Endoglin: Beyond the Endothelium


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Endoglin Structure and Function
2.1. Endoglin Signaling Pathways and Ligands
2.2. Endoglin and Developmental/Tumor Angiogenesis
2.3. Non-Ligand-Dependent Interactions (Integrins/Leukocyte Trafficking)
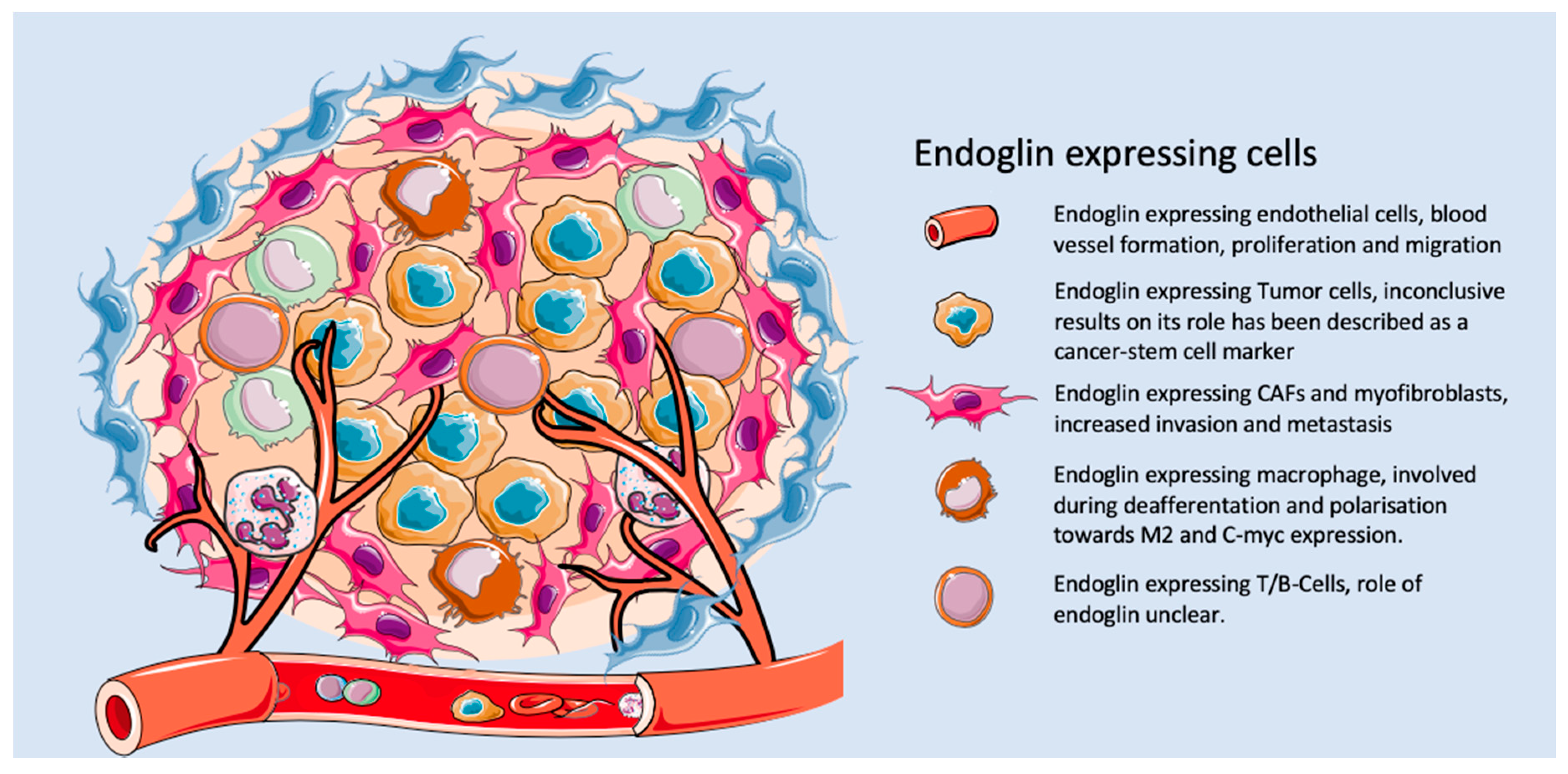
2.4. Endoglin beyond the Endothelium
2.5. Endoglin Expression on Epithelial Cells
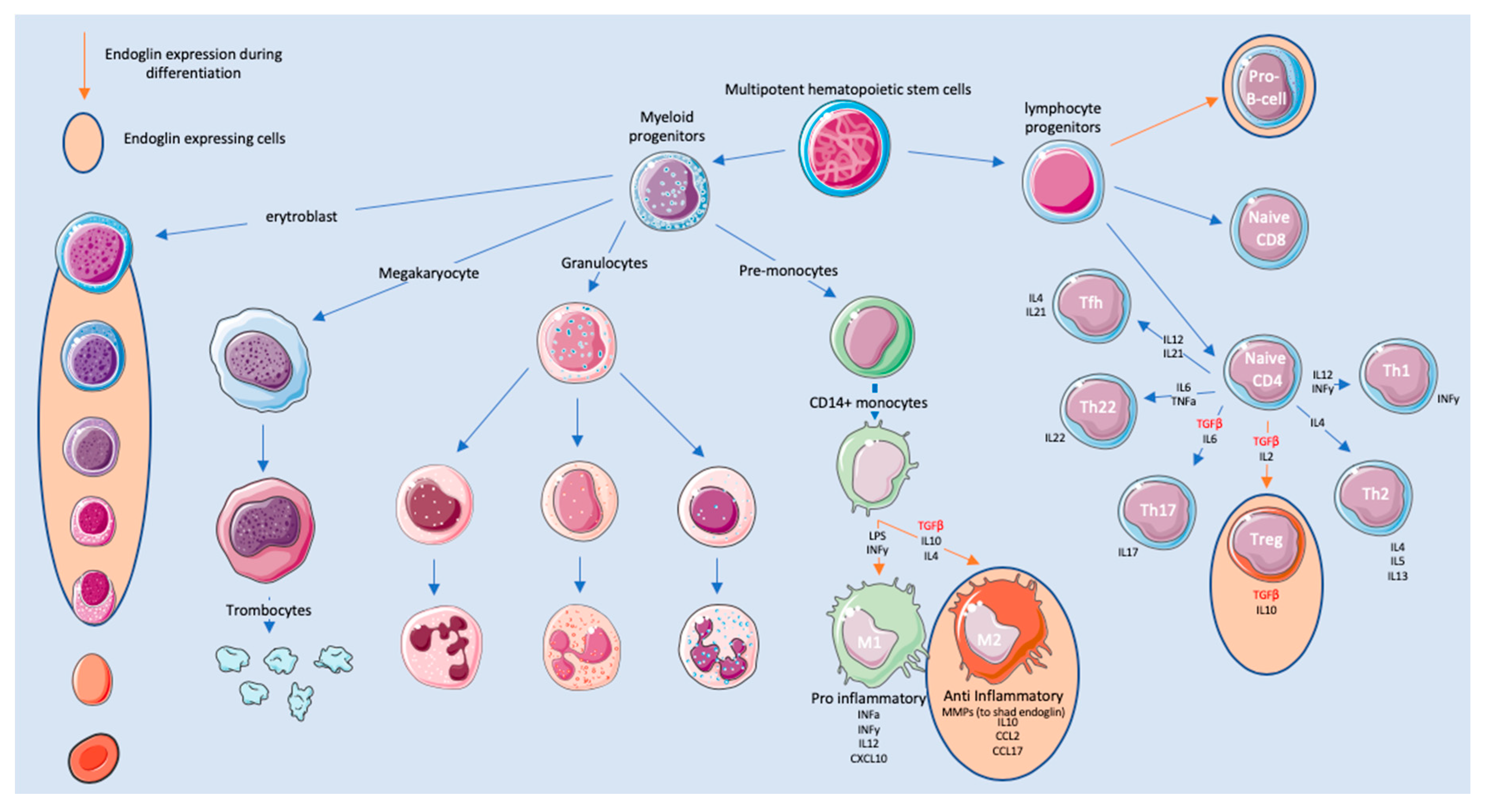
2.6. Endoglin Expression during Haematopoiesis
2.7. Endoglin Expressed on Innate Immune Cells
2.8. Endoglin on Cells of the Adaptive Immune System
2.9. Endoglin Expression on Fibroblasts
2.10. Endoglin on Mesenchymal Stem Cells (MSCs)
2.11. Endoglin-Expressing Fibroblasts in Fibrosis
2.12. Endoglin Expression in Cancer-Associated Fibroblasts (CAFs)
3. Targeting Endoglin in Diseases
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wikstrom, P.; Lissbrant, I.F.; Stattin, P.; Egevad, L.; Bergh, A. Endoglin (CD105) is expressed on immature blood vessels and is a marker for survival in prostate cancer. Prostate 2002, 51, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Arthur, H.M.; Ure, J.; Smith, A.J.; Renforth, G.; Wilson, D.I.; Torsney, E.; Charlton, R.; Parums, D.V.; Jowett, T.; Marchuk, D.A.; et al. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 2000, 217, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Ten Dijke, P. TGF-beta Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed]
- Conley, B.A.; Koleva, R.; Smith, J.D.; Kacer, D.; Zhang, D.; Bernabeu, C.; Vary, C.P. Endoglin controls cell migration and composition of focal adhesions: Function of the cytosolic domain. J. Biol. Chem. 2004, 279, 27440–27449. [Google Scholar] [CrossRef]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef]
- Bautch, V.L. Endoglin moves and shapes endothelial cells. Nat. Cell Biol. 2017, 19, 593–595. [Google Scholar] [CrossRef]
- Kumar, P.; Wang, J.M.; Bernabeu, C. CD105 and angiogenesis. J. Pathol. 1996, 178, 363–366. [Google Scholar] [CrossRef]
- Seon, B.K.; Haba, A.; Matsuno, F.; Takahashi, N.; Tsujie, M.; She, X.; Harada, N.; Uneda, S.; Tsujie, T.; Toi, H.; et al. Endoglin-targeted cancer therapy. Curr. Drug Deliv. 2011, 8, 135–143. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Goumans, M.J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar] [CrossRef]
- Paauwe, M.; ten Dijke, P.; Hawinkels, L.J. Endoglin for tumor imaging and targeted cancer therapy. Expert Opin. Ther. Targets 2013, 17, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Gougos, A.; Letarte, M. Identification of a human endothelial cell antigen with monoclonal antibody 44G4 produced against a pre-B leukemic cell line. J. Immunol. 1988, 141, 1925–1933. [Google Scholar] [PubMed]
- Yamashita, H.; Ichijo, H.; Grimsby, S.; Moren, A.; ten Dijke, P.; Miyazono, K. Endoglin forms a heteromeric complex with the signaling receptors for transforming growth factor-beta. J. Biol. Chem. 1994, 269, 1995–2001. [Google Scholar] [PubMed]
- Luque, A.; Cabanas, C.; Raab, U.; Letamendia, A.; Paez, E.; Herreros, L.; Sanchez-Madrid, F.; Bernabeu, C. The use of recombinant vaccinia virus to generate monoclonal antibodies against the cell-surface glycoprotein endoglin. FEBS Lett. 1997, 413, 265–268. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, E.; St-Jacques, S.; Bellon, T.; Letarte, M.; Bernabeu, C. Assignment of the human endoglin gene (END) to 9q34→qter. Cytogenet Cell Genet. 1993, 64, 204–207. [Google Scholar] [CrossRef]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef]
- Pece, N.; Vera, S.; Cymerman, U.; White, R.I., Jr.; Wrana, J.L.; Letarte, M. Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J. Clin. Investig. 1997, 100, 2568–2579. [Google Scholar] [CrossRef]
- Gougos, A.; Letarte, M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar]
