Prolyl Endopeptidase-Like Facilitates the α-Synuclein Aggregation Seeding, and This Effect Is Reverted by Serine Peptidase Inhibitor PMSF

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.1.1. α-Synuclein
2.1.2. PREPL and POP
2.2. Activity Assays
2.2.1. PREPL Activity Assay with MUGB
2.2.2. POP Activity Assay with Z-GP-MCA
2.2.3. Tests to Detect PREPL Hydrolytic Activity on Amide Bonds
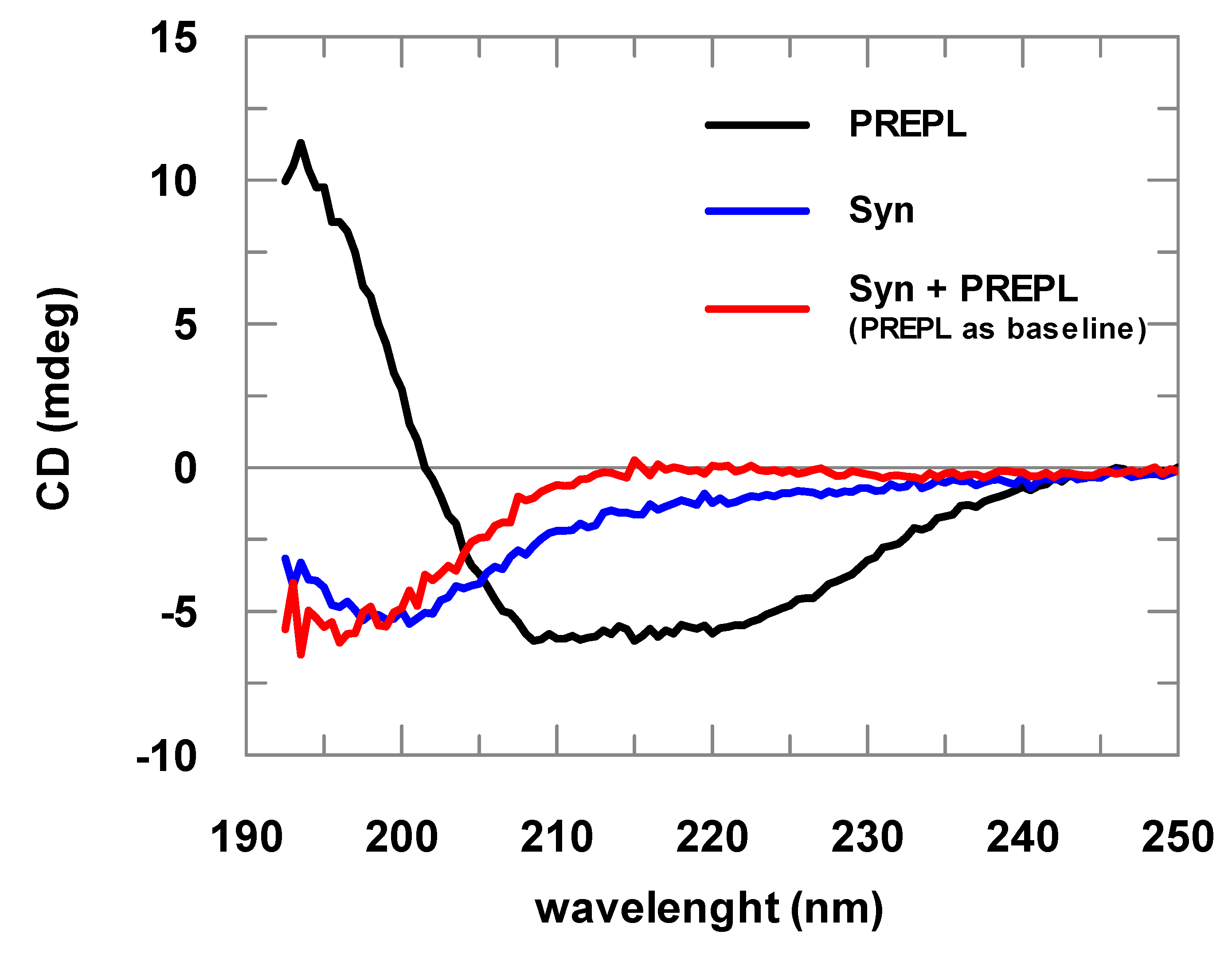
2.3. Circular Dichroism (CD)
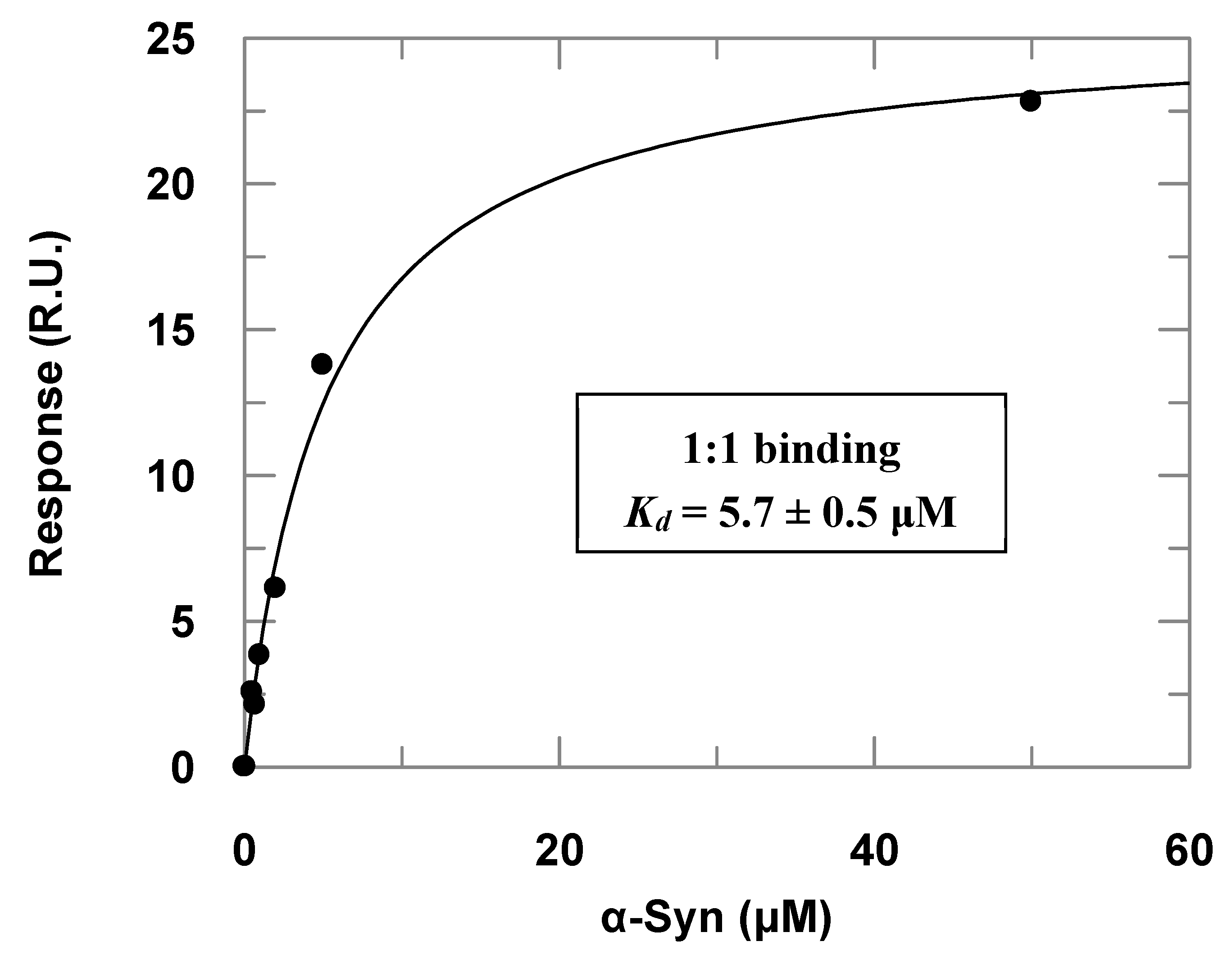
2.4. Surface Plasmon Resonance (SPR)
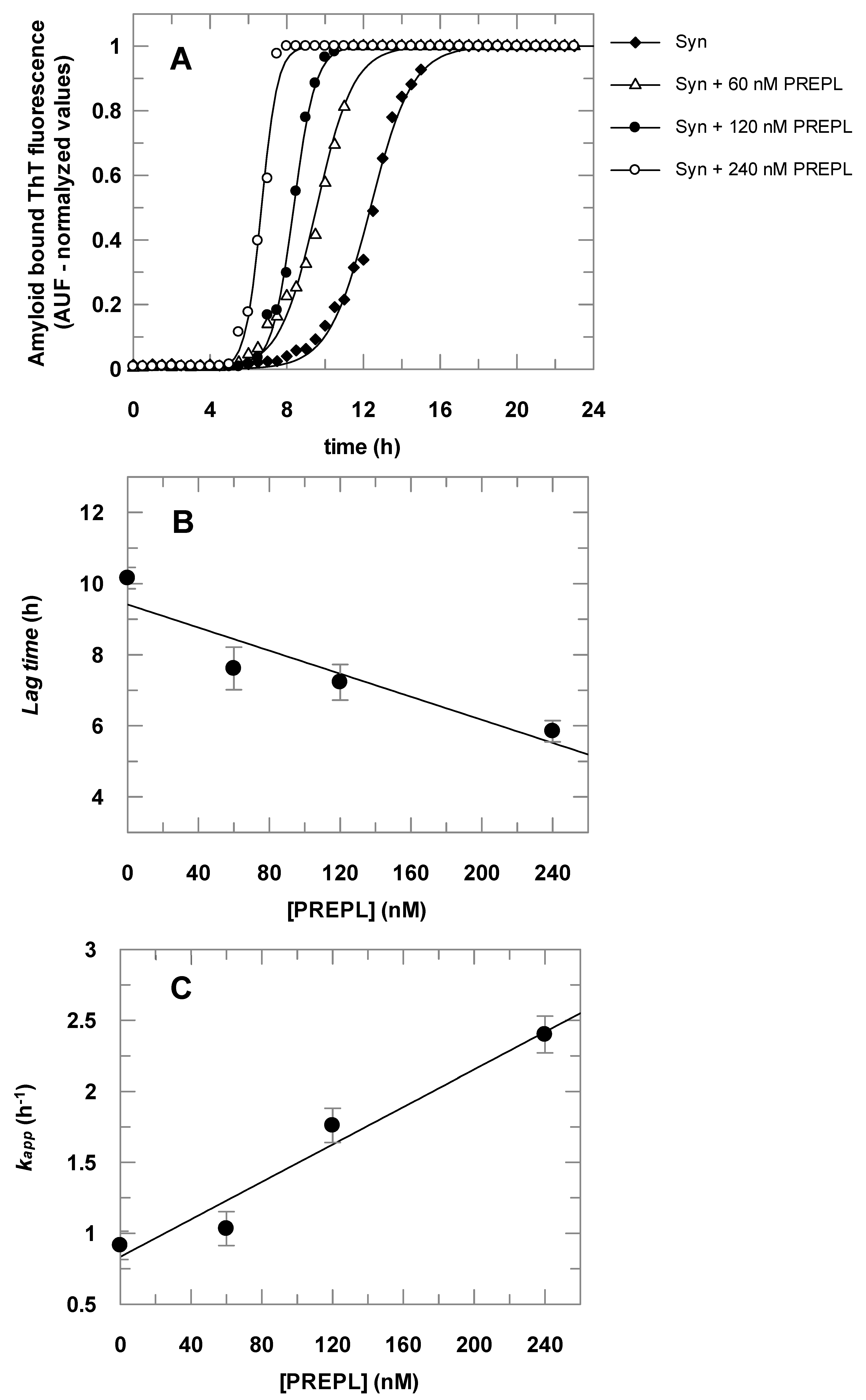
2.5. α-Syn Aggregation Assays
Thioflavin T (ThT) Fluorescence
3. Results and Discussion
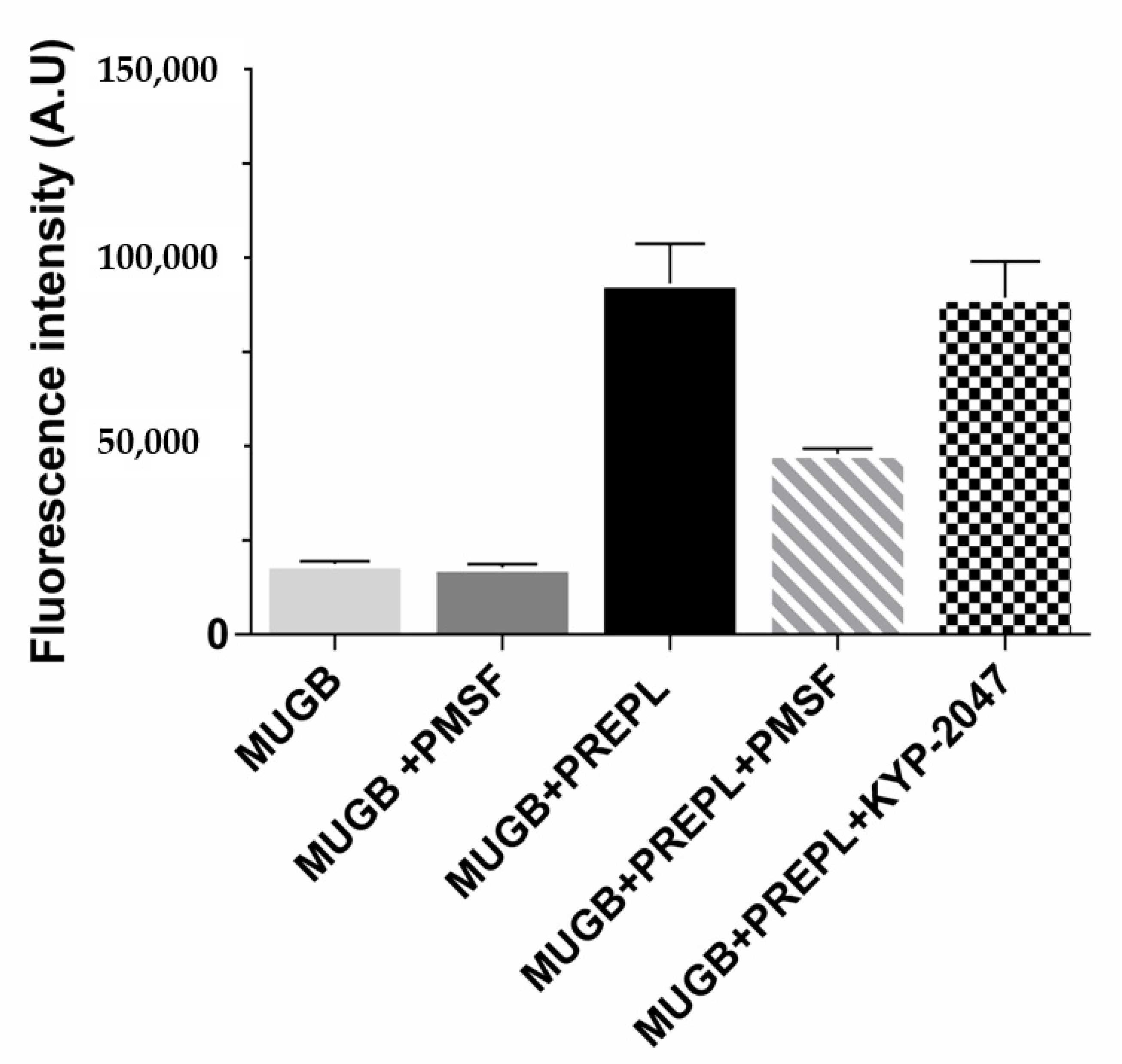
3.1. PREPL Is Active toward an Ester Substrate (MUGB), and It Is Inhibited by PMSF but Not by KYP-2047
3.2. PREPL Does Not Cleave but Interacts with α-Syn
3.3. PREPL Accelerates the α-Syn Oligomerization/Aggregation, and This Effect Is Inhibited by PMSF
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galvin, J.E.; Uryu, K.; Lee, V.M.Y.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, K.; Manabe, Y. The first autopsied case of diffuse Lewy body disease (DLBD): Re-examination by recent immunostaining methods. Neuropathology 2010, 30, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.H.; Galvin, J.E.; Baba, M.; Giasson, B.; Tomita, T.; Leight, S.; Nakajo, S.; Iwatsubo, T.; Trojanowski, J.Q.; Lee, V.M. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann. Neurol. 1998, 44, 415–422. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. α-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Brás, I.C.; Dominguez-Meijide, A.; Gerhardt, E.; Koss, D.; Lázaro, D.F.; Santos, P.I.; Vasili, E.; Xylaki, M.; Outeiro, T.F. Synucleinopathies: Where we are and where we need to go. J. Neurochem. 