Abstract
Background/Objectives: Oxidative stress and iron overload remodel erythrocyte membranes in β-thalassemia, but their systemic metabolic correlates are not well defined. We applied untargeted metabolomics to identify serum biomarkers reflecting these pathophysiological processes. Methods: Thirty-one adults with β-thalassemia [18 transfusion-dependent (TDT), 13 non-transfusion-dependent (NTD)] and 8 age/sex-matched healthy controls were studied. Fasting serum was profiled using untargeted UHPLC–Orbitrap MS. Multivariate modeling (SIMCA-P) and FDR-controlled univariate statistics identified discriminant features, followed by pathway enrichment analysis. Associations with clinical variables (chelation regimen, ferritin, cardiac MRI T2*, and liver iron concentration) were examined. Results: A total of 183 metabolites were detected; versus controls, 124 were decreased, 54 increased, and 5 remained unchanged in patients. Key discriminants included lysophosphatidylcholines (LysoPC 18:1, 18:3), polyunsaturated fatty acid (PUFA)-bearing phosphatidylcholines (PC 20:4/18:0, PC 18:0/20:4), conjugated bile acids (glycocholic acid, glycochenodeoxycholic acid, and glycoursodeoxycholic acid), and bilirubin. Pathway analysis revealed significant enrichment (FDR-corrected) in linoleic acid metabolism (q = 0.024, impact = 1.000) and arachidonic acid metabolism (q = 0.022, impact = 0.433), with supportive nominal signals from glycerophospholipid (impact = 0.401) and porphyrin/heme (impact = 0.242) pathways. No significant metabolic differences were observed between TD and NTD patients. Conclusions: β-thalassemia serum metabolomics reflects oxidative membrane lipid remodeling with a prominent PLA2/LysoPC–arachidonic axis and evidence of heme turnover and altered bile-acid signaling. These data propose a practical biomarker panel-LysoPCs, arachidonic acid-enriched PCs, and conjugated bile acids-warranting targeted validation alongside conventional clinical parameters for disease monitoring and therapeutic assessment.
1. Introduction
β-thalassemia is an autosomal-recessive hemoglobinopathy caused by deficient β-globin synthesis. Homozygous or compound heterozygous mutations produce transfusion-dependent (TD) disease with lifelong anemia, whereas milder genotypes may be non-transfusion-dependent (NTD) with intermittent transfusion needs. Excess unpaired α-chains form hemichromes that catalyze reactive oxygen species, driving lipid/protein oxidation, membrane destabilization, apoptosis of erythroblasts, and ineffective erythropoiesis; peripheral hemolysis further aggravates anemia [1,2]. Beyond hematologic manifestations, chronic hemolysis, endothelial adhesion, inflammation, oxidative stress, and iron overload contribute to multisystem—especially cardiovascular—injury [3,4].
From a genetic and epidemiological perspective, β-thalassemia is an autosomal-recessive disorder caused by pathogenic variants in the HBB gene located within the β-globin gene cluster on chromosome 11 (11p15.5); accordingly, inheritance and population allele frequency are not expected to differ by sex. Age primarily influences clinical expression, cumulative complications, and survival rather than mutation prevalence. Advances in transfusion regimens, iron chelation, and multidisciplinary follow-up have progressively improved survival, resulting in an expanding adult and aging β-thalassemia population in settings with sustained access to modern care [1,2]. Consequently, wide age distributions and occasional sex imbalances observed in contemporary single-center cohorts mainly reflect survivor effects, referral patterns, and sampling variability rather than biologically meaningful differences in disease frequency.
