


Active Sites in Low-Loaded Copper-Exchanged Mordenite: Spectroscopic and Stability Study for Methane Oxidation Using Mild Conditions

 , , , and
, , , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Synthesis of Materials
2.2. Reaction Test: Testing of Activity for Selective Oxidation of Methane
2.3. Materials Characterization
3. Results and Discussion
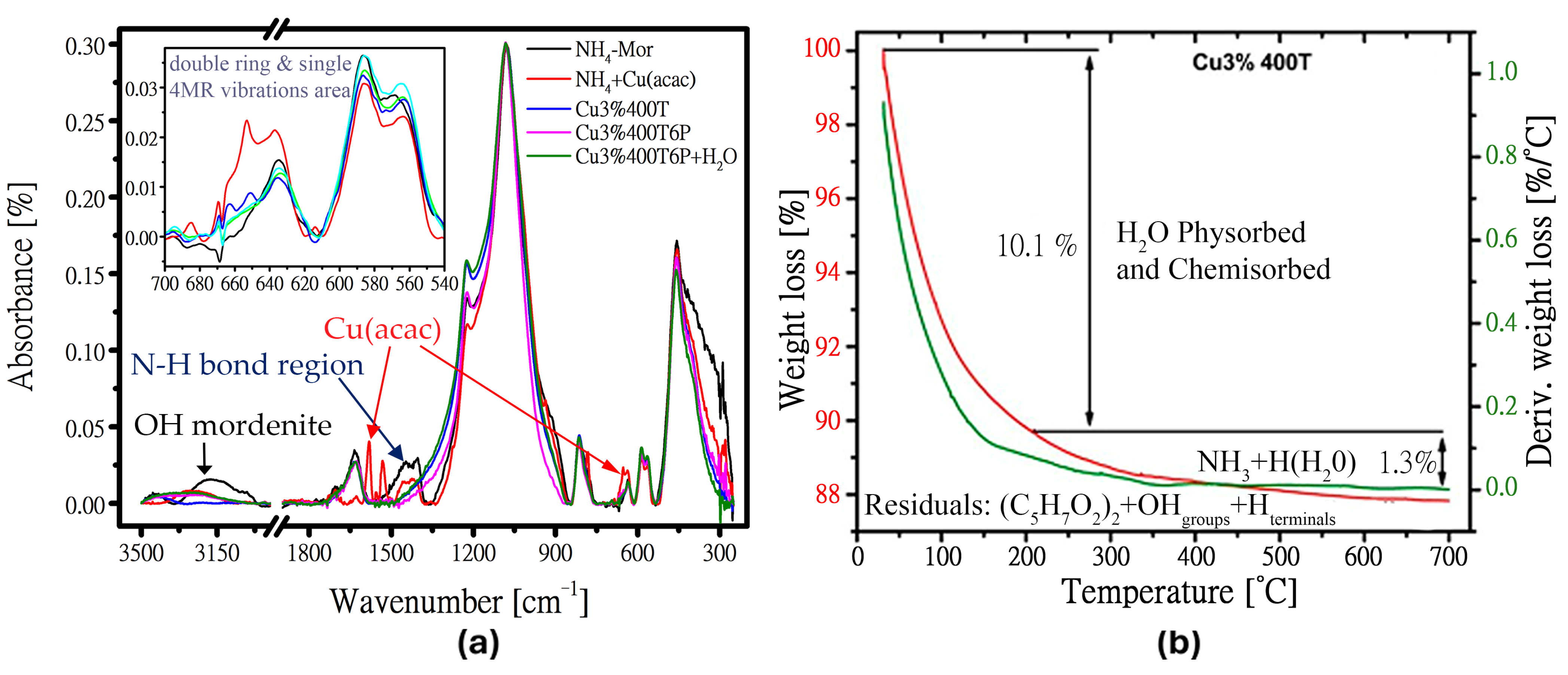
3.1. Sample Characterization—Copper Species Evidence
3.2. Test Reaction: Methane Oxidation Using the Materials Under Study, the Performance of the Active Sites Using Mild Conditions
3.3. Active Sites Characterization After Test Reaction
3.4. EPR Spectra on the Copper Detected Sites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, Z.; Kamarudin, S. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Olivos-Suarez, A.I.; Szécsényi, À.; Hensen, E.J.M.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Malakoff, D. The gas surge. Science 2014, 344, 1464–1467. [Google Scholar] [CrossRef]
- Xu, Z.; Kang, J.; Park, E.D. Continuous gas-phase oxidation of methane into methanol over Cu-mordenite. Microporous Mesoporous Mater. 2023, 360, 112727. [Google Scholar] [CrossRef]
- Passini, R.J.; Picinini, M.; Bueno, J.M.C.; Urquieta-Gonzalez, E.A. Direct methane to methanol stepwise conversion over Cu-oxo species in zeolites—Insights on the Cu-zeolite activation in air or helium from in situ UV-Vis analyses. Mol. Catal. 2022, 530, 112605. [Google Scholar] [CrossRef]
- Olsbye, U.; Svelle, S.; Bjørgen, M.; Beato, P.; Janssens, T.V.W.; Joensen, F.; Bordiga, S.; Lillerud, K.P. Conversion of Methanol to Hydrocarbons: How Zeolite Cavity and Pore Size Controls Product Selectivity. Angew. Chem. Int. Ed. 2012, 51, 5810–5831. [Google Scholar] [CrossRef]
- Wulfers, M.J.; Teketel, S.; Ipek, B.; Lobo, R.F. Conversion of methane to methanol on copper-containing small-pore zeolites and zeotypes. Chem. Commun. 2015, 51, 4447–4450. [Google Scholar] [CrossRef]
- Ikuno, T.; Grundner, S.; Jentys, A.; Li, G.; Pidko, E.; Fulton, J.; Sanchez-Sanchez, M.; Lercher, J.A. Formation of Active Cu-oxo Clusters for Methane Oxidation in Cu-Exchanged Mordenite. J. Phys. Chem. C 2019, 123, 8759–8769. [Google Scholar] [CrossRef]
- Panthi, D.; Adeyiga, O.; Odoh, S.O. DFT Analysis of Methane C-H Activation and Over-Oxidation by [Cu2O]2+ and [Cu2O2 ]2+ Sites in Zeolite Mordenite: Intra- versus Inter-site Over-Oxidation. ChemPhysChem 2021, 22, 2517–2525. [Google Scholar] [CrossRef]