2020, 153, 433–454. [Google Scholar] [CrossRef]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of α-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Südhof, T.C. A broken α-helix in folded α-synuclein. J. Biol. Chem. 2003, 278, 15313–15318. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell, R.; Browne, G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef]
- Surgucheva, I.; Newell, K.L.; Burns, J.; Surguchov, A. New α- and γ-synuclein immunopathological lesions in human brain. Acta Neuropathol. Commun. 2013, 2, 132. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Delenclos, M.; Burgess, J.D.; Lamprokostopoulou, A.; Outeiro, T.F.; Vekrellis, K.; McLean, P.J. Cellular models of alpha-synuclein toxicity and aggregation. J. Neurochem. 2019, 150, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 1–18. [Google Scholar] [CrossRef]
- Grozdanov, V.; Danzer, K.M. Release and uptake of pathologic alpha-synuclein. Cell Tissue Res. 2018, 373, 175–182. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Masaracchia, C.; Hnida, M.; Gerhardt, E.; Lopes da Fonseca, T.; Villar-Pique, A.; Branco, T.; Stahlberg, M.A.; Dean, C.; Fernández, C.O.; Milosevic, I.; et al. Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol. Commun. 2018, 6, 79. [Google Scholar] [CrossRef]
- Burré, J. The synaptic function of α-synuclein. J. Parkinsons. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Surguchov, A. Intracellular Dynamics of Synucleins. In International Review of Cell and Molecular Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 320, pp. 103–169. [Google Scholar]
