study intra-day precision, six standard solutions containing respectively 600 ng/band of celecoxib, 400 ng/band of etoricoxib, 900 ng/band of firocoxib, 800 ng/band of cimicoxib and 1000 ng/band of robenacoxib were prepared, applied to chromatographic plates and developed under the conditions described in Section 2.1.4. Each replicate was created under the same operating conditions and over a short interval time. Inter-day precision was created for the same samples by performing the analysis after a week.
2.2.5. Accuracy
To evaluate the accuracy of the method, recovery studies were carried out by addition of standard substance solution to pre-analyzed sample solutions at three different concentration levels: 80%, 100%, and 120%. The recovery percentages (R [%]) were calculated by formula: R [%] = [(A1 − A2)/A2] × 100%, where: A1—peak area for sample after adding analyte, A2—peak area for sample before adding analyte.
2.2.6. Robustness
The robustness measures the ability of an analytical method to remain intact after small, deliberate changes in method parameters. It provides guidance on the reliability of the analytical method during normal use. The robustness of the proposed method was evaluated by the reliability of the analysis with respect variations in the experimental conditions, such as the sorbent type (TLC 60F254, HPTLC 60F254, TLC 60WF254s, TLC Cellulose F), the chamber type (12 × 11 × 8 cm, 27 × 27 × 7 cm), the distance of development (±0.5 cm), saturation time of the chamber (±5 min), the volume of individual components in used mobile phase (±0.1 mL).
2.3. Stability-Indicating Study
The Food and Drug Administration (FDA) and International Conference on Harmonization (ICH) state the requirement of stability testing data to understand how the quality of a drug substance and drug product changes with time under the influence of various environmental factors [19,39]. Based on the guidelines, the following conditions were investigated: hydrolysis at acid (0.5 M HCl), base (0.5 M NaOH) and buffer solutions (pH 2, 7, 8), thermal degradation (30 °C, 80 °C), photolysis, and oxidation (10% H2O2, v/v). Solutions of individual drugs were prepared by dissolving the weight in an appropriate solution of acid, base, H2O2, buffer or methanol. Aliquots of 1 mL of each drug solution were transferred to minivials and heated at 30 or 80 °C for 1 and 3 h. Sequentially harvested portions of the test solutions were diluted with methanol to the proper volume and applied onto the HPTLC plates and analyzed as described above. A sample powder of each drug was exposed to sunlight for seven days. Next, the substances were dissolved in methanol, and applied onto the chromatographic plates. At the same time, similar determinations were made for neutral samples (methanol solutions).
2.4. Analysis of Pharmaceutical Formulations
Ten tablets, or the content of ten capsules of each commercial product, were separately mixed and weighed. Individual amounts of each product equivalent to 5.0 mg of each drug substance was dissolved in 50.0 mL of methanol. After shaking for 20 min, all solutions were filtered using a 0.2 m Whatman filter paper and used in further research. Standard and sample solutions (6 µL) were applied to the plate and analysis was performed as described above.
3. Results and Discussion
The quality of pharmaceutical products is very important to maintain the overall healthcare of millions of patients. This study presents a new validated isocratic method based on the TLC separation for simultaneous resolution and quantification of selected COX-2 inhibitors, e.g., celecoxib, etoricoxib, firecoxib, robenacoxib and cimicoxib in human and animal pharmaceuticals. The presented procedure was developed taking into account the therapeutic and overdose concentration range. It was validated and verified for the determination of mentioned drugs in raw materials and pharmaceutical formulations.
Several problems are associated with the simultaneous determination of investigated compounds. The first is the choice of separation conditions to ensure efficient drug extraction with minimal interference from the matrix (especially from veterinary preparations). The other is choosing the right chromatographic conditions to achieve the separation of all compounds simultaneously. Next, the method must be sensitive enough to determine the concentrations of tested drugs in their therapeutic range.
To optimize the conditions of the TLC-densitometric method, a number of parameters were varied, such as the type of stationary phase, the mobile phase composition and the developing distance. Based on the literature data and eluotropic series, various ratios of different organic solvents (e.g., methanol, chloroform, acetone, toluene, ethyl acetate, ammonia, glacial acetic acid) were tested. For the simultaneous determination of five coxibs, individual drug solutions were spotted onto the different chromatographic plates and developed at various distances using different mobile phases. The variation in the stationary and mobile phases led to considerable changes in the chromatographic parameters, like a peak symmetry and retardation factor.