- Velasco, S.; Alvarez-Munoz, P.; Pericacho, M.; Dijke, P.T.; Bernabeu, C.; Lopez-Novoa, J.M.; Rodriguez-Barbero, A. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef]
- Bellon, T.; Corbi, A.; Lastres, P.; Cales, C.; Cebrian, M.; Vera, S.; Cheifetz, S.; Massague, J.; Letarte, M.; Bernabeu, C. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur. J. Immunol. 1993, 23, 2340–2345. [Google Scholar] [CrossRef]
- Perez-Gomez, E.; Eleno, N.; Lopez-Novoa, J.M.; Ramirez, J.R.; Velasco, B.; Letarte, M.; Bernabeu, C.; Quintanilla, M. Characterization of murine S-endoglin isoform and its effects on tumor development. Oncogene 2005, 24, 4450–4461. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Grande, M.T.; Langa, C.; Oujo, B.; Velasco, S.; Rodriguez-Barbero, A.; Perez-Gomez, E.; Quintanilla, M.; Lopez-Novoa, J.M.; Bernabeu, C. S-endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ. Res. 2008, 103, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Elsner, T.; Botella, L.M.; Velasco, B.; Langa, C.; Bernabeu, C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J. Biol. Chem. 2002, 277, 43799–43808. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.; ten Dijke, P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P.; et al. MMP-12, Secreted by Pro-Inflammatory Macrophages, Targets Endoglin in Human Macrophages and Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Del Castillo, G.; Sanchez-Blanco, E.; Martin-Villar, E.; Valbuena-Diez, A.C.; Langa, C.; Perez-Gomez, E.; Renart, J.; Bernabeu, C.; Quintanilla, M. Soluble endoglin antagonizes Met signaling in spindle carcinoma cells. Carcinogenesis 2015, 36, 212–222. [Google Scholar] [CrossRef][Green Version]
- Li, C.; Guo, B.; Wilson, P.B.; Stewart, A.; Byrne, G.; Bundred, N.; Kumar, S. Plasma levels of soluble CD105 correlate with metastasis in patients with breast cancer. Int. J. Cancer 2000, 89, 122–126. [Google Scholar] [CrossRef]
- Perez-Gomez, E.; Villa-Morales, M.; Santos, J.; Fernandez-Piqueras, J.; Gamallo, C.; Dotor, J.; Bernabeu, C.; Quintanilla, M. A role for endoglin as a suppressor of malignancy during mouse skin carcinogenesis. Cancer Res. 2007, 67, 10268–10277. [Google Scholar] [CrossRef]
- Li, C.G.; Wilson, P.B.; Bernabeu, C.; Raab, U.; Wang, J.M.; Kumar, S. Immunodetection and characterisation of soluble CD105-TGFbeta complexes. J. Immunol. Methods 1998, 218, 85–93. [Google Scholar] [CrossRef]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Vara, E.; Ruiz-Llorente, L.; Casado-Vela, J.; Ruiz-Rodriguez, M.J.; Lopez-Andres, N.; Pattnaik, A.K.; Quintanilla, M.; Bernabeu, C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells 2019, 8, 1082. [Google Scholar] [CrossRef] [PubMed]
- Lawera, A.; Tong, Z.; Thorikay, M.; Redgrave, R.E.; Cai, J.; van Dinther, M.; Morrell, N.W.; Afink, G.B.; Charnock-Jones, D.S.; Arthur, H.M.; et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 17800–17808. [Google Scholar] [CrossRef] [PubMed]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Alt, A.; Miguel-Romero, L.; Donderis, J.; Aristorena, M.; Blanco, F.J.; Round, A.; Rubio, V.; Bernabeu, C.; Marina, A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE 2012, 7, e29948. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Salmon, R.M.; Jiang, H.; Morrell, N.W. Regulation of the ALK1 ligands, BMP9 and BMP10. Biochem. Soc. Trans. 2016, 44, 1135–1141. [Google Scholar] [CrossRef]