- Männistö, P.T.; García-Horsman, J.A. Mechanism of Action of Prolyl Oligopeptidase (PREP) in Degenerative Brain Diseases: Has Peptidase Activity Only a Modulatory Role on the Interactions of PREP with Proteins? Front. Aging Neurosci. 2017, 9, 27. [Google Scholar] [CrossRef]
- Brandt, I.; Gérard, M.; Sergeant, K.; Devreese, B.; Baekelandt, V.; Augustyns, K.; Scharpé, S.; Engelborghs, Y.; Lambeir, A.-M. Prolyl oligopeptidase stimulates the aggregation of alpha-synuclein. Peptides 2008, 29, 1472–1478. [Google Scholar] [CrossRef]
- Savolainen, M.H.; Yan, X.; Myöhänen, T.T.; Huttunen, H.J. Prolyl oligopeptidase enhances α-synuclein dimerization via direct protein-protein interaction. J. Biol. Chem. 2015, 290, 5117–5126. [Google Scholar] [CrossRef] [PubMed]
- Lambeir, A.-M. Interaction of Prolyl Oligopeptidase with α-Synuclein. CNS Neurol. Disord. Drug Targets 2011, 10, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Svarcbahs, R.; Julku, U.H.; Norrbacka, S.; Myöhänen, T.T. Removal of prolyl oligopeptidase reduces alpha-synuclein toxicity in cells and in vivo. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Myöhänen, T.T.; Hannula, M.J.; Van Elzen, R.; Gerard, M.; Van Der Veken, P.; García-Horsman, J.A.; Baekelandt, V.; Männistö, P.T.; Lambeir, A.M. A prolyl oligopeptidase inhibitor, KYP-2047, reduces α-synuclein protein levels and aggregates in cellular and animal models of Parkinson’s disease. Br. J. Pharmacol. 2012, 166, 1097–1113. [Google Scholar] [CrossRef]
- Fülöp, V.; Böcskei, Z.; Polgár, L. Prolyl oligopeptidase: An unusual beta-propeller domain regulates proteolysis. Cell 1998, 94, 161–170. [Google Scholar] [CrossRef]