Metabolomics, by quantifying small-molecule intermediates and end products [5,6], has begun to delineate the systemic biochemical footprint of these processes across human cohorts and experimental models, but findings remain heterogeneous with respect to matrix, platform, and clinical context. In patients, NMR/GC–MS/LC–MS investigations have reported broad changes spanning organic acids, amino acids, redox/heme catabolites, and lipid classes. For example, urine metabolomics in transfusion-dependent β-thalassemia under deferasirox therapy revealed treatment-linked shifts in small-molecule pathways (organic acids and energy metabolites) [7]. Targeted and untargeted approaches in patient sera have proposed candidate markers of iron load and oxidative damage [8], and untargeted serum profiling has demonstrated significant deviations from healthy controls—including pathways related to carbohydrate, fatty acid, and glycerophospholipid metabolism [9]. Importantly, metabolic re-alignment toward a healthier profile has been observed in hydroxyurea-treated β-thalassemia children, underscoring therapy-responsive biochemical plasticity [10]. Beyond blood and urine, fetal chorionic villi from β-thalassemia pregnancies showed oxidative-stress and inflammatory signatures by metabolomics [11]. At the tissue and immune-cell level, recent multi-omic work in β-thalassemia and sickle models demonstrates pronounced metabolic/proteomic divergence in spleen and liver and between circulating monocytes vs. tissue macrophages, highlighting organ- and cell-type–specific remodeling that plausibly echoes in the circulating metabolome [12,13]. In contemporary patient cohorts, cellular/biochemical heterogeneity has been linked to phenotypic diversity among transfusion-dependent β-thalassemia, reinforcing the need for systemic readouts that integrate red-cell, hepatic, and inflammatory axes [14]. Finally, integrative microbiome–metabolome analyses have implicated gut–liver crosstalk—in particular, taxa influencing bilirubin/stercobilin turnover—as potential modifiers of anemia and iron burden [15].
In β-thalassemia, iron overload and ongoing hemolysis amplify oxidative stress, a relationship captured by metabolomic readouts of iron load and oxidative damage in patient cohorts [8] and by platform-consistent alterations spanning energy and lipid pathways in urine, serum, and fetal tissues [7,9,11,14]. At the red-cell interface, oxidative injury to membrane phospholipids promotes phospholipase A2–mediated remodeling, increasing lysophosphatidylcholines (LysoPCs) and mobilizing polyunsaturated fatty acids for eicosanoid/oxylipin biosynthesis—changes coherent with untargeted serum metabolomics that highlights disruption of glycerophospholipid metabolism and therapy-responsive metabolic plasticity [9,10]. Concomitantly, iron overload suppresses the farnesoid X receptor (FXR) in mouse and human hepatocytes, thereby dysregulating bile-acid homeostasis and potentiating iron-induced hepatotoxicity [16]. This hepatocellular axis likely intersects with gut–liver crosstalk, as microbiome–metabolome analyses in β-thalassemia implicate taxa modulating bilirubin/stercobilin turnover and secondary bile-acid formation as potential modifiers of anemia and iron burden [15]. Together, these observations support a unified model wherein iron-driven oxidative stress couples erythrocyte membrane lipid remodeling with bile-acid dysregulation, motivating our serum metabolomics focus on PLA2/LysoPC–arachidonate and conjugated bile-acid signatures.
Here, we conducted untargeted UHPLC–Orbitrap MS metabolomics of fasting serum from patients with β-thalassemia and age/sex-matched healthy controls, integrating multivariate and FDR-controlled univariate statistics, pathway enrichment, and quantitative associations with ferritin, liver iron concentration, and cardiac T2*. Our explicit novelty relative to prior reports is threefold: (1) a serum-centered, untargeted LC–HRMS analysis that jointly interrogates PLA2/LysoPC–arachidonic remodeling and bile-acid dysregulation within the same cohort; (2) framing these axes into a concise, biomarker panel (LysoPCs, AA-enriched PCs, conjugated bile acids) suitable for targeted validation; and (3) evaluating clinical correlations with standardized iron metrics (ferritin, LIC, cardiac T2*), thereby enhancing translational relevance and paving the way for longitudinal monitoring. By mapping these metabolic derangements, we aim to identify actionable biomarkers and therapeutic targets, facilitating the transformation from a hemoglobin-centered view to a systems-level framework that can guide targeted validation and future therapeutic strategies in β-thalassemia.
2. Materials and Methods
The study included 31 patients with β-thalassemia living in the region of Northwestern Greece and followed at the pediatric and adult thalassemia Units at the University Hospital of Ioannina. The diagnosis was based on clinical, hematological, biosynthetic, and genetic studies. Medical files of the patients were retrieved, and detailed data were recorded and analyzed. Other parameters, such as serum ferritin, cardiac MRI T2*, MRI liver iron concentration (LIC), and chelation regimen, were enrolled in the study. The demographic, laboratory, and clinical data are shown in Table 1. Fourteen patients were male, and the age range was 12–67 years, with a median of 36 years. Eighteen had TD β-thalassemia, and 13 had NTD. Eight apparently healthy individuals, matched to the patient cohort for age and sex distribution, served as controls (age range 13–62 years; median 35.5 years, 4 male). All participants provided written informed consent, and the study was conducted in accordance with the Declaration of Helsinki.