As a result of these experiments, optimal conditions were determined for the analysis of selected coxibs side by side, conducting assays on HPTLC 60F254 chromatographic plates as the stationary phase, and a mixture consisting of chloroform–acetone–toluene in a volume ratio of 12:5:2 as the mobile phase. The selected analysis conditions allowed for the simultaneous determination of all five drug samples.
The obtained chromatograms were subjected to densitometric detection. In the described conditions, the peak shape and resolution were found to be good. The determined values of retardation factors (RF) for individual substances are shown in Table 1. The representative densitogram obtained from a mixed standard solution of analyzed substances is shown in Figure 2.

Table 1.
Retardation (RF), resolution (α) and separation (R) factors.
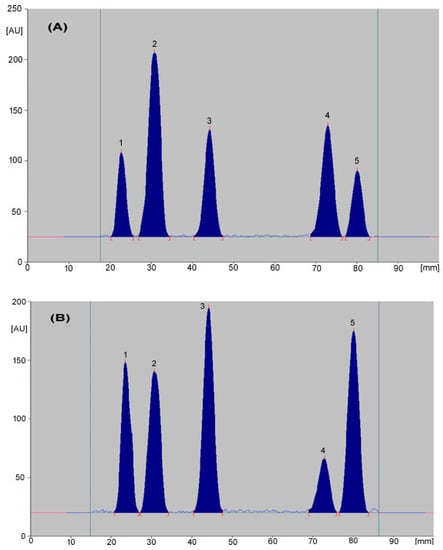
Figure 2.
An example of densitograms obtained for a mixture of standard substances at 254 nm (A) and 290 nm (B) (1—cimicoxib; 2—robenacoxib; 3—etoricoxib; 4—celecoxib; 5—firocoxib).
In addition, the absorption spectra of individual compounds were recorded in the wavelength range from 200 to 400 nm, and compared. All analyzed coxibs show maxima of absorption in the range from 230 to 300 nm (Figure 3); celecoxib and cimicoxib showed considerable absorbance at 254 nm, but etoricoxib, firocoxib and robenacoxib showed a maximum of absorbance at 290 nm. Based on these observations, two wavelengths, 254 and 290 nm, were chosen for the quantification of individual coxibs. The choice of using common wavelengths was considered satisfactory to allow the detection and analysis of all tested drugs with appropriate sensitivity. The identities of each substance on the densitograms were confirmed by comparing the RF values and the absorption spectra of separated peaks with those obtained for standards.
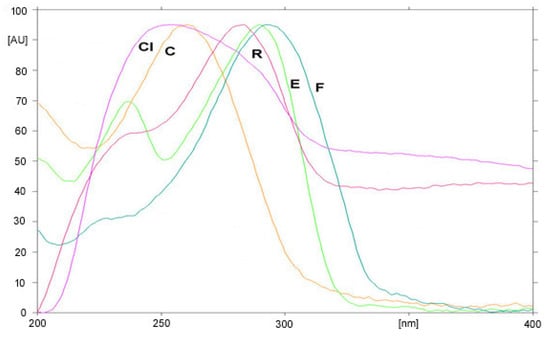
Figure 3.
An absorption spectra registered for analyzed compounds obtained directly from the chromatogram (C—celecoxib; E—etoricoxib; CI—cimicoxib; F—firocoxib; R—robenacoxib).
In order to validate the developed analytical procedure, tests of specificity, linearity, precision, accuracy and robustness of the method were carried out.
The specificity of the method was evaluated regarding possible background impurities and interferences from the excipients in commercial preparations. The obtained chromatograms and registered absorption spectra of all drug solutions did not show and additional peaks compared to the chromatograms and spectra of standard solutions. (Figure 4). This is especially important in the case of veterinary drugs, which contain various additional substances that improve taste and smell. It can be said that the proposed method is selective and specific for the analyzed coxibs.
