Abstract
Zinc is a highly abundant cation in the brain, essential for cellular functions, including transcription, enzymatic activity, and cell signaling. However, zinc can also trigger injurious cascades in neurons, contributing to the pathology of neurodegenerative diseases. Mitochondria, critical for meeting the high energy demands of the central nervous system (CNS), are a principal target of the deleterious actions of zinc. An increasing body of work suggests that intracellular zinc can, under certain circumstances, contribute to neuronal damage by inhibiting mitochondrial energy processes, including dissipation of the mitochondrial membrane potential (MMP), leading to ATP depletion. Additional consequences of zinc-mediated mitochondrial damage include reactive oxygen species (ROS) generation, mitochondrial permeability transition, and excitotoxic calcium deregulation. Zinc can also induce mitochondrial fission, resulting in mitochondrial fragmentation, as well as inhibition of mitochondrial motility. Here, we review the known mechanisms responsible for the deleterious actions of zinc on the organelle, within the context of neuronal injury associated with neurodegenerative processes. Elucidating the critical contributions of zinc-induced mitochondrial defects to neurotoxicity and neurodegeneration may provide insight into novel therapeutic targets in the clinical setting.
1. Introduction
Zinc is a redox-inert divalent cation essential to a large number of biological processes, being the second most abundant trace element in the body, following iron [1]. Overall zinc levels are particularly high in the brain, where total zinc concentration may reach up to 150 µM [2]. Approximately 90% of the zinc present in the brain is protein-bound, contributing to the function of over 2000 proteins [3,4]. Indeed, zinc acts as a cofactor for over 300 enzymes, and, in the hippocampus alone, changes in cytosolic zinc can modulate the expression of over 900 genes [5,6], many of which are linked to the cell cycle, neurite extension, and synaptic growth [6,7,8,9]. Most of the remaining labile, nominally unbound or weakly bound zinc in the brain is present in synaptic vesicles of a large population of excitatory glutamatergic neurons throughout the cerebral cortex, hippocampus, striatum, and auditory brainstem [10,11,12]. Zinc is concentrated within synaptic vesicles by zinc transporter 3 (ZnT3) [13,14,15] and is synaptically released in an activity-dependent manner, acting as a neuromodulator for a number of neurotransmitter receptors [16,17,18,19,20]. Additionally, zinc released at glutamatergic synapses can translocate into postsynaptic cells through calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (CP-AMPARs) [21,22,23], voltage-gated calcium channels (VGCCs) [24,25], and N-methyl-D-aspartate receptors (NMDARs) [26], subsequently activating a number of physiological and pathophysiological signaling processes [19].
With the myriad cellular functions in which zinc plays an important role, disruption of zinc homeostasis can have severe detrimental effects. Nutritional zinc deficiency can lead to impaired brain development and cognitive dysfunction, including memory deficits and impaired learning ability later in life [27,28,29,30]. On the other hand, unregulated zinc release at glutamatergic synapses following traumatic brain injury (TBI), ischemia, and seizures has been implicated in excitotoxic damage and death of postsynaptic neurons [18,31,32,33,34]. Zinc dysregulation has been linked to increased risk for depression [35,36] and has been implicated in several neurodegenerative diseases, including Alzheimer’s disease (AD) [37,38,39,40,41,42], amyotrophic lateral sclerosis (ALS) [43,44], and Parkinson’s Disease (PD) [34,45,46,47]. Intracellular zinc concentrations are normally maintained at tightly controlled levels by a surprisingly large collection of fourteen SLC39a Zrt-/Irt-like protein transporters (ZIPs) and at least ten SLC30a zinc transporters (ZnTs), which transfer zinc into and out of the cytosol, respectively [1,48,49,50]. Intracellular zinc is also modulated by binding to cysteine-rich metallothioneins (MTs), which buffer and traffic the metal within cells [51,52]. Importantly, zinc liberated from MTs during oxidative stress conditions might represent a critical source for the metal in triggering several neurodegenerative changes [53,54,55,56]. Moreover, neuronal mitochondria may transiently contribute to the endogenous zinc pool [57], which can be released in response to injurious stimuli [57,58].
Zinc plays an important role in mitochondrial function, with neurons being notably reliant on this organelle to fulfill the high energy demands of the central nervous system (CNS). Indeed, it has been estimated that individual cortical neurons use approximately 4.7 billion ATP molecules per second compared to the 10 million ATP molecules used by non-neuronal cells in the human brain [59]. Under physiological conditions, zinc likely regulates mitochondrial processes in all tissues, including glycolysis, the tricarboxylic acid (TCA) cycle, and the electron transport chain (ETC) [60,61,62,63]. For instance, zinc increases the production of lactate and glycolytic intermediates in muscle cells and hepatocytes, indicating that it may act as an activating cation for phosphofructokinase (PFK) activity in glycolysis [62,63]. There is also evidence to suggest that hepatic zinc deficiency can inactivate mitochondrial biogenesis and DNA replication, leading to decreased expression of mitochondrial respiratory complexes I, III, and IV and decreased production of reactive oxygen species (ROS) [61]. In fact, dietary zinc protects against oxidative stress and ETC enzyme damage following postnatal protein malnutrition in rats [64]. Furthermore, in a mouse model of AD, dietary zinc supplementation has been shown to restore impaired mitochondrial respiration and increase levels of brain-derived neurotrophic factor (BDNF), which itself is linked to mitochondrial dynamics and oxidative efficiency [37,65,66,67]. The importance of zinc for energy metabolism is further supported by the fact that zinc chelation by N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN) results in a 50% reduction of ATP production in hepatocytes [68].
However, zinc overload by entry through CP-AMPARs, VGCCs, and NMDARs at glutamatergic synapses, as well as release from intracellular zinc-binding proteins [40,53,63], may cause deleterious changes in intracellular zinc levels that contribute to neuronal injury and death, in part by inducing mitochondrial dysfunction [69,70,71,72,73,74,75]. Consequences of zinc-induced disruptions of cellular energy processes include decreased mitochondrial membrane potential (MMP), increased ROS generation, and mitochondrial permeability transition, resulting in the release of pro-apoptotic factors [71,74,76,77,78,79,80]. Synergism between zinc and calcium may exacerbate these effects [34,75,80,81,82,83,84,85]. There is also evidence to suggest that excess zinc may disrupt mitochondrial fusion, fission, and trafficking [86,87]. The overarching purpose of this review is thus to provide a comprehensive update of the current state of knowledge on the multifaceted effects of zinc on mitochondrial function and dynamics within the context of neuronal health, pointing to potential methods of protection against neurodegeneration.
2. Zinc and Bioenergetic Function
Despite constituting only about 2% of human body weight, the brain utilizes approximately 25% of total glucose expenditure to meet its incredible energy demands [88]. The conversion of glucose and oxygen to ATP, carbon dioxide, and water takes place by glycolysis, the TCA cycle, and the ETC [88]. Glycolysis is a ten-step cytosolic pathway that converts glucose to pyruvate, ADP to ATP, and nicotinamide adenine dinucleotide (NAD+) to reduced nicotinamide adenine dinucleotide (NADH) [89,90,91]. Pyruvate is oxidized and utilized in the TCA cycle, a series of chemical reactions in the mitochondrial matrix that releases energy from acetyl-CoA [89,90,91]. This released energy is stored in electron carriers NADH and flavin adenine dinucleotide (FADH2), which are utilized by complexes of the ETC [89,90,91]. NADH donates its electrons to complex I, and FADH2 donates its electrons to complex II. These electrons are transferred in succession to ubiquinone (coenzyme Q), complex III, the mobile electron carrier cytochrome C, complex IV, and the final electron acceptor, oxygen [92]. Energy released from the ETC fuels the pumping of protons from the mitochondrial matrix to the intermembrane space (IMS), which establishes the proton motive force. The flow of protons down their concentration gradient drives ATP synthase, which catalyzes the phosphorylation of ADP to ATP [92]. Excess zinc has been shown to impair energy metabolism by inhibiting components of glycolysis [93], the TCA cycle [94,95], and the ETC [77].
2.1. Glycolysis
Elevated intracellular zinc has been shown to impair the glycolytic enzymes PFK and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [73,96,97]. PFK catalyzes the conversion of fructose 6-phosphate (F6P) and ATP to fructose 1,6-bisphosphate (FBP) and ADP [98]. GAPDH converts glyceraldehyde 3-phosphate (G3P) to 1,3-bisphosphoglycerate (1,3 BPG) and NAD+ to NADH [99]. There is evidence to suggest that zinc may directly inhibit GAPDH. Exposure of cortical neurons to neurotoxic levels of zinc results in a buildup of the upstream intermediates dihydroxyacetone phosphate (DHAP) and FBP, as well as a fall in ATP levels and subsequent cell death [93]. However, zinc inhibition of GAPDH may also occur by an indirect mechanism by which zinc induces a decrease in NAD+ levels, sufficient to inhibit GAPDH in and of itself. This decrease in NAD+ similarly leads to decreased ATP levels and neuronal death in cortical cultures exposed to zinc [93]. In support of this mechanism, the addition of pyruvate, which can regenerate NAD+ through lactic acid fermentation, rescues neurons exposed to toxic levels of zinc [93]. Notably, pyruvate provides protection against zinc toxicity in rat cortical cultures as well as reduced neuronal death and mortality in a rat model of ischemic injury [100].
Although there is no currently known pathology entirely attributed to zinc-mediated GAPDH inhibition, some neurological diseases have been associated with impaired GAPDH function. For instance, inhibition of GAPDH enzymatic activity causes dissociation of NAD+, which destabilizes GAPDH, making it prone to damage by denaturation and aggregation [101]. Denatured GAPDH binds to amyloidogenic proteins, which can contribute to the pathology of neurodegenerative diseases such as PD and AD [101,102,103]. Moreover, GAPDH aggregates in the brain have been proposed as a target for TBI treatment [104]. Of interest, functional GAPDH also promotes mitophagy of damaged mitochondria following cardiac ischemia. As such, disruption of GAPDH function may lead to the accumulation of damaged mitochondria and apoptosis [105], as observed in Huntington’s Disease (HD)-associated pathology [106].
2.2. TCA Cycle
Zinc impairment of the TCA cycle may occur by inhibition of the α-ketoglutarate dehydrogenase complex (KGDHC), a mitochondrial enzyme that catalyzes the conversion of α-ketoglutarate, NAD+, and CoA to succinyl-CoA, carbon dioxide, and NADH [73,94,107]. Zinc inhibition of the KGDHC may have significant pathological consequences, as deficiency of KGDHC activity has been linked to neurodegenerative diseases, including Friedreich’s ataxia, PD, and AD [108,109,110,111,112]. Zinc inhibition of the KGDHC likely occurs via the inactivation of lipoamide dehydrogenase (LADH) [73,78,113]. Indeed, zinc inhibits LADH with an IC50 near 0.15 µM in purified bovine enzyme, consistent with the predicted IC50 value for the KGDHC (0.1–0.4 µM) [94,113]. It is worth noting, however, that studies linking zinc to LADH inhibition were performed using isolated liver mitochondria and thus may not be necessarily translatable in neurons. Indeed, this is an important caveat to consider for all non-neuronal studies described in this review, as brain mitochondria have been shown to respond differently than organelles from other tissues, such as liver [114]. Nonetheless, it is of interest to investigate how zinc binds to and inhibits LADH in a neuronal preparation, and whether it contributes to persistently altered energy metabolism following neuronal injury [115,116].