Table 1.
Demographic, clinical, and laboratory data from 31 thalassemia patients.
Chelation regimen at sampling was recorded as none, deferiprone (DFP), deferasirox (DFX), deferoxamine (DFO), or combination therapy (e.g., DFP + DFX or DFP + DFO). Decisions to initiate or withhold chelation followed standard practice thresholds integrating serum ferritin trends, LIC, and cardiac T2* [17,18]. DFO is a parenteral hexadentate chelator with a short half-life; the iron–DFO complex (ferrioxamine) is mainly eliminated renally, and DFO remains an option for severe iron overload or when oral agents are unsuitable. DFP is an oral bidentate chelator (typically three times a day) that promotes urinary iron excretion and is well recognized for efficacy in myocardial siderosis; idiosyncratic agranulocytosis mandates blood count monitoring. DFX is an oral tridentate, once-daily chelator with predominantly fecal iron excretion and proven efficacy for hepatic iron reduction/maintenance. Combination therapy (most commonly DFP + DFO; less often DFX with another agent) is used in difficult-to-treat overload or cardiac siderosis and can accelerate iron removal [17,18]. Because these agents differ in coordination chemistry, dosing, and excretory pathways (renal vs. biliary/fecal), chelation may modulate systemic metabolites; accordingly, we recorded the regimen per subject (Table 1) and considered chelation as a covariate in sensitivity analyses.
During recruitment, patients with evidence of other chronic illnesses unrelated to thalassemia (such as diabetes mellitus, autoimmune disorders, chronic kidney disease, or active malignancy) were excluded to minimize confounding metabolic alterations. The final study cohort comprised the 31 patients detailed in Table 1. Peripheral blood was drawn after an overnight fast and, for transfused patients, prior to the scheduled transfusion to minimize acute post-transfusion effects. Samples were allowed to clot, centrifuged to obtain serum, aliquoted to avoid repeated freeze–thaw, and stored at −80 °C until analysis. All were performed on ice or at 4 °C (except vortexing) to limit ex vivo metabolic change and oxidation. Sterile pipettes and tips were used, and appropriate precautions were taken during the performance of assays (using a laminar flow instrument, Telstar/Bio II A, Telstar, Terrassa, Spain) in a cell culture room) to protect samples from subsequent environmental contamination. Acetonitrile (ACN), methanol (MeOH), and formic acid (FA) were obtained from Fluka/Riedel-de Haën (Buchs, Switzerland). Millipore’s Milli-Q Plus water purification system (Milford, MA, USA) was utilized to create high-quality purified water.
Sample pretreatment. The serum samples were thawed on ice at 4 °C for 30 to 60 min, depending on the quantity of the available samples. Consequently, an aliquot of 200 μL was drawn from each sample, and 800 μL of MeOH was added, followed by 15 s of vortexing. Afterward, the samples were centrifuged at 12,000× g for 15 min at 4 °C to separate the protein precipitate. The supernatant was transferred to a 1.5 mL Eppendorf tube, vacuum-evaporated (GeneVac). HT-4X EZ-2 series evaporator Lyospeed ENABLED (Genevac. Ltd., Ipswich, UK) at 50 °C, and stored (−80 °C) until analysis. For HRMS analysis, the samples were reconstituted (100 μL 80:20 H2O:ACN (v:v)), vortexed for 30 s, and transferred to 200 μL inserts in appropriate screw-capped autosampler vials. Throughout these processes, the samples were kept on ice before each centrifugation. A pooled quality control (QC) sample was constructed by aliquoting 10 μL from each sample before evaporation. The combined mixture underwent the pretreatment described, adjusted for the volume. Sample handling and analysis are depicted as a workflow in Figure 1.