- Saito, T.; Bokhove, M.; Croci, R.; Zamora-Caballero, S.; Han, L.; Letarte, M.; de Sanctis, D.; Jovine, L. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017, 19, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Mallet, C.; Keramidas, M.; Lamande, N.; Gasc, J.M.; Dupuis-Girod, S.; Plauchu, H.; Feige, J.J.; Bailly, S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008, 102, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Bilandzic, M.; Stenvers, K.L. Reprint of: Betaglycan: A multifunctional accessory. Mol. Cell. Endocrinol. 2012, 359, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Casillas, F.; Cheifetz, S.; Doody, J.; Andres, J.L.; Lane, W.S.; Massague, J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell 1991, 67, 785–795. [Google Scholar] [CrossRef]
- Lopez-Casillas, F.; Wrana, J.L.; Massague, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Sarraj, M.A.; Chua, H.K.; Umbers, A.; Loveland, K.L.; Findlay, J.K.; Stenvers, K.L. Differential expression of TGFBR3 (betaglycan) in mouse ovary and testis during gonadogenesis. Growth Factors 2007, 25, 334–345. [Google Scholar] [CrossRef]
- Compton, L.A.; Potash, D.A.; Brown, C.B.; Barnett, J.V. Coronary vessel development is dependent on the type III transforming growth factor beta receptor. Circ. Res. 2007, 101, 784–791. [Google Scholar] [CrossRef]
- Dong, M.; How, T.; Kirkbride, K.C.; Gordon, K.J.; Lee, J.D.; Hempel, N.; Kelly, P.; Moeller, B.J.; Marks, J.R.; Blobe, G.C. The type III TGF-beta receptor suppresses breast cancer progression. J. Clin. Investig. 2007, 117, 206–217. [Google Scholar] [CrossRef]
- Hempel, N.; How, T.; Cooper, S.J.; Green, T.R.; Dong, M.; Copland, J.A.; Wood, C.G.; Blobe, G.C. Expression of the type III TGF-beta receptor is negatively regulated by TGF-beta. Carcinogenesis 2008, 29, 905–912. [Google Scholar] [CrossRef]
- Hempel, N.; How, T.; Dong, M.; Murphy, S.K.; Fields, T.A.; Blobe, G.C. Loss of betaglycan expression in ovarian cancer: Role in motility and invasion. Cancer Res. 2007, 67, 5231–5238. [Google Scholar] [CrossRef] [PubMed]
- Steiner, W.R. Hereditary Haemorrhagic Telangiectasia, with Report of Three Families and a Review of those previously recorded. Trans. Am. Climatol. Clin. Assoc. 1916, 32, 77–94. [Google Scholar] [PubMed]
- Bourdeau, A.; Dumont, D.J.; Letarte, M. A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 1999, 104, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Tual-Chalot, S.; Oh, S.P.; Arthur, H.M. Mouse models of hereditary hemorrhagic telangiectasia: Recent advances and future challenges. Front Genet. 2015, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.L.; Terzic, A.; Conley, B.A.; Oxburgh, L.H.; Nicola, T.; Vary, C.P. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev. Dyn. 2009, 238, 2479–2493. [Google Scholar] [CrossRef]
- Guilhem, A.; Malcus, C.; Clarivet, B.; Plauchu, H.; Dupuis-Girod, S. Immunological abnormalities associated with hereditary haemorrhagic telangiectasia. J. Int. Med. 2013, 274, 351–362. [Google Scholar] [CrossRef]
- Cirulli, A.; Loria, M.P.; Dambra, P.; Di Serio, F.; Ventura, M.T.; Amati, L.; Jirillo, E.; Sabba, C. Patients with Hereditary Hemorrhagic Telangectasia (HHT) exhibit a deficit of polymorphonuclear cell and monocyte oxidative burst and phagocytosis: A possible correlation with altered adaptive immune responsiveness in HHT. Curr. Pharm. Des. 2006, 12, 1209–1215. [Google Scholar] [CrossRef]
- Gougos, A.; St Jacques, S.; Greaves, A.; O’Connell, P.J.; d’Apice, A.J.; Buhring, H.J.; Bernabeu, C.; van Mourik, J.A.; Letarte, M. Identification of distinct epitopes of endoglin, an RGD-containing glycoprotein of endothelial cells, leukemic cells, and syncytiotrophoblasts. Int. Immunol. 1992, 4, 83–92. [Google Scholar] [CrossRef]
- Lastres, P.; Bellon, T.; Cabanas, C.; Sanchez-Madrid, F.; Acevedo, A.; Gougos, A.; Letarte, M.; Bernabeu, C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur. J. Immunol. 1992, 22, 393–397. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Duwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabanas, C.; Lopez-Novoa, J.M.; Bernabeu, C. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef]