Moreover, LADH is also a component of the pyruvate dehydrogenase complex (PDHC), which converts pyruvate, NAD+, and coenzyme A to acetyl-CoA, carbon dioxide, and NADH [107,117,118]. Interestingly, studies in SN56 cholinergic neuroblastoma cells suggest that zinc may inhibit the PDHC in a concentration-dependent manner, resulting in decreased acetyl-CoA levels and loss of cell viability [119,120,121,122]. Reduced activity of the PDHC has been associated with neuronal death following ischemia [118,123], leaving it open to speculation that zinc inhibition of LADH in the PDHC may play a role in neurodegenerative disease pathology.
Mitochondrial aconitase (mACN), which catalyzes the conversion of citrate to isocitrate in the TCA cycle, is another potential mitochondrial site of zinc inhibition [95]. After toxic zinc exposure, albeit in hepatocytes, there was an observed buildup of citrate and decreased mitochondrial mACN expression, reflective of decreased mACN activity [95]. This is consistent with prior results indicating that zinc shifts equilibrium of the mACN-catalyzed reaction toward the reactant citrate in prostate secretory epithelial cells [124]. The exact mechanism of mACN inhibition is not yet fully elucidated, although it has been suggested to be the result of zinc displacement of iron at Fe-S sites of the enzyme [95]. Because zinc inhibition of other mitochondrial components causes MMP loss and ROS generation [77], we suggest that decreased mACN activity due to zinc may also be the result of these injurious processes. Indeed, mACN inactivation has been linked to MMP loss [125] and ROS accumulation [126]. However, it is worth noting that MMP loss was induced by excess calcium, not zinc, and the study was conducted using a human prostate cancer cell line, which, as noted earlier, may have significantly different biology than neurons [125,126].
2.3. Electron Transport Chain
Zinc has also been shown to inhibit the ETC, of which the mechanism and effects in mitochondria isolated from non-neuronal cell types [127,128] have been previously discussed [73]. Nonetheless, the effects of zinc on the ETC have also been demonstrated in rat brain cells [77]. Sub-micromolar levels of zinc have produced effects consistent with ETC inhibition, such as inhibited oxygen consumption and dissipated MMP [77]. The most probable site of zinc inhibition is complex III, as zinc binds with high affinity to a site in that complex that facilitates the carrying of protons across the inner mitochondrial membrane (IMM) [127,129]. Of relevance, complex III deficits have been linked to the development of numerous neurodegenerative disorders [130,131]. Moreover, complex III inhibition can increase glutamate release at the nerve terminal and contribute to HD-associated excitotoxicity [132,133,134].
Another putative target of zinc to consider is inhibition of complex I [135]. Even partial complex I inhibition can reduce nerve terminal oxygen consumption, increase glutamate release from synaptosomes, and potentially induce calcium-dependent excitotoxicity [136,137]. While zinc has been shown to inhibit complex I in bovine heart mitochondria, it is worth noting that this effect occurred at relatively high concentrations of the metal (10–50 µM), which may not be physiologically relevant [135]. However, one must consider the possibility that zinc inhibition of complex I could be of clinical significance at elevated microdomain concentrations during pathological conditions. It has been well established that complex I inhibition has been linked to neurodegenerative conditions, including brain ischemia/reperfusion injury [138], ALS [139], and PD [140,141,142].
3. Zinc and ROS Generation
One of the consequences of mitochondrial dysfunction is the generation of ROS, which are highly reactive molecules derived from oxygen [143,144]. Being byproducts of aerobic metabolic processes, most ROS products are formed by the reduction of oxygen to superoxide, which not only is heavily implicated in excitotoxic injury [145,146], but also serves as the precursor to other, perhaps more toxic ROS such as hydrogen peroxide, hydroxyl radicals, and peroxynitrite [143,147,148]. Although ROS can be generated by non-mitochondrial sources such as NADPH oxidase (NOX) [149,150], almost 90% of ROS are generated by the mitochondria [151,152,153,154,155]. Overproduction of ROS leads to oxidative stress, which in turn leads to the damage or destruction of proteins, lipids, genetic material, and cellular organelles [156]. Given the evidence for zinc-induced mitochondrial dysfunction and the fact that mitochondria are believed to be major generators of ROS in neurons, it is thus not surprising that zinc has been repeatedly shown to induce ROS production [70,71,77,157,158,159,160,161].
Zinc-induced mitochondrial ROS production has been demonstrated in mouse cortical cultures [71], possibly via inhibition of the LADH component of KGDHC in the TCA cycle [113,162]. LADH typically catalyzes the reduction of NAD+ to NADH. However, zinc inhibits LADH and accelerates the reverse reaction, favoring the oxidation of NADH by available oxygen in the mitochondrial matrix [113]. This reaction produces superoxide, which is converted to hydrogen peroxide by superoxide reductase. ETC components are also potential sources of mitochondrial ROS production. Zinc inhibition of complex III has also been posed as a likely cause of increased ROS levels in rat brain mitochondria [77]. This hypothesis is consistent with the finding that in mitochondria isolated from rat hearts, complex III inhibition seemed to be the main site for mitochondrial ROS production [163]. Inhibition of complex III using antimycin A resulted in increased ROS generation, an effect attenuated by preventing electron flow from complex I and II to complex III [163]. Together, these findings suggest that zinc-induced ROS production may be a result of zinc inhibition of TCA cycle and ETC components.
Non-mitochondrial sources of zinc-induced ROS generation have also been identified, such as the upregulation of NOX [159], a membrane protein that catalyzes the conversion of NADPH and oxygen to NADP+, a hydrogen ion, and superoxide [164]. A study in astrocytes concluded that zinc induces cell death primarily by increasing mitochondrial ROS production, but not by NOX activation, as inhibition of NOX-derived ROS reversed the neurotoxic effects of zinc to a lesser extent than inhibition of mitochondrial-derived ROS [70]. Non-mitochondrial ROS sources are nevertheless important due to the interplay between mitochondrial and non-mitochondrial ROS sources [158,160]. For example, zinc-induced NOX activation triggers mitochondrial ROS production in a HeLa cell model of hypoxia [160,161]. Conversely, zinc-induced mitochondrial ROS production causes NOX upregulation via activation of the NF-κB pathway in vascular smooth muscle cells [158]. Thus, mitochondrial and non-mitochondrial ROS generation may synergistically contribute to zinc-induced oxidative stress.
4. Zinc and Mitochondrial Permeability Transition
Another consequence of mitochondrial dysfunction is mitochondrial permeability transition, caused by the opening of the mitochondrial permeability transition pore (mPTP) [165], a relatively large, nonselective pore on the IMM [166]. mPTP opening occurs due to MMP loss, calcium deregulation, and oxidative stress [167]. Mitochondrial permeability transition causes the release of mitochondrial calcium stores, potentiating excitotoxic damage caused by elevated intracellular calcium [168,169,170] and resulting in the release of pro-apoptotic factors [171]. Mitochondrial permeability transition also allows for the mitochondrial influx of protons and other molecules smaller than 1.5 kD, leading to ETC uncoupling, MMP loss, and ATP depletion [172]. These effects may, in turn, induce ROS generation, amplifying mitochondrial permeability transition-induced cellular damage [167,173,174]. Importantly, excess zinc has been shown to be another trigger for mPTP opening [74,76,175].
Early evidence for zinc-induced mPTP opening showed that 2–10 µM zinc induces swelling of rat liver mitochondria, a marker of mitochondrial permeability transition [76]. In isolated brain mitochondria, zinc concentrations as low as 10 nM have been shown to cause effects consistent with mitochondrial permeability transition, including rapid mitochondrial swelling and release of the pro-apoptotic factors cytochrome C and apoptosis-inducing factor [74]. Although these studies suggest that zinc causes mitochondrial permeability transition by directly binding to and opening the mPTP, it is also possible that zinc may cause mitochondrial permeability transition indirectly by inhibiting enzymes of cellular energy processes and ROS production [78]. Indeed, zinc inhibition of matrix enzymes LADH, a component of KGDHC in the TCA cycle, and glutathione reductase (GR) and thioredoxin reductase (TR), which protect against oxidative stress, precedes changes associated with mPTP opening [78]. The extent and rate of zinc inhibition of these enzymes directly correlate with mPTP opening onset [78], suggesting that oxidative stress resulting from inhibition of these enzymes could induce mPTP opening [78,167].
Interestingly, a study comparing the effects of zinc and calcium on MMP loss in isolated liver mitochondria demonstrated that mPTP modulators reduced calcium-induced MMP loss but, surprisingly, did not attenuate the effects of zinc, suggesting that zinc does not open the mPTP [176]. Supporting this conclusion, the fluorophore calcein, a marker of mitochondrial permeability transition, was released from mitochondria treated with 30 µM calcium, but not with 30 µM zinc [176]. One potential explanation for the lack of mitochondrial permeability transition observed in this study [176] is the presence of zinc contamination in physiological solutions, such that low levels of zinc in control experiments may have already induced mitochondrial permeability transition, making additional zinc-mediated mitochondrial permeability transition difficult to observe [73]. A more likely explanation, however, is the absence of calcium in solutions utilized in experiments reporting a lack of zinc-induced mitochondrial permeability transition, since resting levels of calcium are required to initiate the opening of the mitochondrial calcium uniporter (MCU) [177,178]. The MCU is a likely conduit for zinc influx into the mitochondria, thereby initiating mitochondrial permeability transition [82], as discussed in more detail below.
5. Zinc Transport and Synergy with Calcium
Glutamate-induced calcium entry through NMDARs, CP-AMPARs, and VGCCs leads to changes in intracellular calcium that can result in excitotoxic neuronal cell death [34,179,180,181]. Mitochondria are a major target for this excitotoxic damage [182]. Calcium accumulation in the mitochondria, which occurs through the voltage-dependent anion-selective channel protein 1 (VDAC1) in the outer mitochondrial membrane (OMM) [183,184] and mitochondrial calcium uniporter in the IMM [185], causes impaired mitochondrial respiration, oxidative stress, mitochondrial permeability transition, and release of pro-apoptotic peptides [186]. Interestingly, due to the high affinity of calcium indicators for zinc [187], it has been suggested that some of the excitotoxic effects attributed to calcium may actually be caused by zinc [188]. Due to the development of higher-affinity zinc-selective indicators and low-affinity calcium indicators relatively unaffected by high zinc [189], the effects of these two ions on mitochondrial function can be investigated simultaneously. An increasing body of evidence using these improved detection methods suggests that excitotoxicity-induced mitochondrial dysfunction and subsequent cell death may be a result of the synergistic actions of zinc and calcium [34,75,80,81,82,83,84,85].
5.1. Synaptic Zinc
Zinc released at glutamatergic synapses enters the postsynaptic cell through CP-AMPARs [21,22,23], VGCCs [24,25], and, to some extent, NMDARs [26]. Interestingly, the type of channel through which synaptic zinc enters the cell may influence how the ion interacts with calcium and causes mitochondrial dysfunction. Whereas preferential zinc entry through CP-AMPARs causes acute ROS generation and MMP loss [71,79], smaller zinc loads caused by zinc entry through VGCCs cause more prolonged mitochondrial zinc accumulation and dysfunction [25,83]. The effects of this prolonged mitochondrial zinc accumulation may include mitochondrial swelling and release of pro-apoptotic factors [74]. The extended time durations associated with these mitochondrial defects are consistent with slow cell death after neuronal injury [190]. However, when associated with impaired cytosolic zinc buffering, as exhibited during ischemia, zinc entry through VGCCs can also cause acute mitochondrial dysfunction [85]. The presence of physiological levels of calcium caused greater mitochondrial dysfunction and neurotoxicity than zinc alone, suggesting calcium exacerbation of acute mitochondrial defects caused by zinc entry through VGCCs [85]. Following entry through CP-AMPARs, zinc has been shown to contribute to excitotoxic calcium deregulation and subsequent mitochondrial defects [31,80]. In support of zinc facilitation of calcium deregulation, small elevations in cytosolic zinc precede sharp calcium rises and MMP loss in CA1 neurons during oxygen-glucose deprivation (OGD), leading to subsequent cell death [80]. Furthermore, zinc chelation with TPEN significantly delayed the excitotoxic effects of calcium deregulation and facilitated the recovery of MMP [80].