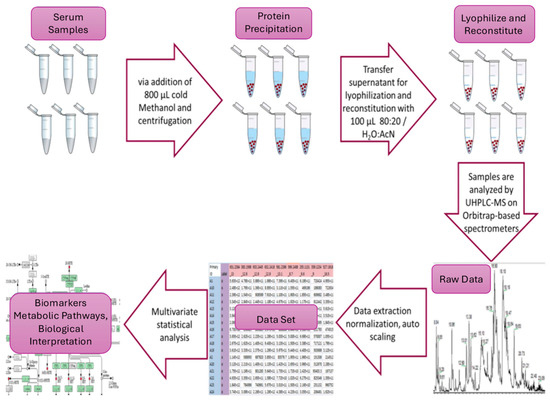
Figure 1.
Untargeted serum metabolomics workflow in β-thalassemia.
Samples were analyzed using an LC-MSn system comprised of an ESI-LTQ-Orbitrap Discovery XL mass spectrometer connected to an Accela UHPLC system (Thermo Scientific, Bremen, Germany). The UHPLC setup included an autosampler, vacuum degasser, binary pump, and a column with controlled temperature. Analysis was conducted using a Waters Corp. ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 μm particle ID, Waters Corporation, Milford, MA, USA). The DDA analysis mode was selected, and the fragmentation was done in the linear ion trap part.
Before conducting the multivariate statistical analysis, the data underwent preprocessing using several tools. Xcalibur® from Thermo Fisher Scientific (San Jose, CA, USA), MZmine 2.10 available at mzmine.sourceforge.net. Microsoft Excel and the web-based MetaboAnalyst 6.0 suite [19] were utilized. Initially, Xcalibur® converted the instrument’s native data files (.raw) to the cdf data format (.cdf) through the Xcalibur® Xconvert program. Subsequently, MZmine 2.10 processed the data, employing baseline correction, peak detection, deconvolution, deisotoping, alignment, and gap-filling procedures. The resulting peak list, displaying accurate mass-tR versuadds intensity, was exported as a .csv file to Microsoft Excel and further manipulated using commands such as CONCATENATE, ROUND, and TRANSPOSE for appropriate adjustments.
For the statistical analyses, both univariate and multivariate statistics have been employed. The online version of Metaboanalyst 6.0 has been used for the univariate analyses, whereas for their multivariate counterparts, SIMCA P+ 10.5 (Umetrics, Umea, Sweden) and EZinfo 2.0 (Umetrics, Umea, Sweden) were also employed. Briefly, PCA was employed for revealing trends and locating putative outliers, whereas PLSDA was used for deciphering the clustering of the thalassemia vs. normal control subjects. The results were also verified employing univariate statistics (t-test and fold-change analyses). The methodology has been included in the Supplementary Material.
3. Results
3.1. Global Serum Metabolome and Data Structure
Untargeted UHPLC–Orbitrap MS detected 183 serum features across patients and controls. Based on combined multivariate and univariate analyses, 124 features were decreased, 54 were increased, and 5 were unchanged in patients versus controls.
Two approaches were adopted for data scaling: UV (auto-scaling) and Pareto scaling for both negative and positive ion modes. UV scaling equalizes the importance of all metabolites but amplifies baseline noise. Conversely, Pareto scaling retains the original measurements to a greater extent but is responsive to significant changes [20]. As depicted in Figure 2 and Figure 3 for positive and negative ion modes, respectively, the samples showed close clustering in PCA score plots. However, the distinction between the two groups was not deemed satisfactory, and therefore supervised methods, such as PLS-DA or OPLS-DA, were employed.
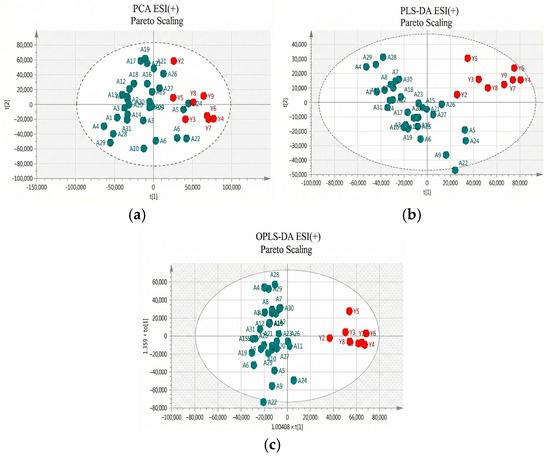
Figure 2.