- Bordeaux, M.; Galarneau, A.; Drone, J. Catalytic, Mild, and Selective Oxyfunctionalization of Linear Alkanes: Current Challenges. Angew. Chem. Int. Ed. 2012, 51, 10712–10723. [Google Scholar] [CrossRef] [PubMed]
- Le, H.V.; Parishan, S.; Sagaltchik, A.; Göbel, C.; Schlesiger, C.; Malzer, W.; Trunschke, A.; Schomäcker, R.; Thomas, A. Solid-State Ion-Exchanged Cu/Mordenite Catalysts for the Direct Conversion of Methane to Methanol. ACS Catal. 2017, 7, 1403–1412. [Google Scholar] [CrossRef]
- Soorholtz, M.; White, R.J.; Zimmermann, T.; Titirici, M.M.; Antonietti, M.; Palkovits, R.; Schüth, F. Direct methane oxidation over Pt-modified nitrogen-doped carbons. Chem. Commun. 2013, 49, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, B.; Zhang, Q.; Deng, W.; Wang, Y.; Yang, Y. Recent advances in heterogeneous selective oxidation catalysis for sustainable chemistry. Chem. Soc. Rev. 2014, 43, 3480. [Google Scholar] [CrossRef]
- Ross, M.O.; Rosenzweig, A.C. A tale of two methane monooxygenases. JBIC J. Biol. Inorg. Chem. 2017, 22, 307–319. [Google Scholar] [CrossRef]
- Que, L.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef]
- Nondestructive Testing, 3. Ultrasonics. In Ullmann’s Encyclopedia of Industrial Chemistry, 1st ed.; Wiley: Hoboken, NJ, USA, 2011. [CrossRef]
- Sainz-Vidal, A.; Balmaseda, J.; Lartundo-Rojas, L.; Reguera, E. Preparation of Cu–mordenite by ionic exchange reaction under milling: A favorable route to form the mono-(μ-oxo) dicopper active species. Microporous Mesoporous Mater. 2014, 185, 113–120. [Google Scholar] [CrossRef]
- Knorpp, A.J.; Pinar, A.B.; Newton, M.A.; Li, T.; Calbry-Muzyka, A.; Van Bokhoven, J.A. Copper-exchanged large-port and small-port mordenite (MOR) for methane-to-methanol conversion. RSC Adv. 2021, 11, 31058–31061. [Google Scholar] [CrossRef]
- Plessers, D.; Heyer, A.J.; Rhoda, H.M.; Bols, M.L.; Solomon, E.I.; Schoonheydt, R.A.; Sels, B.F. Tuning Copper Active Site Composition in Cu-MOR through Co-Cation Modification for Methane Activation. ACS Catal. 2023, 13, 1906–1915. [Google Scholar] [CrossRef]
- Le, H.V.; Ho, P.H.; Trunschke, A.; Schomäcker, R.; Thomas, A. Stepwise conversion of methane to methanol on Cu and Fe/zeolites prepared in solid state: The effect of zeolite type and activation temperature. J. Chem. Technol. Biotechnol. 2023, 98, 2716–2725. [Google Scholar] [CrossRef]
- Hussain, I.; Ganiyu, S.; Alasiri, H.; Alhooshani, K. Highly dispersed Cu-anchored nanoparticles based mordenite zeolite catalyst (Cu-MOR): Influence of the different preparation methods for direct methane oxidation (DMTM) to methanol. J. Energy Inst. 2023, 109, 101269. [Google Scholar] [CrossRef]