The contribution of zinc to calcium deregulation likely takes place following zinc uptake into the mitochondria [80,191]. Numerous studies support the theory that mitochondrial zinc entry through the MCU is a precursor to zinc-induced mitochondrial dysfunction, calcium deregulation, and excitotoxic damage [75,79,80,82,84,192]. Indeed, blocking mitochondrial zinc entry through the MCU using ruthenium red (RR) has been shown to attenuate calcium deregulation [75]. Furthermore, neurons from MCU knockout mice exhibit significantly reduced mitochondrial zinc accumulation and protection against associated mitochondrial defects [82]. This attenuation of mitochondrial dysfunction and neurotoxicity was also demonstrated after wild-type neurons exposed to toxic levels of zinc were treated with RR [82]. In contrast to these data, a study found no evidence of mitochondrial zinc influx prior to mitochondrial depolarization [176]. However, as noted earlier, one potential explanation for the discrepancy is the lack of calcium in the experimental solutions used in this study, a requirement for activation of the MCU [177,178]. The fact that mitochondrial zinc entry is likely dependent on calcium underscores the synergistic relationship between zinc and calcium, where calcium-dependent mitochondrial zinc accumulation induces mitochondrial dysfunction and calcium deregulation [31,75,80]. Because zinc entry through the MCU contributes to calcium-induced excitotoxicity, inhibition of mitochondrial zinc accumulation may be an important potential target for neuroprotection after ischemia [82,192]. The MCU as a target for intervention after ischemia may be of particular promise in CA1 neurons, whose prolonged mitochondrial accumulation after reperfusion may contribute to their greater vulnerability than CA3 neurons to ischemic injury [193]. Of interest, the source of zinc damaging CA1 neurons following ischemia may be intracellular, rather than synaptic, in origin.
5.2. Cytosolic Zinc Pools
In addition to synaptic zinc entry through CP-AMPARs and VGCCs, zinc liberation from cytosolic pools such as MTs [53,54,55,56] can also cause rises in intracellular zinc that potentiate mitochondrial dysfunction and neuronal death [57]. In fact, after 2,2’-dithiodipyridine (DTDP)-induced zinc release from MTs, disrupting intracellular zinc buffering, far lower levels of zinc were required to cause mitochondrial dysfunction, ROS generation, and neuronal cell death [84,85]. As mentioned above, zinc liberation from metallothionein-III (MT-III) has been shown to be a major contributor to the increased susceptibility of CA1 neurons to ischemic injury compared to CA3 neurons [193], providing further support for the importance of intracellular zinc mobilization in neurodegeneration.
Zinc release from MTs is typically thought to be caused by oxidative stress and acidosis [194,195]. Importantly, calcium entry through VGCCs or NMDARs and subsequent mitochondrial ROS generation may serve as an oxidative trigger for the mobilization of these cytosolic zinc pools [72]. Indeed, inhibition of calcium-induced mitochondrial depolarization and ROS formation by cyanidin-3-glucoside protected against glutamate-induced zinc release from intracellular pools in rat hippocampal neurons [196]. Interestingly, mitochondrial calcium entry may also be sufficient to cause zinc release from the mitochondria [83]. However, whether the mitochondria normally serve as an endogenous zinc pool without injury-mediated prior influx remains to be established.
5.3. Mitochondrial Zinc
There is significant colocalization of zinc and mitochondria following zinc loading [57]. Interestingly, in SN56 cholinergic neuroblastoma cells, about 12–20% of cellular zinc localized to the mitochondria, irrespective of zinc treatment conditions [191]. As previously discussed, mitochondrial zinc entry occurs through the MCU [75,79,80,82,84,192]. However, there may also exist MCU-independent mechanisms for mitochondrial zinc accumulation [197]. For instance, MTs have been observed to be localized in the IMS of isolated liver mitochondria [198], although it should be noted that MTs are absent from the IMS of isolated heart mitochondria [58]. However, MTs may localize to the matrix in mouse heart mitochondria, where they directly transfer zinc to mACN [199]. The localization of MTs in neurons has, unfortunately, not been adequately investigated.
An intriguing subject that has been poorly studied is the possible presence of zinc transporters (ZnTs and ZIPs) in neuronal mitochondria. Indeed, mitochondrial localization of ZnT2 has been demonstrated in mouse mammary cells [200], although a conflicting study found no colocalization of ZnT2 with mitochondria in human breast cancer cells [201]. Furthermore, ZnT1, ZnT4, and ZIP8 localize to mitochondria in rat hepatocytes [202], and ZIP7 and ZnT7 in rat cardiomyocytes [203]. Interestingly, a study using HeLa cells demonstrated that mitochondria-localized ZIP1 may associate with the MCU to regulate zinc influx into the mitochondria [204]. However, the distribution of ZnTs and ZIPs is highly tissue-dependent, and some of the mitochondrial zinc transporters investigated in these non-neuronal studies, such as ZnT2, may not be highly expressed in the brain [49,50]. Nonetheless, there are a number of ZnTs and ZIPs known to transport cellular zinc in the brain, such as ZnT1, ZnT3-ZnT6, ZIP1, and ZIP12 [1,48,49,50]. Thus, we believe that the potential mitochondrial localization of neuronal zinc transporters is an important component of neuronal mitochondrial zinc physiology that has yet to be adequately explored.
6. Zinc and Mitochondrial Dynamics
Mitochondria are highly dynamic, endosymbiotically-derived organelles [205] that undergo the opposing yet coordinated processes of fusion and fission [206,207]. Fusion and fission, which often occur quickly, simultaneously, and continuously, influence the morphology, number, and distribution of mitochondria in cells [208,209]. Fusion plays a role in repairing mitochondria during conditions of high energy demand [209,210,211,212,213,214], and fission mediates the selective destruction of damaged mitochondria by the process of mitophagy [215]. Fission also generates new, functional mitochondria [209] and may play a role in reducing oxidative stress-induced mitochondrial swelling and cell death in neurons [216]. Both fusion and fission are controlled by guanosine triphosphatases (GTPases), such as dynamin-related protein 1 (DRP1), mitofusin 1 (MFN1), mitofusin 2 (MFN2), and optic atrophy 1 (OPA1) [208,209,217]. Impaired fusion-fission balance caused by deregulation of these proteins can have detrimental effects on the mitochondrial function, morphology, and trafficking. Loss of fission impairs ETC complex assembly and oxidative phosphorylation [212,218], excess fusion results in mitochondrial elongation, and excess fission results in mitochondrial fragmentation [217,219]. Altered fusion and fission also disrupt mitochondrial transport to dendrites and axon terminals [220,221], where the highest neuronal activity occurs [222]. Driven by the high energy demands of growth cones and dendritic spines, mitochondria are trafficked to these distal areas [223,224,225], where they are required for axonal growth, development, and branching [66,226,227,228,229,230]. Impairments of mitochondrial trafficking cause energy depletion at typically mitochondria-dense sites, resulting in stunted dendritic development, decreased synaptic strength, reduced axonal regeneration, and neuronal cell death [229,231,232,233]. Both mitochondrial fragmentation caused by excess fission and impaired mitochondrial trafficking are implicated in the development of various neurodegenerative diseases [234,235,236,237].
6.1. Fission, Fusion, and Morphology
Zinc causes mitochondrial morphology changes such as mitochondrial fragmentation, evidenced by the formation of small, round mitochondrial puncta [238,239,240,241]. Zinc impairment of fusion–fission dynamics likely plays an important role in causing these morphological changes, as mitochondrial fragmentation and elongation are caused by excess fission and fusion, respectively [217,219]. Activation of DRP1, which plays a critical role in fission [242], has been shown to contribute to mitochondrial fragmentation, and recent studies have demonstrated that zinc causes mitochondrial fragmentation via DRP1 activation in ß cells [239,240,243]. Moreover, zinc induces DRP1-mediated fission, resulting in mitochondrial fragmentation and mitochondrial network disruption in brain endothelial cells [241]. Interestingly, several studies in ß cells, endothelial cells, and neurons suggest that DRP1 activation may be caused by ROS-induced transient receptor potential cation channel, subfamily M, member 2 (TRPM2)-mediated lysosomal zinc release, suggesting the existence of an ROS-zinc-DRP1 pathway that results in mitochondrial fission and fragmentation [240,241,243,244,245]. The mechanism for zinc recruitment of DRP1, however, remains unclear. Additionally, because several of these studies were conducted using non-neuronal cell types, it is of interest to investigate zinc-induced mitochondrial fission and fragmentation in neurons.
Independent from zinc recruitment of DRP1 to cause mitochondrial fragmentation, zinc may also play a role in DRP1-associated mitochondrial surveillance during the fission-mediated process of mitophagy in HeLa cells [204,246,247]. Following DRP1 recruitment, DRP1 interacts with the Zip1 component of an intriguing Zip1-MCU complex, allowing for mitochondrial zinc influx, leading to a reduction in MMP [204]. A novel model of mitochondrial surveillance suggests that this MMP loss is used as a stress test to segregate damaged mitochondria from healthy mitochondria [204]. According to this proposed form of mitochondrial surveillance, mitochondria that can re-establish their MMP are deemed healthy, whereas mitochondria that do not recover are eliminated by mitophagy [204]. The mechanism by which DRP1 interacts with Zip1-MCU to allow mitochondrial zinc influx is unclear. Furthermore, whether excess zinc interferes with this mitochondrial surveillance mechanism has not yet been demonstrated in neurons, although it is an intriguing topic of future study due to the extent that DRP1 activation and mitophagy have been implicated in the pathology of various neurodegenerative diseases [235], such as AD [234,248], PD [249,250,251], HD [252,253], ALS [254], and ischemia [255,256].
6.2. Motility
Excess zinc impairs mitochondrial trafficking in rat neurons [86,225]. Both zinc and pyrithione (a zinc ionophore) treatment and DTDP-induced cytosolic zinc release from MTs caused a significant decrease in mitochondrial movement, suggesting that the zinc sources responsible for these effects may be both extracellular and intracellular [86]. Moreover, zinc chelation with TPEN partially restored mitochondrial movement in cells treated with zinc and pyrithione, suggesting reversibility of this effect [86]. However, it is worth noting that the reversal of zinc inhibition of mitochondrial movement was only partially sensitive to zinc chelation. TPEN failed to restore mitochondrial movement when neurons were exposed to high concentrations of zinc or were exposed to the metal for longer durations. These data suggest the effects of zinc chelation on the restoration of mitochondrial movement are both concentration- and time-dependent, likely reflecting the activation of a downstream signaling cascade [86]. A later study, using primary cortical neurons, supported the idea that elevation of intracellular zinc disrupts mitochondrial transport in neurites [225]. Interestingly, this study showed that there may be spatial differences in the ability of zinc chelation to restore mitochondrial movement, as TPEN restored axonal but not dendritic mitochondrial movement [225]. However, in accordance with previous data, this study used a concentration of zinc that was previously shown to irreversibly impair mitochondrial movement in dendrites [86]. Thus, the extent of zinc inhibition of mitochondrial movement may be concentration-, time-, and location-dependent. Although the mechanism for zinc-induced decrease in mitochondrial movement is not yet fully elucidated, it may involve zinc activation of phosphatidylinositol 3-kinase (PI3K), as PI3K inhibition restored mitochondrial movement following zinc treatment at concentrations and durations insensitive to chelation [86]. It is worth noting that this effect was independent of changes in mitochondrial morphology or function [86].