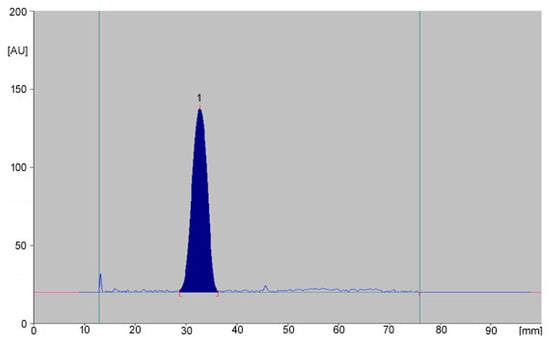
Figure 4.
An example of chromatograms obtained for Onsior preparation containing robenacoxib. (1—cimicoxib).
The separation factor α and resolution factor R, calculated for every two adjacent peaks are presented in Table 1. All obtained values are higher than 1; it can be stated that the developed conditions allow for the correct separation and analysis of selected coxibs.
The concentration of a set of solutions used for the range analysis were the same as in Section 2.2.2. Peak area (P) and drug concentration (C) data were treated by linear regression analysis. The plots P vs. C of each coxib were found to be linear over a range of 0.10–5 mg/mL for celecoxib, 0.02–5 mg/mL for etoricoxib, 0.40–6 mg/mL for firocoxib, 0.05–7 mg/mL for cimicoxib and 0.10–10 mg/mL for robenacoxib. Linear regression equations representing the calibration curves and the regression parameters, such as slope, intercept, standard deviation of the slope and intercept, an estimation error and the correlation coefficients are shown in Table 2. The results showed excellent prediction for the set-in high values of correlation coefficients, ranging between 0.98 and 0.99 for all analytes. This confirms the high predictive ability of the developed method.
Table 2.
Regression equations and validation parameters obtained for analyzed compounds.
Detection and quantification limits were calculated following the formula presented in Section 2.2.3. The calculated LOD and LOQ values are in the range of 29.62 to 93.69 ng/band and from 89.75 to 283.91 ng/band, respectively. The obtained data, presented in Table 2, demonstrated relatively low values, which indicates that the developed method is sufficiently sensitive to analyzed drugs.
Based on the calculated LOQ values and plotted calibration curves, it was found that the standard calibration curves are linear in the respective concentration ranges for individual compounds at both wavelengths (Table 2), with correlation coefficients (r) close to 0.99.
Plotting the residuals helps to identify problems with poor or incorrect curve fitting. If there is a good fit between the data and the regression model, the residuals should be distributed randomly about zero. For this purpose, the correctness of obtained correlations was determined for each regression equation based on residual analysis. Figure 5 displays an example of a scatterplot of residuals for concentration values.
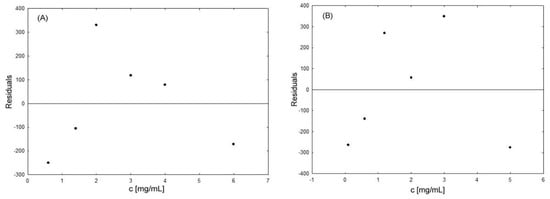
Figure 5.
An example of the plot of residuals for firocoxib at 254 nm (A) and celecoxib at 290 nm (B).
For the obtained results, we can conclude that the residuals locate around zero and are randomly scattered without any patterns. The values of correlation coefficients for individual plots of residuals are presented in Table 2. This random feature confirms that there is no trend in the spread of residuals with concentration, and the assumption of linearity is correct.
The parameter CD, Cook’s distance, was also determined for each point (Table 2). CD is used in regression analysis to find influential outliers in a set of prediction variables. It is a way to identify points that negatively affect a regression model. The obtained values in all cases are convergent and are in the range from 0.2176 to 0.9772. Based on the received parameters for the examined points, no significant deviations that could disturb the received regression were found.
The precision of the proposed method was assessed by analyzing peak areas obtained for all the studied compounds within the same day (for intra-day precision) and after a week (for inter-day precision). The method passed the tests as determined by %RSD of the area of the peaks of six replicate injections at 100% test concentration. The low values of %RSD (not higher than 1.22) indicated good precision of the developed method (Table 3) for all drug substances at both wavelengths.