- Gutman, E.M. Mechanochemistry of Solid Surfaces; World Scientific: Singapore, 1994. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu- and Fe-Exchanged Zeolites. ACS Catal. 2018, 8, 8306–8313. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Safonova, O.V.; Palagin, D.; Newton, M.A.; Van Bokhoven, J.A. Structure of copper sites in zeolites examined by Fourier and wavelet transform analysis of EXAFS. Chem. Sci. 2020, 11, 5299–5312. [Google Scholar] [CrossRef]
- Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J.; Nordlund, P.A. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 1993, 366, 537–543. [Google Scholar] [CrossRef]
- Chan, S.I.; Chen, K.H.C.; Yu, S.S.F.; Chen, C.L.; Kuo, S.S.J. Toward Delineating the Structure and Function of the Particulate Methane Monooxygenase from Methanotrophic Bacteria. Biochemistry 2004, 43, 4421–4430. [Google Scholar] [CrossRef]
- Chan, S.; Wang, V.; Lai, J.; Yu, S.; Chen, P.; Chen, K.; Chen, C.; Chan, M. Redox Potentiometry Studies of Particulate Methane Monooxygenase: Support for a Trinuclear Copper Cluster Active Site. Angew. Chem. Int. Ed. 2007, 46, 1992–1994. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Cation-exchanged zeolites for the selective oxidation of methane to methanol. Catal. Sci. Technol. 2018, 8, 114–123. [Google Scholar] [CrossRef]
- Göltl, F.; Bhandari, S.; Mavrikakis, M. Thermodynamics Perspective on the Stepwise Conversion of Methane to Methanol over Cu-Exchanged SSZ-13. ACS Catal. 2021, 11, 7719–7734. [Google Scholar] [CrossRef]
- Knorpp, A.J.; Pinar, A.B.; Baerlocher, C.; McCusker, L.B.; Casati, N.; Newton, M.A.; Checchia, S.; Meyet, J.; Palagin, D.; Van Bokhoven, J.A. Paired Copper Monomers in Zeolite Omega: The Active Site for Methane-to-Methanol Conversion. Angew. Chem. 2021, 133, 5918–5922. [Google Scholar] [CrossRef]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Nunthakitgoson, W.; Thivasasith, A.; Maihom, T.; Wattanakit, C. Effects of single and double active sites of Cu oxide clusters over the MFI zeolite for direct conversion of methane to methanol: DFT calculations. Phys. Chem. Chem. Phys. 2021, 23, 2500–2510. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhong, L.; Li, S. Catalytic cycle of the partial oxidation of methane to methanol over Cu-ZSM-5 revealed using DFT calculations. Phys. Chem. Chem. Phys. 2021, 23, 4963–4974. [Google Scholar] [CrossRef] [PubMed]