- Rossi, E.; Lopez-Novoa, J.M.; Bernabeu, C. Endoglin involvement in integrin-mediated cell adhesion as a putative pathogenic mechanism in hereditary hemorrhagic telangiectasia type 1 (HHT1). Front. Genet. 2014, 5, 457. [Google Scholar] [CrossRef] [PubMed]
- Quackenbush, E.J.; Letarte, M. Identification of several cell surface proteins of non-T, non-B acute lymphoblastic leukemia by using monoclonal antibodies. J. Immunol. 1985, 134, 1276–1285. [Google Scholar] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, E.; Jerkic, M.; Prieto, M.; Del Castillo, G.; Martin-Villar, E.; Letarte, M.; Bernabeu, C.; Perez-Barriocanal, F.; Quintanilla, M.; Lopez-Novoa, J.M. Impaired wound repair in adult endoglin heterozygous mice associated with lower NO bioavailability. J. Invest Dermatol. 2014, 134, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jovanovic, B.; Pins, M.; Lee, C.; Bergan, R.C. Over expression of endoglin in human prostate cancer suppresses cell detachment, migration and invasion. Oncogene 2002, 21, 8272–8281. [Google Scholar] [CrossRef][Green Version]
- Lakshman, M.; Huang, X.; Ananthanarayanan, V.; Jovanovic, B.; Liu, Y.; Craft, C.S.; Romero, D.; Vary, C.P.; Bergan, R.C. Endoglin suppresses human prostate cancer metastasis. Clin. Exp. Metastasis 2011, 28, 39–53. [Google Scholar] [CrossRef][Green Version]
- Henry, L.A.; Johnson, D.A.; Sarrio, D.; Lee, S.; Quinlan, P.R.; Crook, T.; Thompson, A.M.; Reis-Filho, J.S.; Isacke, C.M. Endoglin expression in breast tumor cells suppresses invasion and metastasis and correlates with improved clinical outcome. Oncogene 2011, 30, 1046–1058. [Google Scholar] [CrossRef]
- Wong, V.C.; Chan, P.L.; Bernabeu, C.; Law, S.; Wang, L.D.; Li, J.L.; Tsao, S.W.; Srivastava, G.; Lung, M.L. Identification of an invasion and tumor-suppressing gene, Endoglin (ENG), silenced by both epigenetic inactivation and allelic loss in esophageal squamous cell carcinoma. Int. J. Cancer 2008, 123, 2816–2823. [Google Scholar] [CrossRef]
- Li, Y.; Zhai, Z.; Liu, D.; Zhong, X.; Meng, X.; Yang, Q.; Liu, J.; Li, H. CD105 promotes hepatocarcinoma cell invasion and metastasis through VEGF. Tumour. Biol. 2015, 36, 737–745. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, B.; Zhang, H.; Li, H. Human epithelial ovarian cancer cells expressing CD105, CD44 and CD106 surface markers exhibit increased invasive capacity and drug resistance. Oncol. Lett. 2019, 17, 5351–5360. [Google Scholar] [CrossRef]
- Hu, J.; Guan, W.; Yan, L.; Ye, Z.; Wu, L.; Xu, H. Cancer Stem Cell Marker Endoglin (CD105) Induces Epithelial Mesenchymal Transition (EMT) but Not Metastasis in Clear Cell Renal Cell Carcinoma. Stem. Cells Int. 2019, 2019, 9060152. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, M.; de Graaf, D.; Monti, S.; Gottgens, B.; Sanchez, M.J.; Lander, E.S.; Golub, T.R.; Green, A.R.; Lodish, H.F. Identification of endoglin as a functional marker that defines long-term repopulating hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 15468–15473. [Google Scholar] [CrossRef]
- Rokhlin, O.W.; Cohen, M.B.; Kubagawa, H.; Letarte, M.; Cooper, M.D. Differential expression of endoglin on fetal and adult hematopoietic cells in human bone marrow. J. Immunol. 1995, 154, 4456–4465. [Google Scholar]
- Moody, J.L.; Singbrant, S.; Karlsson, G.; Blank, U.; Aspling, M.; Flygare, J.; Bryder, D.; Karlsson, S. Endoglin is not critical for hematopoietic stem cell engraftment and reconstitution but regulates adult erythroid development. Stem. Cells 2007, 25, 2809–2819. [Google Scholar] [CrossRef] [PubMed]
- Hettinger, J.; Richards, D.M.; Hansson, J.; Barra, M.M.; Joschko, A.C.; Krijgsveld, J.; Feuerer, M. Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol. 2013, 14, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef]