7. Conclusions
In this review, we have summarized existing knowledge on the myriad effects of zinc on mitochondrial function and dynamics in the context of neuronal health. Notably, zinc disrupts cell metabolism by inhibiting components of glycolysis, the TCA cycle, and the ETC. These zinc-induced impairments in bioenergetics lead to loss of MMP and decreased ATP production, which leads to neuronal cell death. Zinc also causes mitochondrial and non-mitochondrial ROS generation, which may work synergistically to induce oxidative stress. Zinc-induced mPTP opening is another facet of mitochondrial dysfunction, resulting in MMP loss, mitochondrial swelling, and release of apoptosis-promoting factors. It is important to note that many of these previously described mitochondrial defects follow mitochondrial zinc entry through the MCU and that zinc and calcium synergistically induce mitochondrial dysfunction. Finally, zinc impairs mitochondrial dynamics: zinc-induced fission results in mitochondrial fragmentation, and zinc inhibition of mitochondrial movement results in impaired mitochondrial trafficking. Although the potential interactions between these zinc-induced mitochondrial defects and their contributions to neurotoxic cascades are not fully understood, these effects may cause cell death and neurodegeneration. Zinc inhibition of cellular energy processes, subsequent mitochondrial defects, and changes in mitochondrial morphology and trafficking are summarized in Figure 1.
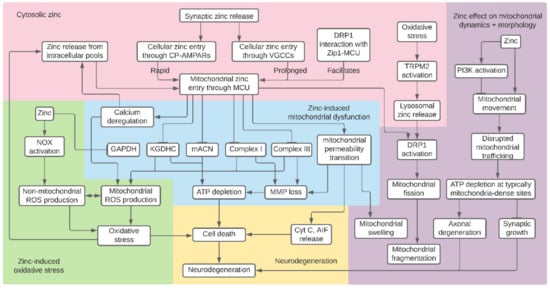
Figure 1.
Summary of proposed mitochondrial effects of zinc. Synaptic zinc entry and release of zinc from cytosolic pools causes rises in intracellular zinc, which accumulates in the mitochondria. Zinc inhibition of components of glycolysis, the tricarboxylic acid (TCA) cycle, and the electron transport chain (ETC) result in mitochondrial membrane potential (MMP) loss and decreased ATP production. Other effects of excess zinc include reactive oxygen species (ROS) generation, mitochondrial permeability transition, and calcium deregulation. Finally, zinc inhibits mitochondrial movement and causes mitochondrial fragmentation by initiating excess fission. It is not clear if these pathways interact to induce their neurotoxic effects. Nevertheless, zinc-induced mitochondrial detriments may collectively result in cell death and neurodegeneration.
While much progress has been made in understanding the effect of zinc on mitochondrial function, the work to elucidate the relationship between zinc, mitochondria, and neurodegeneration is far from over. As previously noted, mitochondrial defects can be cell type-dependent. For instance, brain mitochondria are less susceptible to mitochondrial permeability transition than liver mitochondria [114]. Thus, the findings from any non-neuronal studies described in this review should be interpreted cautiously when applied to neurons. Nevertheless, non-neuronal studies have historically provided insight into similar processes occurring in the brain.
Due to the lack of specificity of certain metal chelators and indicators in the past, there have been difficulties in assigning mitochondrial defects such as mitochondrial permeability transition to either zinc or calcium. In fact, the possible attribution of zinc-induced mitochondrial defects to calcium may underlie the clinical inefficacy of treating ischemia and other neurodegenerative diseases by targeting only calcium-associated excitotoxicity using NMDAR antagonists [257,258,259]. Thus, investigating zinc and calcium in relation to one another may provide new insights into the mechanisms by which these ions work together to contribute to mitochondrial defects such as mitochondrial permeability transition. Excitingly, the development of various novel tools and methods for detecting cytosolic and mitochondrial zinc [260], as well as more selective and fast chelators [20], may allow us to investigate the effects of zinc on mitochondria with greater specificity and accuracy than ever before. Prior use of FluoZin-3 in tandem with the calcium indicator fura-2FF has been particularly useful for determining the simultaneous effects of zinc and calcium [189,190,261]. Other recent detection methods include small molecule fluorophores, such as Zinpyr1 combined with genetically encoded mitochondrial localization sequences, to target fluorescent sensors to the organelle [262]. Fluorescence resonance energy transfer (FRET)-based biosensors, such as ZapCY1 and eCALWY4, which allow for ratiometric zinc measurement, have also been targeted to the mitochondria [263,264,265,266]. Lastly, in recent years, the development of the single fluorescent protein-based zinc sensors GZnP1 and GZnP2 has allowed researchers to visualize mitochondrial zinc dynamics on simpler imaging systems than those required for FRET sensors, and has even enabled estimations of labile zinc concentrations in specific mitochondrial regions such as the matrix and IMS [267,268]. This ability of GZnP2 to measure zinc concentrations across the IMM may be particularly useful in investigations of potential zinc transport across the IMM via ZnTs or ZIPs.
Finally, the contributions of zinc disruption of mitochondrial function, trafficking, and morphology to neurodegenerative disease pathologies are yet to be fully detailed. The role that zinc-induced mitochondrial dysfunction plays in ischemia has already been recognized [190]. However, zinc-induced mitochondrial defects likely play important roles in other neurodegenerative conditions, particularly those already linked to abnormal mitochondrial function and dynamics, such as PD, AD, HD, and ALS [131,235]. Indeed, although zinc-induced mitochondrial dysfunction has not been identified as a therapeutic target for these diseases, there have been new efforts to investigate how zinc relates to mitochondrial dysfunction in neurodegeneration [269]. Understanding the relationship between zinc and neuronal mitochondria provides insight into the myriad ways that zinc plays a role in mitochondrial and neuronal health under normal and pathological conditions. If zinc-induced mitochondrial deficits are found to play a role in neurodegenerative diseases, this may shed light on promising new targets for neuroprotection.
Author Contributions
Writing—original draft preparation, review and editing, H.Y.L., J.R.G., E.A.; writing—review and editing, I.J.R., J.H.W.; figure preparation, H.Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
E.A. is funded by National Institutes of Health grants NS043277, NS111944 and NS117702. J.H.W. is funded by National Institutes of Health grant NS100494.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
I.J.R. is President of YaghPenn Consulting, providing strategic guidance in all aspects of drug discovery for neurological and neurodegenerative diseases.
References
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Ehmann, W.D.; Alauddin, M.; Hossain, T.I. Brain trace element concentrations in aging. Neurobiol. Aging 1984, 5, 19–28. [Google Scholar] [CrossRef]
- Huang, E.P. Metal ions and synaptic transmission: Think zinc. Proc. Natl. Acad. Sci. USA 1997, 94, 13386–13387. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Portbury, S.D.; Adlard, P.A. Zinc signal in brain diseases. Int. J. Mol. Sci. 2017, 18, 2506. [Google Scholar] [CrossRef]
- Sanford, L.; Carpenter, M.C.; Palmer, A.E. Intracellular Zn2+ transients modulate global gene expression in dissociated rat hippocampal neurons. Sci. Rep. 2019, 9, 9411. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Pablo, J.L.; Wang, C.; Presby, M.M.; Pitt, G.S. Polarized localization of voltage-gated Na+ channels is regulated by concerted FGF13 and FGF14 action. Proc. Natl. Acad. Sci. USA 2016, 113, E2665–E2674. [Google Scholar] [CrossRef]
- Wu, Q.-F.; Yang, L.; Li, S.; Wang, Q.; Yuan, X.-B.; Gao, X.; Bao, L.; Zhang, X. Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell 2012, 149, 1549–1564. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef]
- Haug, F.M. Electron microscopical localization of the zinc in hippocampal mossy fibre synapses by a modified sulfide silver procedure. Histochemie 1967, 8, 355–368. [Google Scholar] [CrossRef]
- Rubio, M.E.; Juiz, J.M. Chemical anatomy of excitatory endings in the dorsal cochlear nucleus of the rat: Differential synaptic distribution of aspartate aminotransferase, glutamate, and vesicular zinc. J. Comp. Neurol. 1998, 399, 341–358. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef]
- Wenzel, H.J.; Cole, T.B.; Born, D.E.; Schwartzkroin, P.A.; Palmiter, R.D. Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc. Natl. Acad. Sci. USA 1997, 94, 12676–12681. [Google Scholar] [CrossRef] [PubMed]
- Mcallister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Quinta-Ferreira, M.E.; Sampaio Dos Aidos, F.D.S.; Matias, C.M.; Mendes, P.J.; Dionísio, J.C.; Santos, R.M.; Rosário, L.M.; Quinta-Ferreira, R.M. Modelling zinc changes at the hippocampal mossy fiber synaptic cleft. J. Comput. Neurosci. 2016, 41, 323–337. [Google Scholar] [CrossRef]
- Wolf, C.; Weth, A.; Walcher, S.; Lax, C.; Baumgartner, W. Modeling of zinc dynamics in the synaptic cleft: Implications for CADHERIN mediated adhesion and synaptic plasticity. Front. Mol. Neurosci. 2018, 11, 306. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Maret, W.; Cuajungco, M.P. Zinc and excitotoxic brain injury: A new model. Neuroscientist 2004, 10, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Krall, R.F.; Tzounopoulos, T.; Aizenman, E. The function and regulation of zinc in the brain. Neuroscience 2021. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Z.; Weiss, J.H. Zn2+ permeates Ca2+ permeable AMPA/kainate channels and triggers selective neural injury. Neuroreport 1995, 6, 2553–2556. [Google Scholar] [CrossRef]
- Yin, H.Z.; Ha, D.H.; Carriedo, S.G.; Weiss, J.H. Kainate-stimulated Zn2+ uptake labels cortical neurons with Ca2+-permeable AMPA/kainate channels. Brain Res. 1998, 781, 45–56. [Google Scholar] [CrossRef]
- Jia, Y.; Jeng, J.-M.; Sensi, S.L.; Weiss, J.H. Zn2+ currents are mediated by calcium-permeable AMPA/kainate channels in cultured murine hippocampal neurones. J. Physiol. 2002, 543, 35–48. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Canzoniero, L.M.; Yu, S.P.; Ling, C.; Choi, D.W. Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurones. J. Physiol. 2000, 528, 39–52. [Google Scholar] [CrossRef]