- Grundner, S.; Markovits, M.A.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Curtis, K.; Panthi, D.; Odoh, S.O. Time-Dependent Density Functional Theory Study of Copper(II) Oxo Active Sites for Methane-to-Methanol Conversion in Zeolites. Inorg. Chem. 2021, 60, 1149–1159. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; van Bokhoven, J.A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. Int. Ed. 2016, 55, 5467–5471. [Google Scholar] [CrossRef]
- Tomkins, P.; Ranocchiari, M.; Van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Accounts Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef]
- Mizuno, S.C.; Dulnee, S.; Pereira, T.C.; Passini, R.J.; Urquieta-Gonzalez, E.A.; Gallo, J.M.R.; Santos, J.B.; Bueno, J.M. Stepwise methane to methanol conversion: Effect of copper loading on the formation of active species in copper-exchanged mordenite. Catal. Today 2021, 381, 13–25. [Google Scholar] [CrossRef]
- Ma, C.; Tan, X.; Zhang, H.; Shen, Q.; Sun, N.; Wei, W. Direct conversion of methane to methanol over Cu exchanged mordenite: Effect of counter ions. Chin. Chem. Lett. 2020, 31, 235–238. [Google Scholar] [CrossRef]
- Bogdanov, D.S.; Novikov, R.G.; Pestsov, O.S.; Baranov, D.A.; Shelyapina, M.G.; Tsyganenko, A.A.; Kasatkin, I.A.; Kalganov, V.D.; Silyukov, O.I.; Petranovskii, V. Formation of admixed phase during microwave assisted Cu ion exchange in mordenite. Mater. Chem. Phys. 2021, 261, 124235. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Sushkevich, V.L.; Van Der Wal, L.I.; Bozbag, S.E.; Krumeich, F.; Ranocchiari, M.; Van Bokhoven, J.A. Increasing the activity of copper exchanged mordenite in the direct isothermal conversion of methane to methanol by Pt and Pd doping. Chem. Sci. 2019, 10, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Knorpp, A.J.; Sushkevich, V.L.; Palagin, D.; Van Bokhoven, J.A. Active sites and mechanisms in the direct conversion of methane to methanol using Cu in zeolitic hosts: A critical examination. Chem. Soc. Rev. 2020, 49, 1449–1486. [Google Scholar] [CrossRef] [PubMed]
- Knorpp, A.J.; Newton, M.A.; Mizuno, S.C.M.; Zhu, J.; Mebrate, H.; Pinar, A.B.; Van Bokhoven, J.A. Comparative performance of Cu-zeolites in the isothermal conversion of methane to methanol. Chem. Commun. 2019, 55, 11794–11797. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, T.; Hamill, C.D.; Goguet, A.; Rooney, D.W.; Thompson, J.M. A low temperature, isothermal gas-phase system for conversion of methane to methanol over Cu–ZSM-5. Chem. Commun. 2014, 50, 11053–11055. [Google Scholar] [CrossRef]
- Mojica Molina, H.R.; González Montiel, M.; Navarro Frómeta, A.E. Batch Conversion of Methane to Methanol Using Copper Loaded Mordenite: Influence of the Main Variables of the Process. Ing. Investig. 2021, 41, e87537. [Google Scholar] [CrossRef]