- O’Connell, P.J.; McKenzie, A.; Fisicaro, N.; Rockman, S.P.; Pearse, M.J.; d’Apice, A.J. Endoglin: A 180-kD endothelial cell and macrophage restricted differentiation molecule. Clin. Exp. Immunol. 1992, 90, 154–159. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Aristorena, M.; Blanco, F.J.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Gallardo-Vara, E.; Corbi, A.; Botella, L.M.; Bernabeu, C. Expression of endoglin isoforms in the myeloid lineage and their role during aging and macrophage polarization. J. Cell Sci. 2014, 127, 2723–2735. [Google Scholar] [CrossRef]
- Sundstrom, C.; Nilsson, K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 1976, 17, 565–577. [Google Scholar] [CrossRef]
- Lastres, P.; Letamendia, A.; Zhang, H.; Rius, C.; Almendro, N.; Raab, U.; Lopez, L.A.; Langa, C.; Fabra, A.; Letarte, M.; et al. Endoglin modulates cellular responses to TGF-beta 1. J. Cell. Biol. 1996, 133, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-Fernandez, L.; Recio-Poveda, L.; Aristorena, M.; Lastres, P.; Blanco, F.J.; Sanz-Rodriguez, F.; Gallardo-Vara, E.; de las Casas-Engel, M.; Corbi, A.; Arthur, H.M.; et al. Mice Lacking Endoglin in Macrophages Show an Impaired Immune Response. PLoS Genet. 2016, 12, e1005935. [Google Scholar] [CrossRef] [PubMed]
- Scharpfenecker, M.; Floot, B.; Russell, N.S.; Stewart, F.A. The TGF-beta co-receptor endoglin regulates macrophage infiltration and cytokine production in the irradiated mouse kidney. Radiother Oncol. 2012, 105, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Dingenouts, C.K.; Goumans, M.J.; Bakker, W. Mononuclear cells and vascular repair in HHT. Front. Genet. 2015, 6, 114. [Google Scholar] [CrossRef]
- Chakhachiro, Z.I.; Zuo, Z.; Aladily, T.N.; Kantarjian, H.M.; Cortes, J.E.; Alayed, K.; Nguyen, M.H.; Medeiros, L.J.; Bueso-Ramos, C. CD105 (endoglin) is highly overexpressed in a subset of cases of acute myeloid leukemias. Am. J. Clin. Pathol. 2013, 140, 370–378. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Schmidt-Weber, C.B.; Letarte, M.; Kunzmann, S.; Ruckert, B.; Bernabeu, C.; Blaser, K. TGF-{beta} signaling of human T cells is modulated by the ancillary TGF-{beta} receptor endoglin. Int. Immunol. 2005, 17, 921–930. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound. Care 2013, 22, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Velnar, T.; Bailey, T.; Smrkolj, V. The wound healing process: An overview of the cellular and molecular mechanisms. J. Int. Med. Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef]
- Desmouliere, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar]
- Da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Piatetzky, S., II; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar] [PubMed]
- Caplan, A.I.; Correa, D. The MSC: An injury drugstore. Cell Stem. Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, S.; Bouffi, C.; Djouad, F.; Jorgensen, C.; Noel, D. Immunosuppression by mesenchymal stem cells: Mechanisms and clinical applications. Stem. Cell Res. Ther. 2010, 1, 2. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Barry, F.P.; Boynton, R.E.; Haynesworth, S.; Murphy, J.M.; Zaia, J. The monoclonal antibody SH-2, raised against human mesenchymal stem cells, recognizes an epitope on endoglin (CD105). Biochem. Biophys. Res. Commun. 1999, 265, 134–139. [Google Scholar] [CrossRef]
- Rosu-Myles, M.; Fair, J.; Pearce, N.; Mehic, J. Non-multipotent stroma inhibit the proliferation and differentiation of mesenchymal stromal cells in vitro. Cytotherapy 2010, 12, 818–830. [Google Scholar] [CrossRef]