(a) PCA score plot in Pareto scaling with R2 = 0.610, Q2 = 0.364, and 5 principal components; (b) PLS-DA score plot in Pareto scaling with R2 = 0.853, Q2 = 0.601, and 2 principal components; and (c) OPLS-DA score plot in Pareto scaling with R2 = 0.913, Q2 = 0.715, and 3 principal components for UHPLC-MS serum analysis in positive ionization ESI (+) between controls (red) and patients (green).
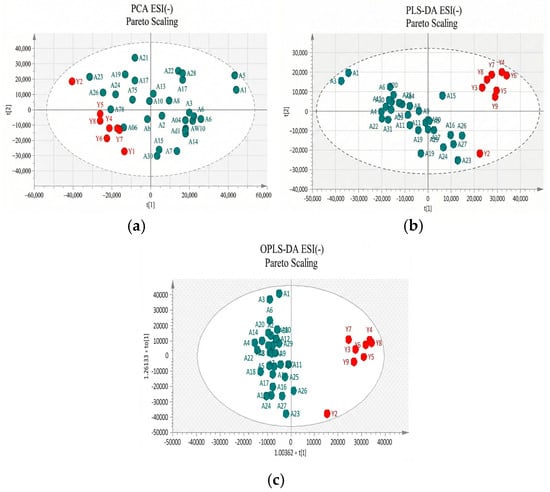
Figure 3.
(a) PCA score plot in Pareto scaling with R2 = 0.302, Q2 = 0.141, and 2 principal components; (b) PLS-DA score plot in Pareto scaling with R2 = 0.799, Q2 = 0.447, and 2 principal components; and (c) OPLS-DA score plot in Pareto scaling with R2 = 0.924, Q2 = 0.514, and 4 principal components for UHPLC-MS serum analysis in negative ionization ESI (−) between controls (red) and patients (green).
Both methods were able to distinguish between the two sets (controls versus patients), displaying distinctive clustering that suggested varying metabolic profiles in both modes, both for UV and Pareto scaling. However, in these supervised models, where users categorize samples into pre-established groups, there is the risk of creating biased clustering or overfitting. Hence, permutation testing involving 100 permutations was used to evaluate the significance of the classification, utilizing the prediction accuracy during training as a statistical test. The results of the permutation test indicated that UV scaling was overfit to the data since R2 and Q2 values derived from the permuted data exceeded the original values on the validation plot. This indicates that relying on the model using UV scaling might not be dependable for subsequent statistical analysis, as it fails to precisely represent the inherent patterns within the data.
3.2. Feature Selection and Identification
The VIP-plot and S-plot in positive (Figure 4a,b) and negative (Figure 4c,d) mode were computed to pinpoint potentially crucial variables that contribute to the distinct clustering observed in the OPLS. VIP values (>1.0) were utilized to pinpoint the metabolites responsible for distinguishing between the two groups.
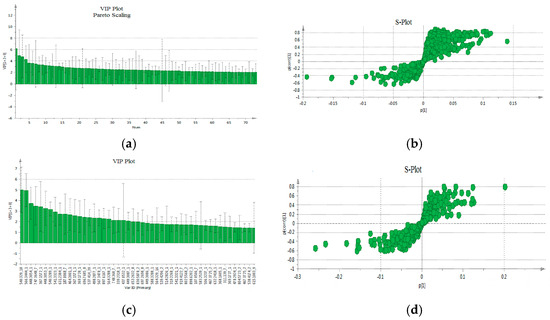
Figure 4.
The VIP-plot and S-plot in positive (a,b) and negative (c,d). (a) VIP-plot and (b) S-plot of OPLS-DA. In the VIP-plot, the variables are ranked from the most statistically significant to the least. In the S-plot, the features positioned at the extremes, farthest from the central cluster, display substantial variation and consistency, thereby holding significant importance when searching for potential biomarkers (characteristics in the lower left quadrant belong to the patients, while those in the upper right quadrant belong to the healthy individuals).
Additionally, to validate the outcomes related to the selection of VIPs from both modes (positive and negative), univariate data analysis was conducted using t-tests, FC monitoring, and their amalgamation in the form of a heatmap plot (Figure 5).