- Rea, D.; Fülöp, V. Prolyl oligopeptidase structure and dynamics. CNS Neurol. Disord. Drug Targets 2011, 10, 306–310. [Google Scholar] [CrossRef]
- Kichik, N.; Tarragó, T.; Claasen, B.; Gairí, M.; Millet, O.; Giralt, E. 15N Relaxation NMR Studies of Prolyl Oligopeptidase, an 80 kDa Enzyme, Reveal a Pre-existing Equilibrium between Different Conformational States. ChemBioChem 2011, 12, 2737–2739. [Google Scholar] [CrossRef]
- López, A.; Herranz-Trillo, F.; Kotev, M.; Gairí, M.; Guallar, V.; Bernadó, P.; Millet, O.; Tarragó, T.; Giralt, E. Active-Site-Directed Inhibitors of Prolyl Oligopeptidase Abolish Its Conformational Dynamics. ChemBioChem 2016, 17, 913–917. [Google Scholar] [CrossRef]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Männistö, P.T.; Maguire-Zeiss, K.A.; Myöhänen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef]
- Kilpeläinen, T.P.; Hellinen, L.; Vrijdag, J.; Yan, X.; Svarcbahs, R.; Vellonen, K.S.; Lambeir, A.M.; Huttunen, H.; Urtti, A.; Wallen, E.A.A.; et al. The effect of prolyl oligopeptidase inhibitors on alpha-synuclein aggregation and autophagy cannot be predicted by their inhibitory efficacy. Biomed. Pharmacother. 2020, 128, 110253. [Google Scholar] [CrossRef] [PubMed]
- Svarcbahs, R.; Jäntti, M.; Kilpeläinen, T.; Julku, U.H.; Urvas, L.; Kivioja, S.; Norrbacka, S.; Myöhänen, T.T. Prolyl oligopeptidase inhibition activates autophagy via protein phosphatase 2A. Pharmacol. Res. 2020, 151, 104558. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Dokleja, L.; Hannula, M.J.; Myöhänen, T.T. Inhibition of prolyl oligopeptidase increases the survival of alpha-synuclein overexpressing cells after rotenone exposure by reducing alpha-synuclein oligomers. Neurosci. Lett. 2014, 583, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Svarcbahs, R.; Julku, U.H.; Myöhänen, T.T. Inhibition of Prolyl Oligopeptidase Restores Spontaneous Motor Behavior in the α-Synuclein Virus Vector-Based Parkinson’s Disease Mouse Model by Decreasing α-Synuclein Oligomeric Species in Mouse Brain. J. Neurosci. 2016, 36, 12485–12497. [Google Scholar] [CrossRef] [PubMed]
- der Veken, P.; Fülöp, V.; Rea, D.; Gerard, M.; Van Elzen, R.; Joossens, J.; Cheng, J.D.; Baekelandt, V.; De Meester, I.; Lambeir, A.M.; et al. P2-substituted N-acylprolylpyrrolidine inhibitors of prolyl oligopeptidase: Biochemical evaluation, binding mode determination, and assessment in a cellular model of synucleinopathy. J. Med. Chem. 2012, 55, 9856–9867. [Google Scholar] [CrossRef]
- Myöhänen, T.T.; Norrbacka, S.; Savolainen, M.H. Prolyl oligopeptidase inhibition attenuates the toxicity of a proteasomal inhibitor, lactacystin, in the alpha-synuclein overexpressing cell culture. Neurosci. Lett. 2017, 636, 83–89. [Google Scholar] [CrossRef]
- Svarcbahs, R.; Julku, U.; Kilpeläinen, T.; Kyyrö, M.; Jäntti, M.; Myöhänen, T.T. New tricks of prolyl oligopeptidase inhibitors—A common drug therapy for several neurodegenerative diseases. Biochem. Pharmacol. 2019, 161, 113–120. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef]
- Jaeken, J.; Martens, K.; François, I.; Eyskens, F.; Lecointre, C.; Derua, R.; Meulemans, S.; Slootstra, J.W.; Waelkens, E.; De Zegher, F.; et al. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am. J. Hum. Genet. 2006, 78, 38–51. [Google Scholar] [CrossRef]