- Weiss, J.H.; Hartley, D.M.; Koh, J.Y.; Choi, D.W. AMPA receptor activation potentiates zinc neurotoxicity. Neuron 1993, 10, 43–49. [Google Scholar] [CrossRef]
- Koh, J.Y.; Choi, D.W. Zinc toxicity on cultured cortical neurons: Involvement of N-methyl-D-aspartate receptors. Neuroscience 1994, 60, 1049–1057. [Google Scholar] [CrossRef]
- Gower-Winter, S.D.; Levenson, C.W. Zinc in the central nervous system: From molecules to behavior. Biofactors 2012, 38, 186–193. [Google Scholar] [CrossRef]
- Sandstead, H.H.; Frederickson, C.J.; Penland, J.G. History of zinc as related to brain function. J. Nutr. 2000, 130, 496S–502S. [Google Scholar] [CrossRef]
- Duncan, J.R.; Hurley, L.S. Thymidine kinase and DNA polymerase activity in normal and zinc deficient developing rat embryos. Proc. Soc. Exp. Biol. Med. 1978, 159, 39–43. [Google Scholar] [CrossRef]
- Halas, E.S.; Hunt, C.D.; Eberhardt, M.J. Learning and memory disabilities in young adult rats from mildly zinc deficient dams. Physiol. Behav. 1986, 37, 451–458. [Google Scholar] [CrossRef]
- Granzotto, A.; Sensi, S.L. Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol. Dis. 2015, 81, 25–37. [Google Scholar] [CrossRef]
- Pochwat, B.; Nowak, G.; Szewczyk, B. Relationship between Zinc (Zn (2+) ) and Glutamate Receptors in the Processes Underlying Neurodegeneration. Neural Plast. 2015, 2015, 591563. [Google Scholar] [CrossRef]
- Weiss, J.H. Ca permeable AMPA channels in diseases of the nervous system. Front. Mol. Neurosci. 2011, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Canzoniero, L.M.T.; Sensi, S.L. A Neurotoxic Ménage-à-trois: Glutamate, calcium, and Zinc in the Excitotoxic cascade. Front. Mol. Neurosci. 2020, 13, 600089. [Google Scholar] [CrossRef] [PubMed]
- Doboszewska, U.; Wlaź, P.; Nowak, G.; Radziwoń-Zaleska, M.; Cui, R.; Młyniec, K. Zinc in the monoaminergic theory of depression: Its relationship to neural plasticity. Neural Plast. 2017, 2017, 3682752. [Google Scholar] [CrossRef]
- Yosaee, S.; Clark, C.C.T.; Keshtkaran, Z.; Ashourpour, M.; Keshani, P.; Soltani, S. Zinc in depression: From development to treatment: A comparative/ dose response meta-analysis of observational studies and randomized controlled trials. Gen. Hosp. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Masciopinto, F.; Silvestri, E.; Viscovo, A.D.; Lattanzio, R.; Sorda, R.L.; Ciavardelli, D.; Goglia, F.; Piantelli, M.; Canzoniero, L.M.T.; et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 2010, 1, e91. [Google Scholar] [CrossRef]
- Lovell, M.A. A potential role for alterations of zinc and zinc transport proteins in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 471–483. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bush, A.I. Metals and alzheimer’s disease: How far have we come in the clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef]
- McCord, M.C.; Aizenman, E. The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 77. [Google Scholar] [CrossRef]
- Religa, D.; Strozyk, D.; Cherny, R.A.; Volitakis, I.; Haroutunian, V.; Winblad, B.; Naslund, J.; Bush, A.I. Elevated cortical zinc in Alzheimer disease. Neurology 2006, 67, 69–75. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of alzheimer’s disease. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Kim, J.; Kim, T.-Y.; Hwang, J.J.; Lee, J.-Y.; Shin, J.-H.; Gwag, B.J.; Koh, J.-Y. Accumulation of labile zinc in neurons and astrocytes in the spinal cords of G93A SOD-1 transgenic mice. Neurobiol. Dis. 2009, 34, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Brewer, G.J.; Kanzer, S.H.; Zimmerman, E.A.; Molho, E.S.; Celmins, D.F.; Heckman, S.M.; Dick, R. Subclinical zinc deficiency in Alzheimer’s disease and Parkinson’s disease. Am. J. Alzheimers Dis. Other Demen. 2010, 25, 572–575. [Google Scholar] [CrossRef]
- Forsleff, L.; Schauss, A.G.; Bier, I.D.; Stuart, S. Evidence of functional zinc deficiency in Parkinson’s disease. J. Altern. Complement. Med. 1999, 5, 57–64. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef]
- Kimura, T.; Kambe, T. The functions of metallothionein and ZIP and Znt transporters: An overview and perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef]
- Takagishi, T.; Hara, T.; Fukada, T. Recent advances in the role of SLC39A/ZIP zinc transporters in vivo. Int. J. Mol. Sci. 2017, 18, 2708. [Google Scholar] [CrossRef]
- Sekler, I.; Sensi, S.L.; Hershfinkel, M.; Silverman, W.F. Mechanism and regulation of cellular zinc transport. Mol. Med. 2007, 13, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Krężel, A.; Maret, W. The functions of metamorphic metallothioneins in zinc and copper metabolism. Int. J. Mol. Sci. 2017, 18, 1237. [Google Scholar] [CrossRef]
- Maret, W. The redox biology of redox-inert zinc ions. Free Radic. Biol. Med. 2019, 134, 311–326. [Google Scholar] [CrossRef]
- Aizenman, E.; Stout, A.K.; Hartnett, K.A.; Dineley, K.E.; McLaughlin, B.; Reynolds, I.J. Induction of neuronal apoptosis by thiol oxidation: Putative role of intracellular zinc release. J. Neurochem. 2000, 75, 1878–1888. [Google Scholar] [CrossRef]
- Carter, R.E.; Aiba, I.; Dietz, R.M.; Sheline, C.T.; Shuttleworth, C.W. Spreading depression and related events are significant sources of neuronal Zn2+ release and accumulation. J. Cereb. Blood Flow Metab. 2011, 31, 1073–1084. [Google Scholar] [CrossRef]
- Suh, S.W.; Hamby, A.M.; Gum, E.T.; Shin, B.S.; Won, S.J.; Sheline, C.T.; Chan, P.H.; Swanson, R.A. Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death. J. Cereb. Blood Flow Metab. 2008, 28, 1697–1706. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Koh, J.Y. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: Evidence against synaptic vesicle origin. J. Neurosci. 2000, 20, RC79. [Google Scholar] [CrossRef]
- Sensi, S.L.; Ton-That, D.; Sullivan, P.G.; Jonas, E.A.; Gee, K.R.; Kaczmarek, L.K.; Weiss, J.H. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl. Acad. Sci. USA 2003, 100, 6157–6162. [Google Scholar] [CrossRef]
- Ye, B.; Maret, W.; Vallee, B.L. Zinc metallothionein imported into liver mitochondria modulates respiration. Proc. Natl. Acad. Sci. USA 2001, 98, 2317–2322. [Google Scholar] [CrossRef]
- Zhu, X.-H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative imaging of energy expenditure in human brain. Neuroimage 2012, 60, 2107–2117. [Google Scholar] [CrossRef]
- Audano, M.; Pedretti, S.; Cermenati, G.; Brioschi, E.; Diaferia, G.R.; Ghisletti, S.; Cuomo, A.; Bonaldi, T.; Salerno, F.; Mora, M.; et al. Zc3h10 is a novel mitochondrial regulator. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G205–G214. [Google Scholar] [CrossRef]
- Tamaki, N.; Ikeda, T.; Funatsuka, A. Zinc as activating cation for muscle glycolysis. J. Nutr. Sci. Vitaminol. 1983, 29, 655–662. [Google Scholar] [CrossRef]
- Rofe, A.M.; Philcox, J.C.; Coyle, P. Activation of glycolysis by zinc is diminished in hepatocytes from metallothionein-null mice. Biol. Trace Elem. Res. 2000, 75, 87–97. [Google Scholar] [CrossRef]
- Adebayo, O.L.; Adenuga, G.A.; Sandhir, R. Selenium and zinc protect brain mitochondrial antioxidants and electron transport chain enzymes following postnatal protein malnutrition. Life Sci. 2016, 152, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Cameron, I.; Franklin, P.; Spedding, M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. Eur. J. Neurosci. 2004, 20, 1189–1196. [Google Scholar] [CrossRef]
- Su, B.; Ji, Y.-S.; Sun, X.; Liu, X.-H.; Chen, Z.-Y. Brain-derived neurotrophic factor (BDNF)-induced mitochondrial motility arrest and presynaptic docking contribute to BDNF-enhanced synaptic transmission. J. Biol. Chem. 2014, 289, 1213–1226. [Google Scholar] [CrossRef]
- Duffey, C.A.; Nicholls, S.; Mantilla, C.B.; Fogarty, M.J.; Sieck, G.C. Effect of BDNF on Mitochondrial Morphology and Protein Expression in NSC-34 Cells. FASEB J. 2018, 32, 743.6. [Google Scholar]
- Yang, X.; Wang, H.; Huang, C.; He, X.; Xu, W.; Luo, Y.; Huang, K. Zinc enhances the cellular energy supply to improve cell motility and restore impaired energetic metabolism in a toxic environment induced by OTA. Sci. Rep. 2017, 7, 14669. [Google Scholar] [CrossRef]
- Sensi, S.L.; Yin, H.Z.; Carriedo, S.G.; Rao, S.S.; Weiss, J.H. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc. Natl. Acad. Sci. USA 1999, 96, 2414–2419. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Stanika, R.I.; Kazanina, G.; Villanueva, I.; Andrews, S.B. The interactive roles of zinc and calcium in mitochondrial dysfunction and neurodegeneration. J. Neurochem. 2014, 128, 592–602. [Google Scholar] [CrossRef]
- Pan, R.; Liu, K.J.; Qi, Z. Zinc causes the death of hypoxic astrocytes by inducing ROS production through mitochondria dysfunction. Biophys. Rep. 2019, 5, 209–217. [Google Scholar] [CrossRef][Green Version]
- Dineley, K.E.; Devinney, M.J.; Zeak, J.A.; Rintoul, G.L.; Reynolds, I.J. Glutamate mobilizes [Zn2+] through Ca2+ -dependent reactive oxygen species accumulation. J. Neurochem. 2008, 106, 2184–2193. [Google Scholar] [CrossRef]
- Dineley, K.E.; Votyakova, T.V.; Reynolds, I.J. Zinc inhibition of cellular energy production: Implications for mitochondria and neurodegeneration. J. Neurochem. 2003, 85, 563–570. [Google Scholar] [CrossRef]
- Jiang, D.; Sullivan, P.G.; Sensi, S.L.; Steward, O.; Weiss, J.H. Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J. Biol. Chem. 2001, 276, 47524–47529. [Google Scholar] [CrossRef]
- Medvedeva, Y.V.; Weiss, J.H. Intramitochondrial Zn2+ accumulation via the Ca2+ uniporter contributes to acute ischemic neurodegeneration. Neurobiol. Dis. 2014, 68, 137–144. [Google Scholar] [CrossRef]
- Wudarczyk, J.; Debska, G.; Lenartowicz, E. Zinc as an inducer of the membrane permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 1999, 363, 1–8. [Google Scholar] [CrossRef]
- Dineley, K.E.; Richards, L.L.; Votyakova, T.V.; Reynolds, I.J. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion 2005, 5, 55–65. [Google Scholar] [CrossRef]
- Gazaryan, I.G.; Krasinskaya, I.P.; Kristal, B.S.; Brown, A.M. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. J. Biol. Chem. 2007, 282, 24373–24380. [Google Scholar] [CrossRef]