Table 3.
Results of the method precision.
To verify the applicability of the method, the recovery experiment of the selected coxibs in pharmaceutical preparations was performed. Before drug determination, the celecoxib, etoricoxib and firocoxib were spiked into their preparations. After shaking, solutions of standard substances and solutions of proper pharmaceutical preparations—the spiked samples—were spotted onto the HPTLC plate and determined in conditions, as described in Section 2.2.5.
Satisfactory recovery demonstrated that the developed method is reliable and sensitive for simultaneous determination of analyzed coxibs. The percent recovery results were satisfactory, ranging from 93.65% to 106.20% with %RSD lower than 2% (Table 4). The obtained results also confirmed that the excipients in commercial formulations do not interfere with the studied compounds.
Table 4.
Results of the accuracy.
The robustness of the TLC-densitometric procedure was evaluated by small changes in experiment conditions, such as sorbent type, chamber type, distance of development, saturation time of the chamber, volumes of the mobile phase components. On the basis of obtained results (analysis of RF values and absorption spectra), it was observed that there were no marked significant changes in chromatographic behavior for all analyzed coxibs, indicating that the method is robust.
Due to the fact that all analyzed compounds show relatively high values of absorbance at selected wavelengths, validation of the developed procedure was executed at two wavelengths, 254 and 290 nm. Obtained results are satisfactory, and indicate the possibility of carrying out determinations of tested coxibs at both wavelengths with appropriate precision in a various concentration ranges.
Summarizing the results, it can be said that the validation process of an analytical method including the TLC-densitometric method is a very powerful tool, necessary in the quantitative determination of analyzed coxibs in their pharmaceutical formulations, like tablets and capsules. The validation report indicates that the developed method fulfills the criteria of an analytical method designated for quantity control of pharmaceuticals in terms of specificity, linearity, limits of detection and quantification, precision, accuracy and robustness.
The chemical stability of pharmaceutical substances is a very important issue because it affects the safety and efficacy of the pharmaceutical product. Forced degradation study of a substance and a product under accelerated conditions shows the chemical behavior of the molecule, which in turn helps in the formulation and packaging. Stress tests are conducted under extreme conditions to demonstrate the specificity of the developed method of measuring changes in drug concentration when little information is available on the potential degradation product, and to confirm the stability of the compound during the given analytical procedure [40]. In studied conditions (hydrolysis at acid, base and buffer solutions, thermal degradation, photolysis and oxidation) no additional peaks for degradation products on chromatograms were obtained. The peak area from individual substances did not decrease, which proves the stability of the tested compounds in the described conditions, and the selectivity of the developed method for the tested coxibs.
In the next step, the developed method was successfully applied for the determination of individual drug contents in their pharmaceutical preparations. The content of all substances in the examined products were determined on the basis of previously described conditions, in ranges of linearity and at the selected analytical wavelength (254 nm for celecoxib and cimicoxib, 290 nm for etoricoxib, firocoxib and robenacoxib). The obtained results of TLC-densitometric analysis of five coxibs with statistical analysis are demonstrated in Table 5.
Table 5.
Results from assay of celecoxib, etoricoxib, firocoxib, cimicoxib and robenacoxib in their pharmaceutical formulations.
The obtained content of each active substance in the examined drugs shows a very good agreement to the amount declared by manufacturers. This indicated acceptably-good accuracy and precision in the analysis of coxibs in tablets and capsules form. The obtained results confirm the suitability of the developed method.
The obtained parameters are similar to the methods created by other researchers in the field of coxib analysis. In Rajmane et al.’s work, where the authors determined etoricoxib in combination with thiocolhicoside by HPTLC, LOQ 33.314 ng/band and recovery near 100% was obtained [32]. Rao et al. analyzed inhibitors COX-2 (celecoxib, rofecoxib, valdecoxib, nimesulide and nabumetone) in pharmaceutical preparations and in human plasma using RP-HPLC. The obtained percentage recoveries were between 97.55%–100.14%, and LOD in the range from 127 to 1040 µg/L [41]. Another method of analyzing COX-2 inhibitors in human plasma by HPLC was developed for celecoxib and rofecoxib, with the linearity range between 20–2000 µg/L and the LOQ, 20 µg/L [42].
