mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
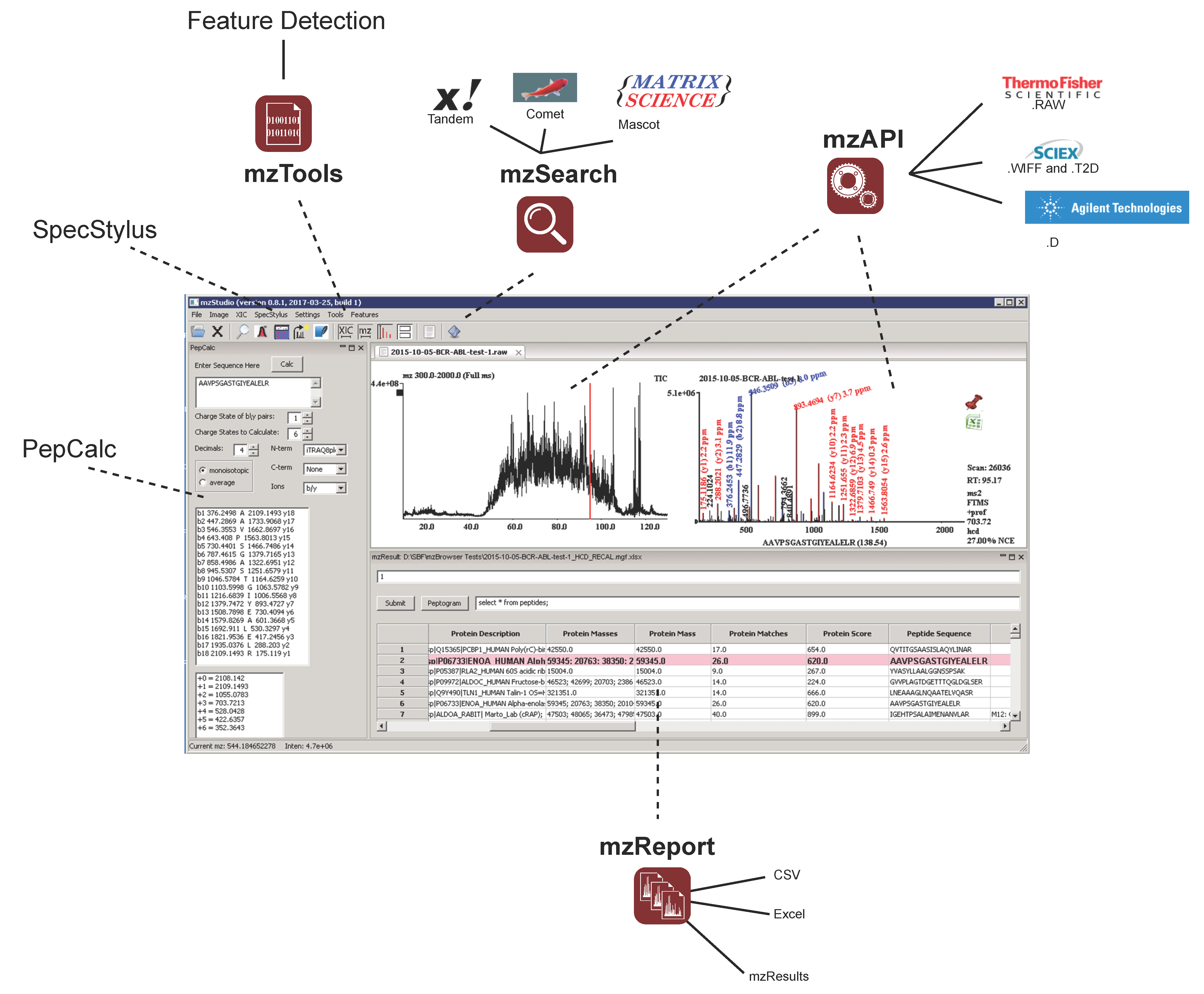
2.1. Architecture
2.2. Results
3. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteom. 2011, 10, M111.012658. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a nad-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Perez-Riverol, Y.; Hermjakob, H.; Vizcaino, J.A. Open source libraries and frameworks for biological data visualisation: A guide for developers. Proteomics 2015, 15, 1356–1374. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.N.; Brusniak, M.Y.; Mani, D.R.; Aebersold, R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 2008, 7, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Maxquant enables high peptide identification rates, individualized p.P.B.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Salomon, A.R. Peptidedepot: Flexible relational database for visual analysis of quantitative proteomic data and integration of existing protein information. Proteomics 2009, 9, 5350–5358. [Google Scholar] [CrossRef] [PubMed]
- Desiere, F.; Deutsch, E.W.; Nesvizhskii, A.I.; Mallick, P.; King, N.L.; Eng, J.K.; Aderem, A.; Boyle, R.; Brunner, E.; Donohoe, S.; et al. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2005, 6, R9. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Fujita, Y.; Parry, H.E.; Yoshizawa, A.C.; Morimoto, K.; Murase, M.; Yamada, Y.; Yao, J.; Utsunomiya, S.I.; Kajihara, S.; et al. Mass++: A visualization and analysis tool for mass spectrometry. J. Proteome Res. 2014, 13, 3846–3853. [Google Scholar] [CrossRef] [PubMed]
- Avtonomov, D.M.; Raskind, A.; Nesvizhskii, A.I. Batmass: A java software platform for lc-ms data visualization in proteomics and metabolomics. J. Proteome Res. 2016, 15, 2500–2509. [Google Scholar] [CrossRef] [PubMed]
- Parikh, J.R.; Askenazi, M.; Ficarro, S.B.; Cashorali, T.; Webber, J.T.; Blank, N.C.; Zhang, Y.; Marto, J.A. Multiplierz: An extensible api based desktop environment for proteomics data analysis. BMC Bioinform. 2009, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.T.; Askenazi, M.; Marto, J.A. Mzresults: An interactive viewer for interrogation and distribution of proteomics results. Mol. Cell. Proteom. 2011, 10, M110.003970. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.M.; Ficarro, S.B.; Adelmant, G.; Marto, J.A. Multiplerz v2.0: A python-based ecosystem for shared access and analysis of native mass spectrometry data. Proteomics 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Askenazi, M.; Parikh, J.R.; Marto, J.A. Mzapi: A new strategy for efficiently sharing mass spectrometry data. Nat. Methods 2009, 6, 240–241. [Google Scholar] [CrossRef] [PubMed]