- Sensi, S.L.; Yin, H.Z.; Weiss, J.H. AMPA/kainate receptor-triggered Zn2+ entry into cortical neurons induces mitochondrial Zn2+ uptake and persistent mitochondrial dysfunction. Eur. J. Neurosci. 2000, 12, 3813–3818. [Google Scholar] [CrossRef]
- Medvedeva, Y.V.; Lin, B.; Shuttleworth, C.W.; Weiss, J.H. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J. Neurosci. 2009, 29, 1105–1114. [Google Scholar] [CrossRef]
- Sharaf, M.S.; van den Heuvel, M.R.; Stevens, D.; Kamunde, C. Zinc and calcium modulate mitochondrial redox state and morphofunctional integrity. Free Radic. Biol. Med. 2015, 84, 142–153. [Google Scholar] [CrossRef]
- Ji, S.G.; Medvedeva, Y.V.; Weiss, J.H. Zn2+ entry through the mitochondrial calcium uniporter is a critical contributor to mitochondrial dysfunction and neurodegeneration. Exp. Neurol. 2020, 325, 113161. [Google Scholar] [CrossRef]
- Sensi, S.L.; Ton-That, D.; Weiss, J.H. Mitochondrial sequestration and Ca2+-dependent release of cytosolic Zn2+ loads in cortical neurons. Neurobiol. Dis. 2002, 10, 100–108. [Google Scholar] [CrossRef]
- Clausen, A.; McClanahan, T.; Ji, S.G.; Weiss, J.H. Mechanisms of rapid reactive oxygen species generation in response to cytosolic Ca2+ or Zn2+ loads in cortical neurons. PLoS ONE 2013, 8, e83347. [Google Scholar] [CrossRef]
- Ji, S.G.; Weiss, J.H. Zn2+-induced disruption of neuronal mitochondrial function: Synergism with Ca2+, critical dependence upon cytosolic Zn2+ buffering, and contributions to neuronal injury. Exp. Neurol. 2018, 302, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Malaiyandi, L.M.; Honick, A.S.; Rintoul, G.L.; Wang, Q.J.; Reynolds, I.J. Zn2+ inhibits mitochondrial movement in neurons by phosphatidylinositol 3-kinase activation. J. Neurosci. 2005, 25, 9507–9514. [Google Scholar] [CrossRef]
- Knies, K.; Lu, Q.; Li, Y. The Effects of Zinc on Mitochondrial Morphology. FASEB J. 2018, 32, 543.17. [Google Scholar]
- Malaiyandi, L.M.; Vergun, O.; Dineley, K.E.; Reynolds, I.J. Direct visualization of mitochondrial zinc accumulation reveals uniporter-dependent and -independent transport mechanisms. J. Neurochem. 2005, 93, 1242–1250. [Google Scholar] [CrossRef]
- Allaman, I.; Magistretti, P.J. Brain Energy Metabolism. In Fundamental Neuroscience; Elsevier: Amsterdam, The Netherlands, 2013; pp. 261–284. [Google Scholar]
- Rigoulet, M.; Bouchez, C.L.; Paumard, P.; Ransac, S.; Cuvellier, S.; Duvezin-Caubet, S.; Mazat, J.P.; Devin, A. Cell energy metabolism: An update. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148276. [Google Scholar] [CrossRef]
- Madeira, V.M.C. Overview of mitochondrial bioenergetics. Methods Mol. Biol. 2018, 1782, 1–6. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Sheline, C.T.; Behrens, M.M.; Choi, D.W. Zinc-induced cortical neuronal death: Contribution of energy failure attributable to loss of NAD+ and inhibition of glycolysis. J. Neurosci. 2000, 20, 3139–3146. [Google Scholar] [CrossRef]
- Brown, A.M.; Kristal, B.S.; Effron, M.S.; Shestopalov, A.I.; Ullucci, P.A.; Sheu, K.F.; Blass, J.P.; Cooper, A.J. Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2000, 275, 13441–13447. [Google Scholar] [CrossRef]
- Lemire, J.; Mailloux, R.; Appanna, V.D. Zinc toxicity alters mitochondrial metabolism and leads to decreased ATP production in hepatocytes. J. Appl. Toxicol. 2008, 28, 175–182. [Google Scholar] [CrossRef]
- Krotkiewska, B.; Banas, T. Interaction of Zn2+, and Cu2+, Ions with glyceraldehyde-3-phosphate dehydrogenase from bovine heart and rabbit muscle. Int. J. Biochem. 1992, 24, 1501–1505. [Google Scholar] [CrossRef]
- Ikeda, T.; Kimura, K.; Morioka, S.; Tamaki, N. Inhibitory effects of Zn2+ on muscle glycolysis and their reversal by histidine. J. Nutr. Sci. Vitaminol. 1980, 26, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Dunaway, G.A. A review of animal phosphofructokinase isozymes with an emphasis on their physiological role. Mol. Cell. Biochem. 1983, 52, 75–91. [Google Scholar] [CrossRef]
- White, M.R.; Garcin, E.D. D-Glyceraldehyde-3-Phosphate Dehydrogenase Structure and Function. Subcell. Biochem. 2017, 83, 413–453. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.H.; Koh, J.Y. Protection by pyruvate against transient forebrain ischemia in rats. J. Neurosci. 2001, 21, RC171. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Barinova, K.V.; Stroylova, Y.Y.; Semenyuk, P.I.; Schmalhausen, E.V. Glyceraldehyde-3-phosphate dehydrogenase: Aggregation mechanisms and impact on amyloid neurodegenerative diseases. Int. J. Biol. Macromol. 2017, 100, 55–66. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Araki, K.; Yagi, N.; Aoyama, K.; Choong, C.-J.; Hayakawa, H.; Fujimura, H.; Nagai, Y.; Goto, Y.; Mochizuki, H. Parkinson’s disease is a type of amyloidosis featuring accumulation of amyloid fibrils of α-synuclein. Proc. Natl. Acad. Sci. USA 2019, 116, 17963–17969. [Google Scholar] [CrossRef] [PubMed]
- Lazarev, V.F.; Dutysheva, E.A.; Komarova, E.Y.; Mikhaylova, E.R.; Guzhova, I.V.; Margulis, B.A. GAPDH-targeted therapy—A new approach for secondary damage after traumatic brain injury on rats. Biochem. Biophys. Res. Commun. 2018, 501, 1003–1008. [Google Scholar] [CrossRef]
- Yogalingam, G.; Hwang, S.; Ferreira, J.C.B.; Mochly-Rosen, D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cδ (PKCδ) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J. Biol. Chem. 2013, 288, 18947–18960. [Google Scholar] [CrossRef]
- Hwang, S.; Disatnik, M.-H.; Mochly-Rosen, D. Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol. Med. 2015, 7, 1307–1326. [Google Scholar] [CrossRef]
- Sheu, K.F.; Blass, J.P. The alpha-ketoglutarate dehydrogenase complex. Ann. N. Y. Acad. Sci. 1999, 893, 61–78. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Besnard, A.M. Thiamine-dependent enzyme changes in temporal cortex of patients with Alzheimer’s disease. Metab. Brain Dis. 1990, 5, 179–184. [Google Scholar] [CrossRef]
- Mastrogiacoma, F.; Lindsay, J.G.; Bettendorff, L.; Rice, J.; Kish, S.J. Brain protein and alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. Ann. Neurol. 1996, 39, 592–598. [Google Scholar] [CrossRef]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P.; Baker, A.; Carlson, K.C.; Harding, B.; Perrino, P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch. Neurol. 1988, 45, 836–840. [Google Scholar] [CrossRef]
- Mizuno, Y.; Matuda, S.; Yoshino, H.; Mori, H.; Hattori, N.; Ikebe, S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 1994, 35, 204–210. [Google Scholar] [CrossRef]
- Blass, J.P.; Kark, R.A.; Menon, N.K. Low activities of the pyruvate and oxoglutarate dehydrogenase complexes in five patients with Friedreich’s ataxia. N. Engl. J. Med. 1976, 295, 62–67. [Google Scholar] [CrossRef]
- Gazaryan, I.G.; Krasnikov, B.F.; Ashby, G.A.; Thorneley, R.N.F.; Kristal, B.S.; Brown, A.M. Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J. Biol. Chem. 2002, 277, 10064–10072. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.B.; Watkins, S.C.; Hastings, T.G. Quantitative biochemical and ultrastructural comparison of mitochondrial permeability transition in isolated brain and liver mitochondria: Evidence for reduced sensitivity of brain mitochondria. Exp. Neurol. 2000, 164, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Baranovicova, E.; Kalenska, D.; Tomascova, A.; Lehotsky, J. Metabolomic study of altered energy metabolism during global forebrain ischemia and ischemic precoditioning in blood plasma in homocysteine treated rats. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Xiong, Y.; Peterson, P.L.; Lee, C.P. Alterations in cerebral energy metabolism induced by traumatic brain injury. Neurol. Res. 2001, 23, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Korotchkina, L.G.; Sidhu, S. Interaction of E1 and E3 components with the core proteins of the human pyruvate dehydrogenase complex. J. Mol. Catal. B. Enzym. 2009, 61, 2–6. [Google Scholar] [CrossRef]
- Park, S.; Jeon, J.H.; Min, B.K.; Ha, C.M.; Thoudam, T.; Park, B.Y.; Lee, I.K. Role of the pyruvate dehydrogenase complex in metabolic remodeling: Differential pyruvate dehydrogenase complex functions in metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef]
- Ronowska, A.; Dyś, A.; Jankowska-Kulawy, A.; Klimaszewska-Łata, J.; Bielarczyk, H.; Romianowski, P.; Pawełczyk, T.; Szutowicz, A. Short-term effects of zinc on acetylcholine metabolism and viability of SN56 cholinergic neuroblastoma cells. Neurochem. Int. 2010, 56, 143–151. [Google Scholar] [CrossRef]
- Ronowska, A.; Gul-Hinc, S.; Bielarczyk, H.; Pawełczyk, T.; Szutowicz, A. Effects of zinc on SN56 cholinergic neuroblastoma cells. J. Neurochem. 2007, 103, 972–983. [Google Scholar] [CrossRef]
- Zyśk, M.; Bielarczyk, H.; Gul-Hinc, S.; Dyś, A.; Gapys, B.; Ronowska, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A. Phenotype-Dependent Interactions between N-acetyl-L-Aspartate and Acetyl-CoA in Septal SN56 Cholinergic Cells Exposed to an Excess of Zinc. J. Alzheimers Dis. 2017, 56, 1145–1158. [Google Scholar] [CrossRef]
- Ronowska, A.; Gul-Hinc, S.; Michno, A.; Bizon-Zygmańska, D.; Zyśk, M.; Bielarczyk, H.; Szutowicz, A.; Gapys, B.; Jankowska-Kulawy, A. Aggravated effects of coexisting marginal thiamine deficits and zinc excess on SN56 neuronal cells. Nutr. Neurosci. 2019, 1–11. [Google Scholar] [CrossRef]
- Zaidan, E.; Sims, N.R. Reduced activity of the pyruvate dehydrogenase complex but not cytochrome c oxidase is associated with neuronal loss in the striatum following short-term forebrain ischemia. Brain Res. 1997, 772, 23–28. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B.; Liu, Y.; Kennedy, M.C. Zinc causes a shift toward citrate at equilibrium of the m-aconitase reaction of prostate mitochondria. J. Inorg. Biochem. 2000, 78, 161–165. [Google Scholar] [CrossRef]
- Tabuchi, A.; Funaji, K.; Nakatsubo, J.; Fukuchi, M.; Tsuchiya, T.; Tsuda, M. Inactivation of aconitase during the apoptosis of mouse cerebellar granule neurons induced by a deprivation of membrane depolarization. J. Neurosci. Res. 2003, 71, 504–515. [Google Scholar] [CrossRef]
- Xue, Y.-N.; Liu, Y.-N.; Su, J.; Li, J.-L.; Wu, Y.; Guo, R.; Yu, B.-B.; Yan, X.-Y.; Zhang, L.-C.; Sun, L.-K.; et al. Zinc cooperates with p53 to inhibit the activity of mitochondrial aconitase through reactive oxygen species accumulation. Cancer Med. 2019, 8, 2462–2473. [Google Scholar] [CrossRef]