- Szeltner, Z.; Alshafee, I.; Juhász, T.; Parvari, R.; Polgár, L. The PREPL A protein, a new member of the prolyl oligopeptidase family, lacking catalytic activity. Cell. Mol. Life Sci. 2005, 62, 2376–2381. [Google Scholar] [CrossRef] [PubMed]
- Boonen, K.; Regal, L.; Jaeken, J.; WM Creemers, J. PREPL, a Prolyl Endopeptidase-Like Enzyme by Name Only? – Lessons from Patients. CNS Neurol. Disord. Drug Targets 2011, 10, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Lone, A.M.; Bachovchin, D.A.; Westwood, D.B.; Speers, A.E.; Spicer, T.P.; Fernandez-Vega, V.; Chase, P.; Hodder, P.S.; Rosen, H.; Cravatt, B.F.; et al. A substrate-free activity-based protein profiling screen for the discovery of selective PREPL inhibitors. J. Am. Chem. Soc. 2011, 133, 11665–11674. [Google Scholar] [CrossRef][Green Version]
- Laugwitz, L.; Redler, S.; Buchert, R.; Sturm, M.; Zeile, I.; Schara, U.; Wieczorek, D.; Haack, T.; Distelmaier, F. Isolated PREPL deficiency associated with congenital myasthenic syndrome-22. Klin. Padiatr. 2018, 230, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Régal, L.; Mårtensson, E.; Maystadt, I.; Voermans, N.; Lederer, D.; Burlina, A.; Juan Fita, M.J.; Hoogeboom, A.J.M.; Olsson Engman, M.; Hollemans, T.; et al. PREPL deficiency: Delineation of the phenotype and development of a functional blood assay. Genet. Med. 2018, 20, 109–118. [Google Scholar] [CrossRef]
- Régal, L.; Shen, X.M.; Selcen, D.; Verhille, C.; Meulemans, S.; Creemers, J.W.M.; Engel, A.G. PREPL deficiency with or without cystinuria causes a novel myasthenic syndrome. Neurology 2014, 82, 1254–1260. [Google Scholar] [CrossRef]