- Ficarro, S.B.; Browne, C.M.; Card, J.D.; Alexander, W.M.; Zhang, T.; Park, E.; McNally, R.; Dhe-Paganon, S.; Seo, H.S.; Lamberto, I.; et al. Leveraging gas-phase fragmentation pathways for improved identification and selective detection of targets modified by covalent probes. Anal. Chem. 2016, 88, 12248–12254. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tien, A.C.; Alberta, J.A.; Ficarro, S.B.; Griveau, A.; Sun, Y.; Deshpande, J.S.; Card, J.D.; Morgan-Smith, M.; Michowski, W.; et al. A sequentially priming phosphorylation cascade activates the gliomagenic transcription factor olig2. Cell Rep. 2017, 18, 3167–3177. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Meijer, D.H.; Alberta, J.A.; Mehta, S.; Kane, M.F.; Tien, A.-C.; Fu, H.; Petryniak, M.A.; Potter, G.B.; Liu, Z.; et al. Phosphorylation state of olig2 regulates proliferation of neural progenitors. Neuron 2011, 69, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Fiorelli, R.; Kupp, R.; Rajan, S.; Szeto, E.; Lo Cascio, C.; Maire, C.L.; Sun, Y.; Alberta, J.A.; Eschbacher, J.M.; et al. Post-translational modifications of olig2 regulate glioma invasion through the tgf-β pathway. Cell Rep. 2016, 16, 950–966. [Google Scholar] [CrossRef] [PubMed]

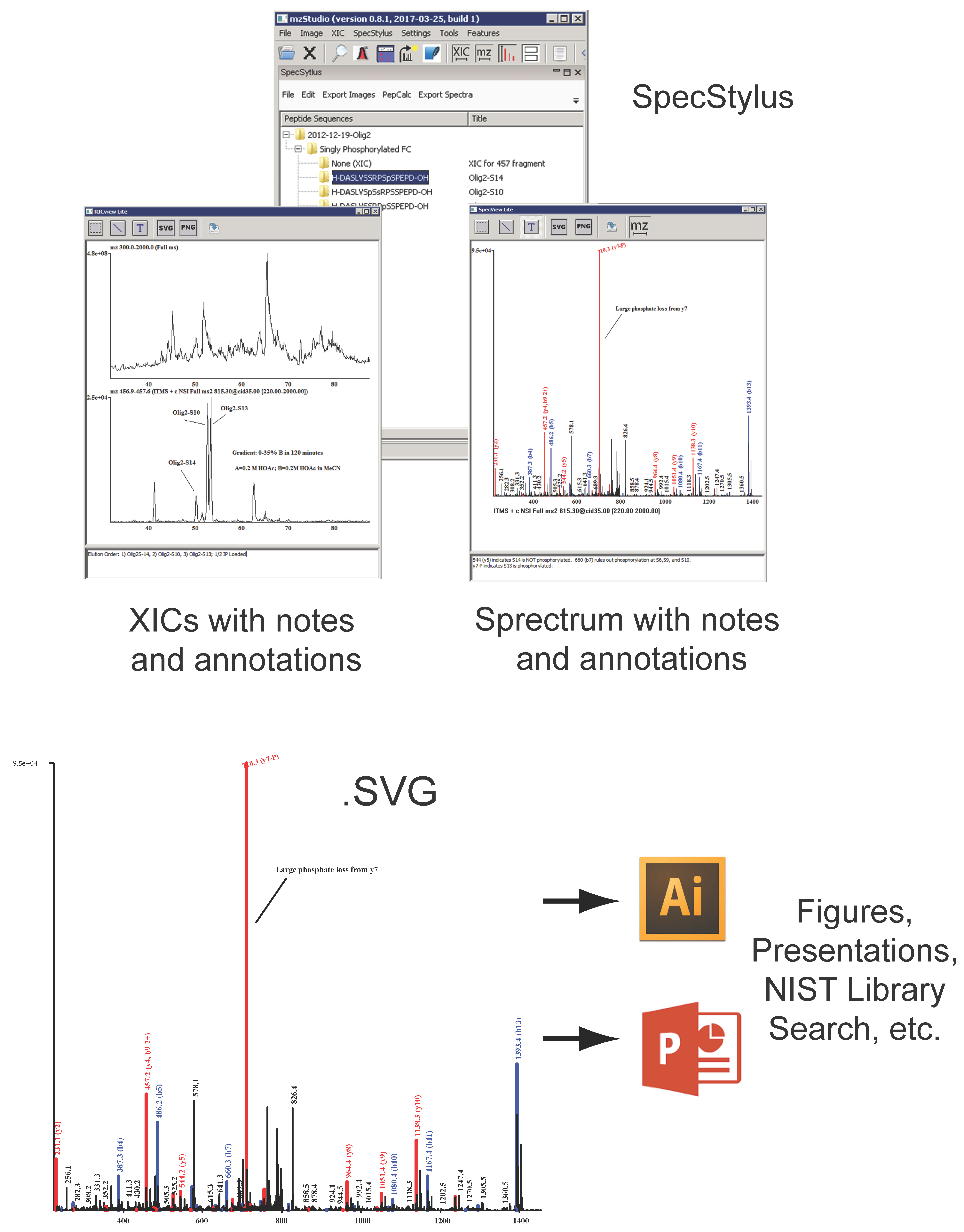
 and importing search results
and importing search results  , users can click individual PSMs to view annotated peaks
, users can click individual PSMs to view annotated peaks  . This particular MS/MS spectrum was obtained during analysis of peptides modified with the CDK7 kinase inhibitor THZ1 and yields a low-confidence Mascot score due to the presence of several inhibitor-related ions
. This particular MS/MS spectrum was obtained during analysis of peptides modified with the CDK7 kinase inhibitor THZ1 and yields a low-confidence Mascot score due to the presence of several inhibitor-related ions  and non-canonical fragmentation pathways. Using mzStudio’s built-in spectral processing tools
and non-canonical fragmentation pathways. Using mzStudio’s built-in spectral processing tools  , users can easily experiment with different processing algorithms (i.e., filter inhibitor related ions, reduce charge of highly charged species, and score ions from inhibitor related fragmentation pathways), and assess the impact on peptide sequence scores through the integrated search tool