The presented newly developed method also offers many advantages, such as a low cost, the lack of need for any pre-treatment, and the analysis of several samples for all analyzed compounds simultaneously during one analysis (on one plate). It can be used in wide concentration ranges of celecoxib, etoricoxib, firocoxib, cimicoxib and robenacoxib with high stability, sensitivity and reproducibility. The method did not show any notable deviations in results from acceptable limits. Despite the fact that the analyzed coxibs are not used simultaneously, the presented method allows for quick and easy checking, taking into account which substance is used and in what quantity in the analyzed sample.
4. Conclusions
Under the described optimized conditions, the complete separation of five coxibs, i.e., celecoxib, etoricoxib, firocoxib, cimicoxib and robenacoxib, was observed. The performed analysis indicated that none of the excipients presented in drug formulations interfered with the proposed assay method. This offers possibilities for the simultaneous assay of five similar substances in one analytical procedure condition with good separation and resolution of the chromatogram peaks. The developed TLC-densitometric method is fast, reliable and cost-effective and can be used for the analysis of the studied drugs in commercial pharmaceutical products with good selectivity, precision and accuracy. The use of TLC has advantages over, e.g., the HPLC method, such as: a lack of need to thoroughly clean samples, the ability to perform analyses of several samples at the same time, the subsequent storage of plates, as well as a greater number of detection methods. The LOQ, small sample volume and short chromatography time per analysis—without the need for special sample preparation and costly solvents—makes it advantageous to be adapted to routine assay requirements in quality control laboratories, and stability studies.
Author Contributions
Investigations, P.G., M.D. and M.S.; writing-original draft, P.G.; writing—review and editing, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Katori, M.; Majima, M. Cyclooxygenase-2: Its rich diversity of roles and possible application if it’s selective inhibitors. Inflamm. Res. 2000, 49, 367–392. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef]
- Curtis, E.; Fuggle, N.; Shaw, S.; Spooner, L.; Ntani, G.; Parsons, C.; Corp, N.; Honvo, G.; Baird, J.; Maggi, S.; et al. Safety of cyclooxygenase-2 inhibitors in osteoarthritis: Outcomes of a systematic review and meta-analysis. Drugs Aging 2019, 36, 25–44. [Google Scholar] [CrossRef]
- Martín Arias, L.H.; Martín González, A.; Sanz Fadrique, R.; Salgueiro Vázquez, E. Gastrointestinal safety of coxibs: Systematic review and meta-analysis of observational studies on selective inhibitors of cyclo-oxygenase 2. Fundam. Clin. Pharmacol. 2019, 33, 134–147. [Google Scholar] [CrossRef]
- Fu, J.Y.; Masferrer, J.L.; Seibert, K.; Raz, A.; Needleman, P. The induction and suppression of prostaglandin Hz synthase (cyclooxygenase) in human monocytes. J. Biol. Chem. 1990, 265, 16737–16740. [Google Scholar]
- Bjorkman, D.J. One hundred years of NSAID gastropathy: Are coxibs the answer? Rev. Gastroenterol. Disord. 2001, 1, 121–127. [Google Scholar]
- McMurray, R.W.; Hardy, K.J. COX-2 inhibitors: Today and tomorrow. Am. J. Med. Sci. 2002, 323, 181–189. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Dingle, J.T. The effect of NSAID on the matrix of human articular cartilages. Z. Rheumatol. 1999, 58, 125–129. [Google Scholar] [CrossRef]
- Merck Announces Voluntary Worldwide Withdrawal of VIOXX®. Available online: http://www.vioxx.com/vioxx/documents/english/vioxx_press_release.pdf (accessed on 17 November 2004).