- Levi, B.; Wan, D.C.; Glotzbach, J.P.; Hyun, J.; Januszyk, M.; Montoro, D.; Sorkin, M.; James, A.W.; Nelson, E.R.; Li, S.; et al. CD105 protein depletion enhances human adipose-derived stromal cell osteogenesis through reduction of transforming growth factor beta1 (TGF-beta1) signaling. J. Biol. Chem. 2011, 286, 39497–39509. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Carrillo-Galvez, A.B.; Garcia-Perez, A.; Cobo, M.; Martin, F. CD105 (endoglin)-negative murine mesenchymal stromal cells define a new multipotent subpopulation with distinct differentiation and immunomodulatory capacities. PLoS ONE 2013, 8, e76979. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; van der Schaft, D.W.; Wiercinska, E.; Gorter, A.; Hogendoorn, P.C.; Griffioen, A.W.; ten Dijke, P. Critical role of endoglin in tumor cell plasticity of Ewing sarcoma and melanoma. Oncogene 2011, 30, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Postiglione, L.; Di Domenico, G.; Caraglia, M.; Marra, M.; Giuberti, G.; Del Vecchio, L.; Montagnani, S.; Macri, M.; Bruno, E.M.; Abbruzzese, A.; et al. Differential expression and cytoplasm/membrane distribution of endoglin (CD105) in human tumour cell lines: Implications in the modulation of cell proliferation. Int. J. Oncol. 2005, 26, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Pohlers, D.; Brenmoehl, J.; Loffler, I.; Muller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-beta and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, C.; Brembilla, N.C.; Montanari, E.; Truchetet, M.E. Fibrosis and immune dysregulation in systemic sclerosis. Autoimmun. Rev. 2011, 10, 276–281. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Hawinkels, L.J.; Ten Dijke, P. Exploring anti-TGF-beta therapies in cancer and fibrosis. Growth Factors 2011, 29, 140–152. [Google Scholar] [CrossRef]
- Finnson, K.W.; Philip, A. Endoglin in liver fibrosis. J. Cell Commun. Signal 2012, 6, 1–4. [Google Scholar] [CrossRef]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef]
- Shyu, K.G. The Role of Endoglin in Myocardial Fibrosis. Acta Cardiol. Sin. 2017, 33, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Felix, J.M.; Oujo, B.; Lopez-Novoa, J.M. The role of endoglin in kidney fibrosis. Expert Rev. Mol. Med. 2014, 16, e18. [Google Scholar] [CrossRef] [PubMed]
- Oujo, B.; Munoz-Felix, J.M.; Arevalo, M.; Nunez-Gomez, E.; Perez-Roque, L.; Pericacho, M.; Gonzalez-Nunez, M.; Langa, C.; Martinez-Salgado, C.; Perez-Barriocanal, F.; et al. L-Endoglin overexpression increases renal fibrosis after unilateral ureteral obstruction. PLoS ONE 2014, 9, e110365. [Google Scholar] [CrossRef] [PubMed]
- Van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef]
- Diez-Marques, L.; Ortega-Velazquez, R.; Langa, C.; Rodriguez-Barbero, A.; Lopez-Novoa, J.M.; Lamas, S.; Bernabeu, C. Expression of endoglin in human mesangial cells: Modulation of extracellular matrix synthesis. Biochim. Biophys. Acta 2002, 1587, 36–44. [Google Scholar] [CrossRef]
- Morris, E.; Chrobak, I.; Bujor, A.; Hant, F.; Mummery, C.; Ten Dijke, P.; Trojanowska, M. Endoglin promotes TGF-beta/Smad1 signaling in scleroderma fibroblasts. J. Cell Physiol. 2011, 226, 3340–3348. [Google Scholar] [CrossRef]
- Meurer, S.K.; Tihaa, L.; Lahme, B.; Gressner, A.M.; Weiskirchen, R. Identification of endoglin in rat hepatic stellate cells: New insights into transforming growth factor beta receptor signaling. J. Biol. Chem. 2005, 280, 3078–3087. [Google Scholar] [CrossRef]
- Alsamman, M.; Sterzer, V.; Meurer, S.K.; Sahin, H.; Schaeper, U.; Kuscuoglu, D.; Strnad, P.; Weiskirchen, R.; Trautwein, C.; Scholten, D. Endoglin in human liver disease and murine models of liver fibrosis-A protective factor against liver fibrosis. Liver Int. 2018, 38, 858–867. [Google Scholar] [CrossRef]
- Kapur, N.K.; Wilson, S.; Yunis, A.A.; Qiao, X.; Mackey, E.; Paruchuri, V.; Baker, C.; Aronovitz, M.J.; Karumanchi, S.A.; Letarte, M.; et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 2012, 125, 2728–2738. [Google Scholar] [CrossRef]