- Alayon, E.M.; Nachtegaal, M.; Ranocchiari, M.; Van Bokhoven, J.A. Catalytic conversion of methane to methanol over Cu–mordenite. Chem. Commun. 2012, 48, 404–406. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; van Bokhoven, J.A. The Effect of the Active-Site Structure on the Activity of Copper Mordenite in the Aerobic and Anaerobic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2018, 57, 8906–8910. [Google Scholar] [CrossRef]
- Flores-Valenzuela, J.; Leal-Perez, J.E.; Almaral-Sanchez, J.L.; Hurtado-Macias, A.; Borquez-Mendivil, A.; Vargas-Ortiz, R.A.; Garcia-Grajeda, B.A.; Duran-Perez, S.A.; Cortez-Valadez, M. Structural Analysis of Cu+ and Cu2+ Ions in Zeolite as a Nanoreactor with Antibacterial Applications. ACS Omega 2023, 23, 30563–30568. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Krylova, E.A.; Zhukov, Y.M.; Zvereva, I.A.; Rodriguez-Iznaga, I.; Petranovskii, V.; Fuentes-Moyado, S. Comprehensive Analysis of the Copper Exchange Implemented in Ammonia and Protonated Forms of Mordenite Using Microwave and Conventional Methods. Molecules 2019, 23, 4216. [Google Scholar] [CrossRef]
- Lamberti, C.; Bordiga, S.; Zecchina, A.; Salvalaggio, M.; Geobaldo, F.; Otero Areán, C. XANES, EXAFS and FTIR characterization of copper-exchanged mordenite. J. Chem. Soc. Faraday Trans. 1998, 94, 1519–1525. [Google Scholar] [CrossRef]
- Long, R.Q.; Yang, R.T. Selective Catalytic Reduction of NO with Ammonia over Fe3+-Exchanged Mordenite (Fe–MOR): Catalytic Performance, Characterization, and Mechanistic Study. J. Catal. 2002, 23, 274–285. [Google Scholar] [CrossRef]
- Schroeder, C.; Hansen, M.R.; Koller, H. Spatial Proximities between Brønsted Acid Sites, AlOH Groups, and Residual NH4+ Cations in Zeolites Mordenite and Ferrierite. J. Phys. Chem. C 2023, 23, 736–745. [Google Scholar] [CrossRef]
- Barreca, D.; Gasparotto, A.; Tondello, E. CVD Cu2O and CuO Nanosystems Characterized by XPS. Surf. Sci. Spectra 2007, 14, 41–51. [Google Scholar] [CrossRef]
- Artiglia, L.; Sushkevich, V.L.; Palagin, D.; Knorpp, A.J.; Roy, K.; Van Bokhoven, J.A. In Situ X-ray Photoelectron Spectroscopy Detects Multiple Active Sites Involved in the Selective Anaerobic Oxidation of Methane in Copper-Exchanged Zeolites. ACS Catal. 2019, 9, 6728–6737. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology. NIST X-Ray Photoelectron Spectroscopy Database. NIST Standard Reference Database Number 20. Available online: https://srdata.nist.gov/xps/ (accessed on 10 October 2024).
