Since the accidental discovery of carbon quantum dots (cQDs) in 2004, a class of nanomaterials with sizes in the range of 2–10 nm, as well as their consequential attractive properties, the field has rapidly expanded to now include cQDs made from neat starting materials such as citrate, urea and diethylamine, or from waste products, biomass and plant-based carbon sources such as watermelon peel, grass, banana and strawberry juice, ginger, garlic, coriander and even cyanobacteria [
1].
The expansion of the cQD “library” co-relates with the expansion of its applications relative to inorganic quantum dots (QDs). Due to the higher bio-compatibility (compared to their inorganic counterparts) as well as their excellent photoluminescence, cQDs have found widespread applications in the areas of electrochemical immuno-sensing, biological imaging, theranostics and as fluorescent probes. cQDs with sp
2-hybridized carbons are effective in cancer mitigation [
1]. A recent and landmark study has extended the role of cQDs to the ambit of neurodegenerative disorders [
2]. Graphene-based quantum dots (GQDs) were able to inhibit the fibrillization of α-synuclein, a protein associated with the onset of synucleinopathies such as Parkinson’s disease [
2]. The study also revealed that GQDs were also able to interact directly with mature fibrils and caused their dissolution—suggesting promise in both prophylaxis and therapeutics.
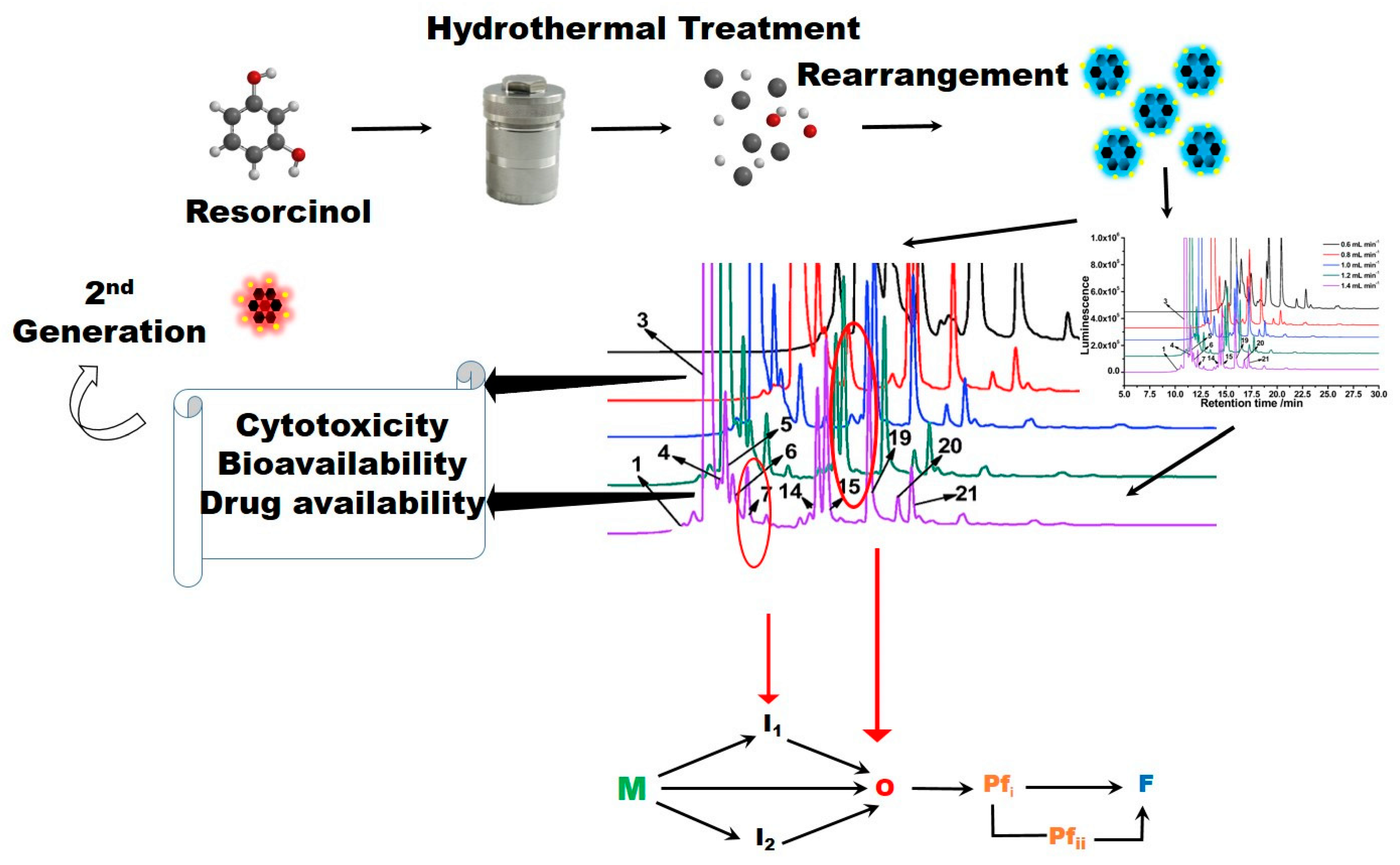
The transitioning of cQDs from its applications in electronics, catalysis and environmental sensing to the biological realm and across the blood–brain barrier represents an important crossover point. Factors such as toxicity, bioavailability, high homogeneity, side effects, druggability and specificity to the disease of interest become important in biomedical applications. In this context, it is important to recognize that the synthesis of cQDs even via the mildest of preparations potentially generates a heterogeneous reaction mixture comprising several distinct cQD and non-cQD constituents [
3].