- Clemente, M.; Nunez, O.; Lorente, R.; Rincon, D.; Matilla, A.; Salcedo, M.; Catalina, M.V.; Ripoll, C.; Iacono, O.L.; Banares, R.; et al. Increased intrahepatic and circulating levels of endoglin, a TGF-beta1 co-receptor, in patients with chronic hepatitis C virus infection: Relationship to histological and serum markers of hepatic fibrosis. J. Viral. Hepat. 2006, 13, 625–632. [Google Scholar] [CrossRef]
- Preativatanyou, K.; Honsawek, S.; Chongsrisawat, V.; Vejchapipat, P.; Theamboonlers, A.; Poovorawan, Y. Correlation of circulating endoglin with clinical outcome in biliary atresia. Eur. J. Pediatr. Surg. 2010, 20, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Yagmur, E.; Rizk, M.; Stanzel, S.; Hellerbrand, C.; Lammert, F.; Trautwein, C.; Wasmuth, H.E.; Gressner, A.M. Elevation of endoglin (CD105) concentrations in serum of patients with liver cirrhosis and carcinoma. Eur. J. Gastroenterol. Hepatol. 2007, 19, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Charytan, D.M.; Helfand, A.M.; MacDonald, B.A.; Cinelli, A.; Kalluri, R.; Zeisberg, E.M. Circulating endoglin concentration is not elevated in chronic kidney disease. PLoS ONE 2011, 6, e23718. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pena, A.; Eleno, N.; Duwell, A.; Arevalo, M.; Perez-Barriocanal, F.; Flores, O.; Docherty, N.; Bernabeu, C.; Letarte, M.; Lopez-Novoa, J.M. Endoglin upregulation during experimental renal interstitial fibrosis in mice. Hypertension 2002, 40, 713–720. [Google Scholar] [CrossRef]
- Munoz-Felix, J.M.; Perez-Roque, L.; Nunez-Gomez, E.; Oujo, B.; Arevalo, M.; Ruiz-Remolina, L.; Cuesta, C.; Langa, C.; Perez-Barriocanal, F.; Bernabeu, C.; et al. Overexpression of the short endoglin isoform reduces renal fibrosis and inflammation after unilateral ureteral obstruction. Biochim. Biophys. Acta 2016, 1862, 1801–1814. [Google Scholar] [CrossRef]
- Shyu, K.G.; Wang, B.W.; Chen, W.J.; Kuan, P.; Hung, C.R. Mechanism of the inhibitory effect of atorvastatin on endoglin expression induced by transforming growth factor-beta1 in cultured cardiac fibroblasts. Eur. J. Heart Fail 2010, 12, 219–226. [Google Scholar] [CrossRef]
- Chen, K.; Mehta, J.L.; Li, D.; Joseph, L.; Joseph, J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ. Res. 2004, 95, 1167–1173. [Google Scholar] [CrossRef]
- Rodriguez-Barbero, A.; Obreo, J.; Alvarez-Munoz, P.; Pandiella, A.; Bernabeu, C.; Lopez-Novoa, J.M. Endoglin modulation of TGF-beta1-induced collagen synthesis is dependent on ERK1/2 MAPK activation. Cell. Physiol. Biochem. 2006, 18, 135–142. [Google Scholar] [CrossRef]
- Kramer, C.J.H.; Vangangelt, K.M.H.; van Pelt, G.W.; Dekker, T.J.A.; Tollenaar, R.; Mesker, W.E. The prognostic value of tumour-stroma ratio in primary breast cancer with special attention to triple-negative tumours: A review. Breast Cancer Res. Treat. 2019, 173, 55–64. [Google Scholar] [CrossRef]
- Barth, P.J.; Ebrahimsade, S.; Ramaswamy, A.; Moll, R. CD34+ fibrocytes in invasive ductal carcinoma, ductal carcinoma in situ, and benign breast lesions. Virchows. Arch. 2002, 440, 298–303. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 1795. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Harryvan, T.J.; Verdegaal, E.M.E.; Hardwick, J.C.H.; Hawinkels, L.; van der Burg, S.H. Targeting of the Cancer-Associated Fibroblast-T-Cell Axis in Solid Malignancies. J. Clin. Med. 2019, 8, 1989. [Google Scholar] [CrossRef]
- Romero, D.; O’Neill, C.; Terzic, A.; Contois, L.; Young, K.; Conley, B.A.; Bergan, R.C.; Brooks, P.C.; Vary, C.P. Endoglin regulates cancer-stromal cell interactions in prostate tumors. Cancer Res. 2011, 71, 3482–3493. [Google Scholar] [CrossRef]