- Braun, J.; Baraliakos, X.; Westhoff, T. Nonsteroidal anti-inflammatory drugs and cardiovascular risk—A matter of indication. Semin. Arthritis Rheum. 2020, 50, 285–288. [Google Scholar] [CrossRef]
- Meek, I.L.; Van de Laar, M.A.F.J.; Vonkeman, H.E. Non-steroidal anti-inflammatory drugs: An overview of cardiovascular risks. Pharmaceuticals 2010, 3, 2146–2162. [Google Scholar] [CrossRef]
- Sgambati, S.A. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial: Commentary. Dis. Colon Rectum 2005, 48, 1330–1331. [Google Scholar]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Eng. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Bergh, M.S.; Budsberg, S.C. The Coxib NSAIDs: Potential clinical and pharmacologic importance in veterinary medicine. J. Vet. Intern. Med. 2005, 9, 633–643. [Google Scholar] [CrossRef]
- Jung, M.; Lees, P.; Seewald, W.; King, J.N. Analytical determination and pharmacokinetics of robenacoxib in the dog. J. Vet. Pharmacol. Ther. 2009, 32, 41–48. [Google Scholar] [CrossRef]
- Kim, T.W.; Giorgi, M. A brief overview of the coxib drugs in the veterinary field. Am. J. Anim. Vet. Sci. 2013, 8, 89–97. [Google Scholar] [CrossRef]
- Kongara, K.; Chambers, P. Robenacoxib in the treatment of pain in cats and dogs: Safety, efficacy, and place in therapy. Vet. Med. Res. Rep. 2018, 9, 53–61. [Google Scholar] [CrossRef]
- FDA Guidance, Analytical Procedures and Method Validation: Chemistry, Manufacturing, and Controls Documentation, Draft Guidance, Food and Drug Administration. 2000. Available online: http://www.fda.gov/downloads/Drugs/.../Guidance/ucm122858.pdf (accessed on 20 January 2020).
- Pollmeier, M.; Toulemonde, C.; Fleishman, C.; Hanson, P.D. Clinical evaluation of firocoxib and carprofen for the treatment of dogs with osteoarthritis. Vet. Res. 2006, 159, 547–551. [Google Scholar] [CrossRef]
- Hanson, P.D.; Brooks, K.C.; Case, J.; Conzemius, M.; Gordon, W.; Schuessler, J.; Shelley, B.; Sifferman, R.; Drag, M.; Alva, R.; et al. Efficacy and safety of firacoxib in the management of canine osteoarthritis under field conditions. Vet. Ther. 2006, 7, 127–140. [Google Scholar]
- Üner, M.; Yener, G.; Ergüven, M. Design of colloidal drug carriers of celecoxib for use in treatment of breast cancer and leukemia. Mater. Sci. Eng. C 2019, 103. [Google Scholar] [CrossRef]
- Regulski, M.; Regulska, K.; Prukała, W.; Piotrowska, H.; Stanisz, B.; Murias, M. COX-2 inhibitors: A novel strategy in the management of breast cancer. Drug Discov. Today 2016, 21, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Yarla, N.S.; Madka, V.; Rao, C.V. Clinically relevant anti-inflammatory agents for chemoprevention of colorectal cancer: New perspectives. Int. J. Mol. Sci. 2018, 19, 2332. [Google Scholar] [CrossRef] [PubMed]
- Aasy, N.K.A.; Ragab, D.; Sallam, M.A.; Abdelmonsif, D.A.; Aly, R.G.; Elkhodairy, K.A. A comparative study: The prospective influence of nanovectors in leveraging the chemopreventive potential of COX-2 inhibitors against skin cancer. Int. J. Nanomed. 2019, 14, 7561–7581. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.; Villarino, N.; Sommardahl, C.; Kvaternick, V.; Zarabadipour, C.; Siger, L.; Yarbrough, J.; Amicucci, A.; Reed, K.; Breeding, D.; et al. Disposition of firocoxib in equine plasma after an oral loading dose and a multiple dose regimen. Vet. J. 2013, 198, 382–385. [Google Scholar]
- Oh, H.A.; Kim, D.; Lee, S.H.; Jung, B.H. Simultaneous quantitative determination of celecoxib and its two metabolites using liquid chromatography-tandem mass spectrometry in alternating polarity switching mode. J. Pharm. Biomed. Anal. 2015, 107, 32–39. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, N.; Ji, W.; Wen, Q. Rapid quantitative analysis of etoricoxib in human plasma by UPLC-MS/MS and application to a pharmacokinetic study in Chinese healthy volunteers. Biomed. Chromatogr. 2019, 33, e4414. [Google Scholar] [CrossRef]