- Bergeret, G.; Gallezot, P.; Imelik, B. X-ray study of the activation, reduction, and reoxidation of palladium in Y-type zeolites. J. Phys. Chem. 1981, 85, 411–416. [Google Scholar] [CrossRef]
- Biesinger, M.C. Advanced analysis of copper X-ray photoelectron spectra. Surf. Interface Anal. 2017, 49, 1325–1334. [Google Scholar] [CrossRef]
- Grundner, S.; Luo, W.; Sanchez-Sanchez, M.; Lercher, J.A. Synthesis of single-site copper catalysts for methane partial oxidation. Chem. Commun. 2016, 52, 2553–2556. [Google Scholar] [CrossRef]
- Smeets, P.J.; Hadt, R.G.; Woertink, J.S.; Vanelderen, P.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Oxygen Precursor to the Reactive Intermediate in Methanol Synthesis by Cu-ZSM-5. J. Am. Chem. Soc. 2010, 132, 14736–14738. [Google Scholar] [CrossRef]
- Vanelderen, P.; Snyder, B.E.R.; Tsai, M.L.; Hadt, R.G.; Vancauwenbergh, J.; Coussens, O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. [Google Scholar] [CrossRef]
- Vanelderen, P.; Vancauwenbergh, J.; Tsai, M.; Hadt, R.G.; Solomon, E.I.; Schoonheydt, R.A.; Sels, B.F. Spectroscopy and Redox Chemistry of Copper in Mordenite. ChemPhysChem 2014, 15, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.E.R.; Vanelderen, P.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Second-Sphere Effects on Methane Hydroxylation in Cu-Zeolites. J. Am. Chem. Soc. 2018, 140, 9236–9243. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzis, K.D.; Li, G.; Hensen, E.J.M.; Gagliardi, L.; Pidko, E.A. Electronic Structure of the [Cu3(μ-O)3]2+ Cluster in Mordenite Zeolite and Its Effects on the Methane to Methanol Oxidation. J. Phys. Chem. C 2017, 121, 22295–22302. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Krylova, E.A.; Mazur, A.S.; Tsyganenko, A.A.; Shergin, Y.V.; Satikova, E.A.; Petranovskii, V. Active Sites in H-Mordenite Catalysts Probed by NMR and FTIR. Catalysts 2023, 13, 344. [Google Scholar] [CrossRef]
- Delabie, A.; Pierloot, K.; Groothaert, M.H.; Weckhuysen, B.M.; Schoonheydt, R.A. The siting of Cu(II) in mordenite: A theoretical spectroscopic study. Phys. Chem. Chem. Phys. 2002, 4, 134–145. [Google Scholar] [CrossRef]
- Narsimhan, K.; Michaelis, V.K.; Mathies, G.; Gunther, W.R.; Griffin, R.G.; Román-Leshkov, Y. Methane to Acetic Acid over Cu-Exchanged Zeolites: Mechanistic Insights from a Site-Specific Carbonylation Reaction. J. Am. Chem. Soc. 2015, 137, 1825–1832. [Google Scholar] [CrossRef]
- Ferrer, S.; Lloret, F.; Pardo, E.; Clemente-Juan, J.M.; Liu-González, M.; García-Granda, S. Antisymmetric Exchange in Triangular Tricopper(II) Complexes: Correlation among Structural, Magnetic, and Electron Paramagnetic Resonance Parameters. Inorg. Chem. 2012, 51, 985–1001. [Google Scholar] [CrossRef]
- Yoon, J.; Mirica, L.M.; Stack, T.D.P.; Solomon, E.I. Variable-Temperature, Variable-Field Magnetic Circular Dichroism Studies of Tris-Hydroxy- and μ3-Oxo-Bridged Trinuclear Cu(II) Complexes: Evaluation of Proposed Structures of the Native Intermediate of the Multicopper Oxidases. J. Am. Chem. Soc. 2005, 127, 13680–13693. [Google Scholar] [CrossRef]
- De Raedt, H.; Miyashita, S.; Michielsen, K.; Machida, M. Dzyaloshinskii-Moriya interactions and adiabatic magnetization dynamics in molecular magnets. Phys. Rev. B 2004, 70, 064401. [Google Scholar] [CrossRef]