- Chabrol, B.; Martens, K.; Meulemans, S.; Cano, A.; Jaeken, J.; Matthijs, G.; Creemers, J.W.M. Deletion of C2orf34, PREPL and SLC3A1 causes atypical hypotonia-cystinuria syndrome. J. Med. Genet. 2008, 45, 314–318. [Google Scholar] [CrossRef]
- Chabrol, B.; Martens, K.; Meulemans, S.; Cano, A.; Jaeken, J.; Matthijs, G.; Creemers, J.W.M. Deletion of C2orf34, PREPL and SLC3A1 causes atypical hypotonia-cystinuria syndrome. BMJ Case Rep. 2009. [Google Scholar] [CrossRef]
- Parvari, R.; Gonen, Y.; Alshafee, I.; Buriakovsky, S.; Regev, K.; Hershkovitz, E. The 2p21 deletion syndrome: Characterization of the transcription content. Genomics 2005, 86, 195–211. [Google Scholar] [CrossRef]
- Morawski, M.; Nuytens, K.; Juhasz, T.; Zeitschel, U.; Seeger, G.; Waelkens, E.; Regal, L.; Schulz, I.; Arendt, T.; Szeltner, Z.; et al. Cellular and ultra structural evidence for cytoskeletal localization of prolyl endopeptidase-like protein in neurons. Neuroscience 2013, 242, 128–139. [Google Scholar] [CrossRef]
- Coelho-Cerqueira, E.; Carmo-Gonçalves, P.; Sá Pinheiro, A.; Cortines, J.; Follmer, C. α-Synuclein as an intrinsically disordered monomer—Fact or artefact? FEBS J. 2013, 280, 4915–4927. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- Willander, H.; Presto, J.; Askarieh, G.; Biverstål, H.; Frohm, B.; Knight, S.D.; Johansson, J.; Linse, S. BRICHOS domains efficiently delay fibrillation of amyloid β-peptide. J. Biol. Chem. 2012, 287, 31608–31617. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, A.J.; Leikas, J.V.; Forsberg, M.M. KYP-2047 penetrates mouse brain and effectively inhibits mouse prolyl oligopeptidase. Basic Clin. Pharmacol. Toxicol. 2014, 114, 460–463. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample and Condition | kapp (h−1) | Lag Time (h) |