- Jedziniak, P.; Szprengier-Juszkiewicz, T.; Pietruk, K.; Ledziska, E.; Żmudzki, J. Determination of non-steroidal anti-inflammatory drugs and their metabolites in milk by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 2955–2963. [Google Scholar] [CrossRef]
- Starek, M.; Komsta, Ł.; Krzek, J. Reversed-phase thin-layer chromatography technique for the comparison of the lipophilicity of selected non-steroidal anti-inflammatory drugs. J. Pharm. Biomed. Anal. 2013, 85, 132–137. [Google Scholar] [CrossRef]
- Kumar, S.; Joshi, A.; Thakur, R.S.; Pathak, A.K. Simultaneous estimation of etoricoxib and thiocolchicoside by RP-HPLC method in combined dosage forms. Acta Pol. Pharm. 2011, 68, 839–843. [Google Scholar]
- Rajmane, V.S.; Gandhi, S.V.; Patil, U.P.; Sengar, M.R. High-performance thin-layer chromatographic determination of etoricoxib and thiocolchicoside in combined tablet dosage form. J. AOAC Int. 2010, 93, 783–787. [Google Scholar]
- Singh, S.; Mishra, A.; Verma, A.; Ghosh, A.K.; Mishra, A.K. A simple ultraviolet spectrophotometric method for the determination of etoricoxib in dosage formulations. J. Adv. Pharm. Technol. Res. 2012, 3, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Sangoi, M.S.; Wrasse-Sangoi, M.; Oliveira, P.R.; Bernardi, L.S. Determination of lumiracoxib by a validated stability-indicating MEKC method and identification of its degradation products by LC-ESI-MS studies. J. Sep. Sci. 2011, 34, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, L.; Bao, Y.; Yang, Q.; Zhou, L.; Hao, H.; Xie, C. Confirmation of more stable polymorphic form of etoricoxib at room temperature. J. Pharm. Sci. 2018, 107, 1903–1910. [Google Scholar] [CrossRef]
- Giorgi, M.; Kim, T.-W.; Saba, A.; Rouini, M.-R.; Yun, H.; Ryschanova, R.; Owen, H. Detection and quantification of cimicoxib, a novel COX-2 inhibitor, in canine plasma by HPLC with spectrofluorimetric detection: Development and validation of a new methodology. J. Pharm. Biomed. Anal. 2013, 83, 28–33. [Google Scholar] [CrossRef]
- Knych, H.K.; Stanley, S.D.; Arthur, R.M.; Mitchell, M.M. Detection and pharmacokinetics of three formulations of firocoxib following multiple administrations to horses. Equine Vet. J. 2014, 46, 734–738. [Google Scholar] [CrossRef]
- Triñanes, S.; Casais, M.C.; Mejuto, M.C.; Cela, R. Selective determination of COXIBs in environmental water samples by mixed-mode solid phase extraction and liquid chromatography quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2015, 1420, 35–45. [Google Scholar] [CrossRef]
- ICH Guide Q2 (R1). Validation of Analytical Procedures-Text and Methodology; International Conference on Harmonization: Geneva, Switzerland, 2005; Available online: https://www.ich.org/page/quality-guidelines (accessed on 8 January 2020).
- Bakshi, M.; Singh, S. Development of validated stability-indicating assay methods—Critical review. J. Pharm. Biomed. Anal. 2002, 28, 1011–1040. [Google Scholar] [CrossRef]
- Rao, R.N.; Meena, S.; Nagaraju, D.; Rao, A.R.R. Development and validation of a reversed-phase liquid chromatographic method for separation and simultaneous determination of COX-2 inhibitors in pharmaceuticals and its application to biological fluids. Biomed. Chromatogr. 2005, 19, 362–368. [Google Scholar]
- Hamama, A.K.; Ray, J.; Day, R.O.; Brien, E. Simultaneous determination of rofecoxib and celecoxib in human plasma by high-performance liquid chromatography. J. Chromatogr. Sci. 2005, 43, 351–354. [Google Scholar] [CrossRef][Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).