- Paauwe, M.; Schoonderwoerd, M.J.A.; Helderman, R.; Harryvan, T.J.; Groenewoud, A.; van Pelt, G.W.; Bor, R.; Hemmer, D.M.; Versteeg, H.H.; Snaar-Jagalska, B.E.; et al. Endoglin Expression on Cancer-Associated Fibroblasts Regulates Invasion and Stimulates Colorectal Cancer Metastasis. Clin. Cancer Res. 2018, 24, 6331–6344. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lovrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Numakura, S.; Uozaki, H.; Kikuchi, Y.; Watabe, S.; Togashi, A.; Watanabe, M. Mesenchymal Stem Cell Marker Expression in Gastric Cancer Stroma. Anticancer Res. 2019, 39, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; Zhou, K.; Zhang, G.; Wang, F.; Ren, J. Isolation and characterization of CD105+/CD90+ subpopulation in breast cancer MDA-MB-231 cell line. Int. J. Clin. Exp. Pathol. 2015, 8, 5105–5112. [Google Scholar]
- Fonsatti, E.; Altomonte, M.; Arslan, P.; Maio, M. Endoglin (CD105): A target for anti-angiogenetic cancer therapy. Curr. Drug Targets 2003, 4, 291–296. [Google Scholar] [CrossRef]
- Karzai, F.H.; Apolo, A.B.; Cao, L.; Madan, R.A.; Adelberg, D.E.; Parnes, H.; McLeod, D.G.; Harold, N.; Peer, C.; Yu, Y.; et al. A phase I study of TRC105 anti-endoglin (CD105) antibody in metastatic castration-resistant prostate cancer. BJU Int. 2015, 116, 546–555. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ma, C.; Ulahannan, S.V.; Rahma, O.E.; Makarova-Rusher, O.; Cao, L.; Yu, Y.; Kleiner, D.E.; Trepel, J.; Lee, M.J.; et al. Phase I and Preliminary Phase II Study of TRC105 in Combination with Sorafenib in Hepatocellular Carcinoma. Clin. Cancer Res. 2017, 23, 4633–4641. [Google Scholar] [CrossRef]
- Dorff, T.B.; Longmate, J.A.; Pal, S.K.; Stadler, W.M.; Fishman, M.N.; Vaishampayan, U.N.; Rao, A.; Pinksi, J.K.; Hu, J.S.; Quinn, D.I.; et al. Bevacizumab alone or in combination with TRC105 for patients with refractory metastatic renal cell cancer. Cancer 2017, 123, 4566–4573. [Google Scholar] [CrossRef]
- Gordon, M.S.; Robert, F.; Matei, D.; Mendelson, D.S.; Goldman, J.W.; Chiorean, E.G.; Strother, R.M.; Seon, B.K.; Figg, W.D.; Peer, C.J.; et al. An open-label phase Ib dose-escalation study of TRC105 (anti-endoglin antibody) with bevacizumab in patients with advanced cancer. Clin. Cancer Res. 2014, 20, 5918–5926. [Google Scholar] [CrossRef]
- Rosen, L.S.; Hurwitz, H.I.; Wong, M.K.; Goldman, J.; Mendelson, D.S.; Figg, W.D.; Spencer, S.; Adams, B.J.; Alvarez, D.; Seon, B.K.; et al. A phase I first-in-human study of TRC105 (Anti-Endoglin Antibody) in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 4820–4829. [Google Scholar] [CrossRef]
- Apolo, A.B.; Karzai, F.H.; Trepel, J.B.; Alarcon, S.; Lee, S.; Lee, M.J.; Tomita, Y.; Cao, L.; Yu, Y.; Merino, M.J.; et al. A Phase II Clinical Trial of TRC105 (Anti-Endoglin Antibody) in Adults with Advanced/Metastatic Urothelial Carcinoma. Clin. Genitourin. Cancer 2017, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Paauwe, M.; Heijkants, R.C.; Oudt, C.H.; van Pelt, G.W.; Cui, C.; Theuer, C.P.; Hardwick, J.C.; Sier, C.F.; Hawinkels, L.J. Endoglin targeting inhibits tumor angiogenesis and metastatic spread in breast cancer. Oncogene 2016, 35, 4069–4079. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoonderwoerd, M.J.A.; Goumans, M.-J.T.H.; Hawinkels, L.J.A.C. Endoglin: Beyond the Endothelium. Biomolecules 2020, 10, 289. https://doi.org/10.3390/biom10020289
Schoonderwoerd MJA, Goumans M-JTH, Hawinkels LJAC. Endoglin: Beyond the Endothelium. Biomolecules. 2020; 10(2):289. https://doi.org/10.3390/biom10020289
Chicago/Turabian StyleSchoonderwoerd, Mark J.A., Marie-Jose T.H. Goumans, and Lukas J.A.C. Hawinkels. 2020. "Endoglin: Beyond the Endothelium" Biomolecules 10, no. 2: 289. https://doi.org/10.3390/biom10020289
APA StyleSchoonderwoerd, M. J. A., Goumans, M.-J. T. H., & Hawinkels, L. J. A. C. (2020). Endoglin: Beyond the Endothelium. Biomolecules, 10(2), 289. https://doi.org/10.3390/biom10020289