, users can easily experiment with different processing algorithms (i.e., filter inhibitor related ions, reduce charge of highly charged species, and score ions from inhibitor related fragmentation pathways), and assess the impact on peptide sequence scores through the integrated search tool  .
and importing search results , users can click individual PSMs to view annotated peaks . This particular MS/MS spectrum was obtained during analysis of peptides modified with the CDK7 kinase inhibitor THZ1 and yields a low-confidence Mascot score due to the presence of several inhibitor-related ions and non-canonical fragmentation pathways. Using mzStudio’s built-in spectral processing tools , users can easily experiment with different processing algorithms (i.e., filter inhibitor related ions, reduce charge of highly charged species, and score ions from inhibitor related fragmentation pathways), and assess the impact on peptide sequence scores through the integrated search tool .
.
and importing search results , users can click individual PSMs to view annotated peaks . This particular MS/MS spectrum was obtained during analysis of peptides modified with the CDK7 kinase inhibitor THZ1 and yields a low-confidence Mascot score due to the presence of several inhibitor-related ions and non-canonical fragmentation pathways. Using mzStudio’s built-in spectral processing tools , users can easily experiment with different processing algorithms (i.e., filter inhibitor related ions, reduce charge of highly charged species, and score ions from inhibitor related fragmentation pathways), and assess the impact on peptide sequence scores through the integrated search tool .

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ficarro, S.B.; Alexander, W.M.; Marto, J.A. mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data. Proteomes 2017, 5, 20. https://doi.org/10.3390/proteomes5030020
Ficarro SB, Alexander WM, Marto JA. mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data. Proteomes. 2017; 5(3):20. https://doi.org/10.3390/proteomes5030020
Chicago/Turabian StyleFicarro, Scott B., William M. Alexander, and Jarrod A. Marto. 2017. "mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data" Proteomes 5, no. 3: 20. https://doi.org/10.3390/proteomes5030020
APA StyleFicarro, S. B., Alexander, W. M., & Marto, J. A. (2017). mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data. Proteomes, 5(3), 20. https://doi.org/10.3390/proteomes5030020