|---|---|---|
| α-Syn | 1.18 ± 0.06 | 10.3 ± 0.3 |
| α-Syn + PMSF | 1.11 ± 0.09 | 10.1 ± 0.5 |
| α-Syn + KYP-2047 | 1.2 ± 0.1 | 9.9 ± 0.5 |
| α-Syn + PREPLa | 1.76 ± 0.06 | 7.4 ± 0.5 |
| α-Syn + PREPLa + PMSF | 1.2 ± 0.1 | 15.7 ± 0.5 |
| α-Syn + POPa | 1.7 ± 0.1 | 7.7 ± 0.7 |
| α-Syn + POPa + PMSF | 1.0 ± 0.1 | 13.2 ± 0.4 |
| α-Syn + POPa + KYP-2047 | 1.2 ± 0.1 | 12.2 ± 0.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, G.S.; Oyadomari, W.Y.; Carvalho, E.A.; Torquato, R.S.; Oliveira, V. Prolyl Endopeptidase-Like Facilitates the α-Synuclein Aggregation Seeding, and This Effect Is Reverted by Serine Peptidase Inhibitor PMSF. Biomolecules 2020, 10, 962. https://doi.org/10.3390/biom10060962
Santos GS, Oyadomari WY, Carvalho EA, Torquato RS, Oliveira V. Prolyl Endopeptidase-Like Facilitates the α-Synuclein Aggregation Seeding, and This Effect Is Reverted by Serine Peptidase Inhibitor PMSF. Biomolecules. 2020; 10(6):962. https://doi.org/10.3390/biom10060962
Chicago/Turabian StyleSantos, Gabriel S., William Y. Oyadomari, Elizangela A. Carvalho, Ricardo S. Torquato, and Vitor Oliveira. 2020. "Prolyl Endopeptidase-Like Facilitates the α-Synuclein Aggregation Seeding, and This Effect Is Reverted by Serine Peptidase Inhibitor PMSF" Biomolecules 10, no. 6: 962. https://doi.org/10.3390/biom10060962
APA StyleSantos, G. S., Oyadomari, W. Y., Carvalho, E. A., Torquato, R. S., & Oliveira, V. (2020). Prolyl Endopeptidase-Like Facilitates the α-Synuclein Aggregation Seeding, and This Effect Is Reverted by Serine Peptidase Inhibitor PMSF. Biomolecules, 10(6), 962. https://doi.org/10.3390/biom10060962