- Mimmi, M.C.; Gullotti, M.; Santagostini, L.; Battaini, G.; Monzani, E.; Pagliarin, R.; Zoppellaro, G.; Casella, L. Models for biological trinuclear copper clusters. Characterization and enantioselective catalytic oxidation of catechols by the copper(II) complexes of a chiral ligand derived from (S)-(-)-1,1′-binaphthyl-2,2′-diamine. Dalton Trans. 2004, 2192–2201. [Google Scholar] [CrossRef]
- Mathivathanan, L.; Boudalis, A.K.; Turek, P.; Pissas, M.; Sanakis, Y.; Raptis, R.G. Interactions between H-bonded [CuII3(μ3-OH)] triangles; a combined magnetic susceptibility and EPR study. Phys. Chem. Chem. Phys. 2018, 20, 17234–17244. [Google Scholar] [CrossRef]
- Meyet, J.; Ashuiev, A.; Noh, G.; Newton, M.A.; Klose, D.; Searles, K.; Van Bavel, A.P.; Horton, A.D.; Jeschke, G.; Van Bokhoven, J.A.; et al. Methane-to-Methanol on Mononuclear Copper(II) Sites Supported on Al2O3: Structure of Active Sites from Electron Paramagnetic Resonance**. Angew. Chem. Int. Ed. 2021, 60, 16200–16207. [Google Scholar] [CrossRef] [PubMed]
- Carl, P.J.; Larsen, S.C. Variable-Temperature Electron Paramagnetic Resonance Studies of Copper-Exchanged Zeolites. J. Catal. 1999, 182, 208–218. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Artsiusheuski, M.; Klose, D.; Jeschke, G.; Van Bokhoven, J.A. Identification of Kinetic and Spectroscopic Signatures of Copper Sites for Direct Oxidation of Methane to Methanol. Angew. Chem. Int. Ed. 2021, 60, 15944–15953. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EPR Spectrum | Experimental Sample | g‖ Value | g⊥ Value | A‖ Hyperfine Constant (cm−1) |
|---|---|---|---|---|
| c | Cu3%400T (activated) | 2.385 | 2.099 | 138.63 × 10−4 |
| d | Cu3%400T6P (reacted with methane) | 2.379 | 2.097 | 138.46 × 10−4 |
| e | Cu3%400T6P + H2O (washed with water) | 2.391 | 2.114 | 136.00 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mojica, R.; González-Montiel, M.; Ramírez-Rosales, D.; Crespo-Barrera, P.M.; Navarro-Frómeta, A.E. Active Sites in Low-Loaded Copper-Exchanged Mordenite: Spectroscopic and Stability Study for Methane Oxidation Using Mild Conditions. Processes 2025, 13, 1795. https://doi.org/10.3390/pr13061795
Mojica R, González-Montiel M, Ramírez-Rosales D, Crespo-Barrera PM, Navarro-Frómeta AE. Active Sites in Low-Loaded Copper-Exchanged Mordenite: Spectroscopic and Stability Study for Methane Oxidation Using Mild Conditions. Processes. 2025; 13(6):1795. https://doi.org/10.3390/pr13061795
Chicago/Turabian StyleMojica, Rodrigo, Marlene González-Montiel, Daniel Ramírez-Rosales, Paula M. Crespo-Barrera, and Amado Enrique Navarro-Frómeta. 2025. "Active Sites in Low-Loaded Copper-Exchanged Mordenite: Spectroscopic and Stability Study for Methane Oxidation Using Mild Conditions" Processes 13, no. 6: 1795. https://doi.org/10.3390/pr13061795
APA StyleMojica, R., González-Montiel, M., Ramírez-Rosales, D., Crespo-Barrera, P. M., & Navarro-Frómeta, A. E. (2025). Active Sites in Low-Loaded Copper-Exchanged Mordenite: Spectroscopic and Stability Study for Methane Oxidation Using Mild Conditions. Processes, 13(6), 1795. https://doi.org/10.3390/pr13061795