- Link, T.A.; von Jagow, G. Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group. J. Biol. Chem. 1995, 270, 25001–25006. [Google Scholar] [CrossRef]
- Lorusso, M.; Cocco, T.; Sardanelli, A.M.; Minuto, M.; Bonomi, F.; Papa, S. Interaction of Zn2+ with the bovine-heart mitochondrial bc1 complex. Eur. J. Biochem. 1991, 197, 555–561. [Google Scholar] [CrossRef]
- Berry, E.A.; Zhang, Z.; Bellamy, H.D.; Huang, L. Crystallographic location of two Zn2+-binding sites in the avian cytochrome bc(1) complex. Biochim. Biophys. Acta 2000, 1459, 440–448. [Google Scholar] [CrossRef]
- Panchal, K.; Tiwari, A.K. Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 2019, 47, 151–173. [Google Scholar] [CrossRef]
- Cabral-Costa, J.V.; Kowaltowski, A.J. Neurological disorders and mitochondria. Mol. Asp. Med. 2020, 71, 100826. [Google Scholar] [CrossRef]
- Kilbride, S.M.; Gluchowska, S.A.; Telford, J.E.; O’Sullivan, C.; Davey, G.P. High-level inhibition of mitochondrial complexes III and IV is required to increase glutamate release from the nerve terminal. Mol. Neurodegener. 2011, 6, 53. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Sharpley, M.S.; Hirst, J. The inhibition of mitochondrial complex I (NADH:ubiquinone oxidoreductase) by Zn2+. J. Biol. Chem. 2006, 281, 34803–34809. [Google Scholar] [CrossRef]
- Telford, J.E.; Kilbride, S.M.; Davey, G.P. Complex I is rate-limiting for oxygen consumption in the nerve terminal. J. Biol. Chem. 2009, 284, 9109–9114. [Google Scholar] [CrossRef]
- Kilbride, S.M.; Telford, J.E.; Tipton, K.F.; Davey, G.P. Partial inhibition of complex I activity increases Ca-independent glutamate release rates from depolarized synaptosomes. J. Neurochem. 2008, 106, 826–834. [Google Scholar] [CrossRef]
- Kahl, A.; Stepanova, A.; Konrad, C.; Anderson, C.; Manfredi, G.; Zhou, P.; Iadecola, C.; Galkin, A. Critical Role of Flavin and Glutathione in Complex I-Mediated Bioenergetic Failure in Brain Ischemia/Reperfusion Injury. Stroke 2018, 49, 1223–1231. [Google Scholar] [CrossRef]
- Straub, I.R.; Janer, A.; Weraarpachai, W.; Zinman, L.; Robertson, J.; Rogaeva, E.; Shoubridge, E.A. Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet. 2018, 27, 178–189. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Sherer, T.B.; Betarbet, R.; Panov, A.V. Complex I and Parkinson’s disease. IUBMB Life 2001, 52, 135–141. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Patel, M.; Day, B.J.; Crapo, J.D.; Fridovich, I.; McNamara, J.O. Requirement for superoxide in excitotoxic cell death. Neuron 1996, 16, 345–355. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl Radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem. 1991, 266, 4244–4250. [Google Scholar] [CrossRef]
- Tammariello, S.P.; Quinn, M.T.; Estus, S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J. Neurosci. 2000, 20, RC53. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Boveris, A.; Ragan, C.I.; Stoppani, A.O. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977, 180, 248–257. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Sensi, S.L.; Yin, H.Z.; Weiss, J.H. Glutamate triggers preferential Zn2+ flux through Ca2+ permeable AMPA channels and consequent ROS production. Neuroreport 1999, 10, 1723–1727. [Google Scholar] [CrossRef]
- Salazar, G.; Huang, J.; Feresin, R.G.; Zhao, Y.; Griendling, K.K. Zinc regulates Nox1 expression through a NF-κB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic. Biol. Med. 2017, 108, 225–235. [Google Scholar] [CrossRef]
- Bishop, G.M.; Dringen, R.; Robinson, S.R. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radic. Biol. Med. 2007, 42, 1222–1230. [Google Scholar] [CrossRef]
- Slepchenko, K.G.; Lu, Q.; Li, Y.V. Zinc wave during the treatment of hypoxia is required for initial reactive oxygen species activation in mitochondria. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 44–51. [Google Scholar]
- Slepchenko, K.G.; Lu, Q.; Li, Y.V. Cross talk between increased intracellular zinc (Zn2+) and accumulation of reactive oxygen species in chemical ischemia. Am. J. Physiol. Cell Physiol. 2017, 313, C448–C459. [Google Scholar] [CrossRef]
- Zündorf, G.; Kahlert, S.; Bunik, V.I.; Reiser, G. α-Ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience 2009, 158, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.M.; Koh, J.Y. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J. Neurosci. 2000, 20, RC111. [Google Scholar] [CrossRef] [PubMed]
- Doczi, J.; Turiák, L.; Vajda, S.; Mándi, M.; Töröcsik, B.; Gerencser, A.A.; Kiss, G.; Konràd, C.; Adam-Vizi, V.; Chinopoulos, C. Complex contribution of cyclophilin D to Ca2+-induced permeability transition in brain mitochondria, with relation to the bioenergetic state. J. Biol. Chem. 2011, 286, 6345–6353. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef]
- Schinder, A.F.; Olson, E.C.; Spitzer, N.C.; Montal, M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 1996, 16, 6125–6133. [Google Scholar] [CrossRef]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.M.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef]
- White, R.J.; Reynolds, I.J. Mitochondrial depolarization in glutamate-stimulated neurons: An early signal specific to excitotoxin exposure. J. Neurosci. 1996, 16, 5688–5697. [Google Scholar] [CrossRef]
- Büki, A.; Okonkwo, D.O.; Wang, K.K.; Povlishock, J.T. Cytochrome c release and caspase activation in traumatic axonal injury. J. Neurosci. 2000, 20, 2825–2834. [Google Scholar] [CrossRef]
- Ichas, F.; Mazat, J.-P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta BBA Bioenerg. 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Peng, T.-I.; Jou, M.-J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Akopova, O.V.; Kolchynskayia, L.Y.; Nosar, V.Y.; Smyrnov, A.N.; Malisheva, M.K.; Man’kovskaia, Y.N.; Sahach, V.F. The effect of permeability transition pore opening on reactive oxygen species production in rat brain mitochondria. Ukr Biokhim Zh (1999) 2011, 83, 46–55. [Google Scholar]
- Liu, X.-R.; Li, J.-H.; Zhang, Y.; Ge, Y.-S.; Tian, F.-F.; Dai, J.; Jiang, F.-L.; Liu, Y. Mitochondrial permeability transition induced by different concentrations of zinc. J. Membr. Biol. 2011, 244, 105–112. [Google Scholar] [CrossRef]
- Devinney, M.J.; Malaiyandi, L.M.; Vergun, O.; DeFranco, D.B.; Hastings, T.G.; Dineley, K.E. A comparison of Zn2+- and Ca2+-triggered depolarization of liver mitochondria reveals no evidence of Zn2+-induced permeability transition. Cell Calcium 2009, 45, 447–455. [Google Scholar] [CrossRef]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef]
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys. Acta 2015, 1853, 2006–2011. [Google Scholar] [CrossRef]
- Randall, R.D.; Thayer, S.A. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 1992, 12, 1882–1895. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxicity: Still hammering the ischemic brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 1985, 58, 293–297. [Google Scholar] [CrossRef]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial Calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358, 147–155. [Google Scholar] [CrossRef]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta 2007, 1768, 2510–2515. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef]
- Cheng, C.; Reynolds, I.J. Calcium-sensitive fluorescent dyes can report increases in intracellular free zinc concentration in cultured forebrain neurons. J. Neurochem. 1998, 71, 2401–2410. [Google Scholar] [CrossRef]
- Stork, C.J.; Li, Y.V. Intracellular zinc elevation measured with a “calcium-specific” indicator during ischemia and reperfusion in rat hippocampus: A question on calcium overload. J. Neurosci. 2006, 26, 10430–10437. [Google Scholar] [CrossRef]
- Devinney, M.J.; Reynolds, I.J.; Dineley, K.E. Simultaneous detection of intracellular free calcium and zinc using fura-2FF and FluoZin-3. Cell Calcium 2005, 37, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.G.; Medvedeva, Y.V.; Wang, H.-L.; Yin, H.Z.; Weiss, J.H. Mitochondrial Zn2+ accumulation: A potential trigger of hippocampal ischemic injury. Neuroscientist 2019, 25, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Zyśk, M.; Gapys, B.; Ronowska, A.; Gul-Hinc, S.; Erlandsson, A.; Iwanicki, A.; Sakowicz-Burkiewicz, M.; Szutowicz, A.; Bielarczyk, H. Protective effects of voltage-gated calcium channel antagonists against zinc toxicity in SN56 neuroblastoma cholinergic cells. PLoS ONE 2018, 13, e0209363. [Google Scholar] [CrossRef]
- Yin, H.Z.; Wang, H.-L.; Ji, S.G.; Medvedeva, Y.V.; Tian, G.; Bazrafkan, A.K.; Maki, N.Z.; Akbari, Y.; Weiss, J.H. Rapid intramitochondrial Zn2+ accumulation in CA1 hippocampal pyramidal neurons after transient global ischemia: A possible contributor to mitochondrial disruption and cell death. J. Neuropathol. Exp. Neurol. 2019, 78, 655–664. [Google Scholar] [CrossRef]
- Medvedeva, Y.V.; Ji, S.G.; Yin, H.Z.; Weiss, J.H. Differential Vulnerability of CA1 versus CA3 Pyramidal Neurons After Ischemia: Possible Relationship to Sources of Zn2+ Accumulation and Its Entry into and Prolonged Effects on Mitochondria. J. Neurosci. 2017, 37, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metallothionein redox biology in the cytoprotective and cytotoxic functions of zinc. Exp. Gerontol. 2008, 43, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. The function of zinc metallothionein: A link between cellular zinc and redox state. J. Nutr. 2000, 130, 1455S–1458S. [Google Scholar] [CrossRef]
- Yang, J.S.; Perveen, S.; Ha, T.J.; Kim, S.Y.; Yoon, S.H. Cyanidin-3-glucoside inhibits glutamate-induced Zn2+ signaling and neuronal cell death in cultured rat hippocampal neurons by inhibiting Ca2+-induced mitochondrial depolarization and formation of reactive oxygen species. Brain Res. 2015, 1606, 9–20. [Google Scholar] [CrossRef]
- Costello, L.C.; Guan, Z.; Franklin, R.B.; Feng, P. Metallothionein can function as a chaperone for zinc uptake transport into prostate and liver mitochondria. J. Inorg. Biochem. 2004, 98, 664–666. [Google Scholar] [CrossRef]
- Feng, W.; Cai, J.; Pierce, W.M.; Franklin, R.B.; Maret, W.; Benz, F.W.; Kang, Y.J. Metallothionein transfers zinc to mitochondrial aconitase through a direct interaction in mouse hearts. Biochem. Biophys. Res. Commun. 2005, 332, 853–858. [Google Scholar] [CrossRef]