A recent study involving cQD synthesis using
m-resorcinol as the source resulted in a mixture of at least 21 cQD and non-cQD compounds. The study only reinforced the complexity of cQD chemistry and more importantly, demonstrated the need to unequivocally identify each constituent prior to biomedical use [
3]. The resolution of cQD chemical composition is only possible by separating the cQD mixture and then identifying the chemical structure of each cQD constituent (including the assignment of chiral centers). Other factors such as the purity, cytotoxicity, bioavailability and druggability are equally important and need to be determined for each cQD within the ensemble prior to any biological use. A case in point is related to the toxicity issues associated with the chemistry driving cQD product formation. cQDs often produced via the Maillard-like reaction may be toxic and/or mutagenic to cells [
4]. The early stage condensation reactions remain the backbone of many cQDs, where the elimination of water between an amino group and a carbonyl group results in the formation of a Schiff base. However, it is likely succeeded by a ketosamine that is the product of an Amadori rearrangement reaction. The degradation of Amadori products to reactive carbonyls such as keto-aldehydes, dicarbonyls and reductones can result in the formation of toxic products such as glyoxal, methylglyoxal, 3-deoxyglucosone (3-DG), hetero-cyclic amines and acryl amides.
Other than toxicity-related issues, an atomic- and molecular-level understanding of cQD–receptor interactions is critical for tailored therapies [
5,
6,
7]. For example, Graphene quantum dots in alveolar macrophages were analyzed in nuclei, nuclear responses and DNA, and docking studies have been previously reported using the crystal structure of DNA (PDB entry code: 1DJD). π–π stacking (
Figure 1A) interactions play a dominant role in the interactions between AG-QDs and DNA chains as ascertained by molecular docking simulations [
5].
In CB1-marijuana (Tetrahydrocannabinol) interactions, π–π stacking plays a dominant role as well. Therefore, cQDs targeting receptors within the brain need to engage with specificity and high affinity (
Figure 1B,C) as shown in a hypothetical cQD prepared from marijuana and then docked to the receptor [
8]. The anticipated results are significant because they permit an in silico-based design and modelling of CQDs onto the receptor. The experimental validation would require the steps in
Scheme 1 to be executed.
The need to resolve the specifics of cQD intervention is further illustrated by the aforementioned ability of graphene quantum dots (GQDs) to not only inhibit α-synuclein fibrillization but also dissolve mature fibrils of the Parkinson’s-disease-associated amyloid protein [
2] The noticeably different fibril-inhibiting tendencies of pristine GQDs, nano-graphene oxide and reduced GQDs observed in that study, suggested variations in the mechanism of action and therefore differences in efficacy. Compounding this knowledge gap is the fact that pristine (pure; undoped) GQDs are an ensemble of structures that differ in molecular mass, chemical activity, likely ability to penetrate the blood–brain barrier, toxicity, etc. [
1,
2,
3,
6,
7]. In order to further advance GQD and cQD intervention in neurodegenerative disorders, the critical first-step is the resolution, with atomic precision, of the experimental mechanism of their intervention in the disease-specific amyloidogenic trajectory (
Scheme 1).
To illustrate this point, consider a typical amyloid-forming landscape involving the transformation of soluble monomeric amyloid into fibrils (
Scheme 1). The process involves the generation of toxic intermediates known as oligomers and proto-fibrils that populate the amyloidogenic trajectory. Given their association with the onset and pathogenesis of neurodegenerative disorders, oligomers and protofibrils are established targets in drug-design efforts. Other intermediates that are on-pathway to these toxic species would also qualify as targets for arresting the identifying pathogenesis. There may be dead-end species that are of little interest therapeutically and serve little purpose and only lead to the futile consumption of any cQDs interacting with them.
The advance of successful small molecules from molecular dynamics simulations to in vitro and in vivo assays and eventually to clinical trials requires (i) establishing the purity of the small-molecule, (ii) the knowledge of its chemical structure, (iii) the dose-response (and therefore knowledge of its molar mass), (iv) determining in which step the molecule may interfere in the trajectory and (v) how the molecule engages the pathogenic target, etc. [
Scheme 1]. Based on the mode of action, structure–activity relationships will permit the development of second-generation candidates that may exhibit enhanced inhibition efficacies, better bioavailability, lesser cytotoxicity, etc.
The holy grail in the transition of cQDs to pre-clinical and clinical trials would be the necessary separation and purification of the cQD ensembles into individual chemical entities. This would need to be followed by resolving their structure at an atomic level. The mechanism of intervention of the cQD in the desired pathology would need to be addressed. Finally, a toxicological profile of each cQD to be used biomedically must be established. An Angstrom-level characterization would liberate cQDs from their “quantum” confinement and catapult their use across wider domains.



{kind=link}
{kind=link}