- Seo, Y.A.; Lopez, V.; Kelleher, S.L. A histidine-rich motif mediates mitochondrial localization of ZnT2 to modulate mitochondrial function. Am. J. Physiol. Cell Physiol. 2011, 300, C1479–C1489. [Google Scholar] [CrossRef]
- Golan, Y.; Berman, B.; Assaraf, Y.G. Heterodimerization, altered subcellular localization, and function of multiple zinc transporters in viable cells using bimolecular fluorescence complementation. J. Biol. Chem. 2015, 290, 9050–9063. [Google Scholar] [CrossRef]
- Sun, Q.; Zhong, W.; Zhang, W.; Li, Q.; Sun, X.; Tan, X.; Sun, X.; Dong, D.; Zhou, Z. Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G757–G766. [Google Scholar] [CrossRef]
- Tuncay, E.; Bitirim, C.V.; Olgar, Y.; Durak, A.; Rutter, G.A.; Turan, B. Zn2+-transporters ZIP7 and ZnT7 play important role in progression of cardiac dysfunction via affecting sarco(endo)plasmic reticulum-mitochondria coupling in hyperglycemic cardiomyocytes. Mitochondrion 2019, 44, 41–52. [Google Scholar] [CrossRef]
- Cho, H.M.; Ryu, J.R.; Jo, Y.; Seo, T.W.; Choi, Y.N.; Kim, J.H.; Chung, J.M.; Cho, B.; Kang, H.C.; Yu, S.-W.; et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 2019, 73, 364–376.e8. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp. Soc. Exp. Biol. 1975, 21–38. [Google Scholar]
- Spinazzi, M.; Cazzola, S.; Bortolozzi, M.; Baracca, A.; Loro, E.; Casarin, A.; Solaini, G.; Sgarbi, G.; Casalena, G.; Cenacchi, G.; et al. A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function. Hum. Mol. Genet. 2008, 17, 3291–3302. [Google Scholar] [CrossRef]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; van der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Yoneda, M.; Miyatake, T.; Attardi, G. Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol. Cell. Biol. 1994, 14, 2699–2712. [Google Scholar] [CrossRef]
- Nakada, K.; Inoue, K.; Ono, T.; Isobe, K.; Ogura, A.; Goto, Y.I.; Nonaka, I.; Hayashi, J.I. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 2001, 7, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Long, Q.; Liu, J.; Tang, H.; Li, Y.; Bao, F.; Qin, D.; Pei, D.; Liu, X. Mitochondrial fusion provides an “initial metabolic complementation” controlled by mtDNA. Cell Mol. Life Sci. 2015, 72, 2585–2598. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnauné-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Acín-Peréz, R.; Geghman, K.D.; Manfredi, G.; Lu, B.; Li, C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. USA 2011, 108, 12920–12924. [Google Scholar] [CrossRef]
- Hom, J.; Yu, T.; Yoon, Y.; Porter, G.; Sheu, S.-S. Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim. Biophys. Acta 2010, 1797, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef]
- Fukumitsu, K.; Hatsukano, T.; Yoshimura, A.; Heuser, J.; Fujishima, K.; Kengaku, M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol. Cell. Neurosci. 2016, 71, 56–65. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989, 12, 94–101. [Google Scholar] [CrossRef]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.W.; Honick, A.S.; Reynolds, I.J. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 2006, 26, 7035–7045. [Google Scholar] [CrossRef] [PubMed]
- Spillane, M.; Ketschek, A.; Merianda, T.T.; Twiss, J.L.; Gallo, G. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 2013, 5, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Courchet, J.; Lewis, T.L.; Lee, S.; Courchet, V.; Liou, D.-Y.; Aizawa, S.; Polleux, F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 2013, 153, 1510–1525. [Google Scholar] [CrossRef]
- Morris, R.L.; Hollenbeck, P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993, 104 Pt 3, 917–927. [Google Scholar]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Development 2016, 143, 1981–1992. [Google Scholar] [CrossRef]
- Chada, S.R.; Hollenbeck, P.J. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr. Biol. 2004, 14, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Qiao, H.; Pan, P.-Y.; Chen, Y.; Sheng, Z.-H. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 2013, 4, 413–419. [Google Scholar] [CrossRef]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef]
- Fukumitsu, K.; Fujishima, K.; Yoshimura, A.; Wu, Y.K.; Heuser, J.; Kengaku, M. Synergistic action of dendritic mitochondria and creatine kinase maintains ATP homeostasis and actin dynamics in growing neuronal dendrites. J. Neurosci. 2015, 35, 5707–5723. [Google Scholar] [CrossRef] [PubMed]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial fission in human diseases. Handb. Exp. Pharmacol. 2017, 240, 159–188. [Google Scholar] [CrossRef]
- Billur, D.; Tuncay, E.; Okatan, E.N.; Olgar, Y.; Durak, A.T.; Degirmenci, S.; Can, B.; Turan, B. Interplay between cytosolic free Zn2+ and mitochondrion morphological changes in rat ventricular cardiomyocytes. Biol. Trace Elem. Res. 2016, 174, 177–188. [Google Scholar] [CrossRef]
- Abuarab, N.; Li, F.; Sivaprasadarao, A. Ionic signalling and mitochondrial dynamics. Mol. Cell. Oncol. 2017, 4, e1373889. [Google Scholar] [CrossRef][Green Version]
- Li, F.; Munsey, T.S.; Sivaprasadarao, A. TRPM2-mediated rise in mitochondrial Zn2+ promotes palmitate-induced mitochondrial fission and pancreatic β-cell death in rodents. Cell Death Differ. 2017, 24, 1999–2012. [Google Scholar] [CrossRef]
- Qi, Z.; Shi, W.; Zhao, Y.; Ji, X.; Liu, K.J. Zinc accumulation in mitochondria promotes ischemia-induced BBB disruption through Drp1-dependent mitochondria fission. Toxicol. Appl. Pharmacol. 2019, 377, 114601. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Abuarab, N.; Munsey, T.S.; Jiang, L.-H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, W.; Jiang, L.-H. Alteration in Intracellular Zn2+ Homeostasis as a Result of TRPM2 Channel Activation Contributes to ROS-Induced Hippocampal Neuronal Death. Front. Mol. Neurosci. 2017, 10, 414. [Google Scholar] [CrossRef]
- Li, X.; Jiang, L.-H. A critical role of the transient receptor potential melastatin 2 channel in a positive feedback mechanism for reactive oxygen species-induced delayed cell death. J. Cell Physiol. 2019, 234, 3647–3660. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.M.; Sun, W. The coordinated regulation of mitochondrial structure and function by Drp1 for mitochondrial quality surveillance. BMB Rep. 2019, 52, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E. Think we understand the role of DRP1 in mitochondrial biology? zinc again! Mol. Cell 2019, 73, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.-H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of drp1 ameliorates synaptic depression, aβ deposition, and cognitive impairment in an alzheimer’s disease model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.-T.; Wang, Z.-Z.; Yuan, Y.-H.; Wang, X.-L.; Sun, H.-M.; Chen, N.-H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, W.; Shao, Q.; Yang, Y.; Xu, Z.; Chen, J.; Zhang, X.; Ge, X. Drp1, a potential therapeutic target for Parkinson’s disease, is involved in olfactory bulb pathological alteration in the Rotenone-induced rat model. Toxicol. Lett. 2020, 325, 1–13. [Google Scholar] [CrossRef]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.A.; Carafoli, E.; Scorrano, L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Flippo, K.H.; Lin, Z.; Dickey, A.S.; Zhou, X.; Dhanesha, N.A.; Walters, G.C.; Liu, Y.; Merrill, R.A.; Meller, R.; Simon, R.P.; et al. Deletion of a Neuronal Drp1 Activator Protects against Cerebral Ischemia. J. Neurosci. 2020, 40, 3119–3129. [Google Scholar] [CrossRef]
- Flippo, K.H.; Gnanasekaran, A.; Perkins, G.A.; Ajmal, A.; Merrill, R.A.; Dickey, A.S.; Taylor, S.S.; McKnight, G.S.; Chauhan, A.K.; Usachev, Y.M.; et al. AKAP1 Protects from Cerebral Ischemic Stroke by Inhibiting Drp1-Dependent Mitochondrial Fission. J. Neurosci. 2018, 38, 8233–8242. [Google Scholar] [CrossRef]
- Hoyte, L.; Barber, P.A.; Buchan, A.M.; Hill, M.D. The rise and fall of NMDA antagonists for ischemic stroke. Curr. Mol. Med. 2004, 4, 131–136. [Google Scholar] [CrossRef]
- Lipton, S.A. Failures and successes of NMDA receptor antagonists: Molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 2004, 1, 101–110. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef]
- Pratt, E.P.S.; Damon, L.J.; Anson, K.J.; Palmer, A.E. Tools and techniques for illuminating the cell biology of zinc. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118865. [Google Scholar] [CrossRef]
- Zhao, J.; Bertoglio, B.A.; Gee, K.R.; Kay, A.R. The zinc indicator FluoZin-3 is not perturbed significantly by physiological levels of calcium or magnesium. Cell Calcium 2008, 44, 422–426. [Google Scholar] [CrossRef]
- Tomat, E.; Nolan, E.M.; Jaworski, J.; Lippard, S.J. Organelle-specific zinc detection using zinpyr-labeled fusion proteins in live cells. J. Am. Chem. Soc. 2008, 130, 15776–15777. [Google Scholar] [CrossRef]
- Miranda, J.G.; Weaver, A.L.; Qin, Y.; Park, J.G.; Stoddard, C.I.; Lin, M.Z.; Palmer, A.E. New alternately colored FRET sensors for simultaneous monitoring of Zn2+ in multiple cellular locations. PLoS ONE 2012, 7, e49371. [Google Scholar] [CrossRef]
- Carter, K.P.; Carpenter, M.C.; Fiedler, B.; Jimenez, R.; Palmer, A.E. Critical Comparison of FRET-Sensor Functionality in the Cytosol and Endoplasmic Reticulum and Implications for Quantification of Ions. Anal. Chem. 2017, 89, 9601–9608. [Google Scholar] [CrossRef]
- Park, J.G.; Qin, Y.; Galati, D.F.; Palmer, A.E. New sensors for quantitative measurement of mitochondrial Zn2+. ACS Chem. Biol. 2012, 7, 1636–1640. [Google Scholar] [CrossRef]
- Chabosseau, P.; Tuncay, E.; Meur, G.; Bellomo, E.A.; Hessels, A.; Hughes, S.; Johnson, P.R.V.; Bugliani, M.; Marchetti, P.; Turan, B.; et al. Mitochondrial and ER-targeted eCALWY probes reveal high levels of free Zn2+. ACS Chem. Biol. 2014, 9, 2111–2120. [Google Scholar] [CrossRef]
- Qin, Y.; Sammond, D.W.; Braselmann, E.; Carpenter, M.C.; Palmer, A.E. Development of an optical Zn2+ probe based on a single fluorescent protein. ACS Chem. Biol. 2016, 11, 2744–2751. [Google Scholar] [CrossRef]
- Fudge, D.H.; Black, R.; Son, L.; LeJeune, K.; Qin, Y. Optical Recording of Zn2+ Dynamics in the Mitochondrial Matrix and Intermembrane Space with the GZnP2 Sensor. ACS Chem. Biol. 2018, 13, 1897–1905. [Google Scholar] [CrossRef]
- Park, J.-S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).