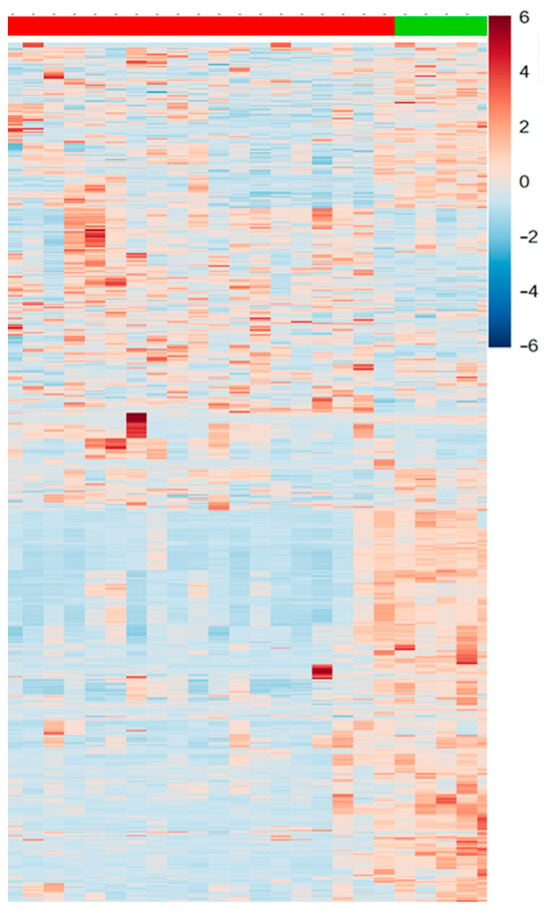
Figure 5.
Heatmap representation of significant differences between patients (red) and controls (green).
By merging the findings from both univariate and multivariate analyses, a compilation of features that differentiate the two groups was assembled for structural identification. The identification strategy involved comparing the selected features with online databases based on mass accuracy, isotopic patterns, RDBeq information, and potential low-energy or in-source fragmentation data for assessment. As a result, 20 metabolites (Table 2) were recognized via Orbitrap-based analysis.
Table 2.
List of the most significant up-regulated metabolites in β-thalassemia patients in comparison to healthy controls.
3.3. Metabolic Pathway Analysis
Pathway analysis (MetaboAnalyst/KEGG) [19] showed significant enrichment for arachidonic acid metabolism (q = 0.022, impact = 0.433) and linoleic acid metabolism (q = 0.024, impact = 1.000) (Figure 6). Glycerophospholipid metabolism (q = 0.220, impact = 0.401) and porphyrin metabolism (q = 0.145, impact = 0.242) reached nominal significance but did not survive FDR correction. These patterns, together with LysoPC up-regulation at the feature level, support PUFA (polyunsaturated fatty acid)- and membrane-lipid-centered remodeling under oxidative stress, with eicosanoid precursor flux as a plausible downstream consequence. Primary bile-acid biosynthesis and α-linolenic acid metabolism were not significant after correction in this dataset.
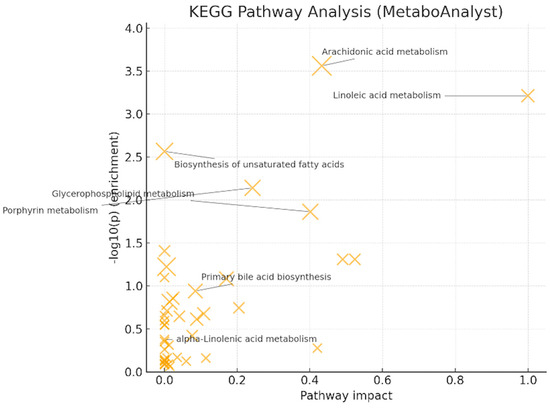
Figure 6.
KEGG pathway analysis (MetaboAnalyst 6.0) [–log10(p) vs. pathway impact; bubble size = hits. Significant after FDR: linoleic acid (q = 0.0245, impact = 1.000) and arachidonic acid (q = 0.0220, impact = 0.433). Glycerophospholipid and porphyrin are only nominal].
3.4. Subgroup and Clinical Correlations
Within the β-thalassemia cohort, no significant differences were observed between TDT and NTDT groups in global multivariate structure or in individual metabolites. This lack of separation persisted after considering demographic, clinical, and laboratory covariates provided in Table 1.
3.5. Study Limitations
Limitations of the study include modest sample size; class imbalance (31 patients vs. 8 controls); wide age range; potential confounding by transfusion timing, chelation, diet, and microbiota being not fully controlled; and the inherent constraints of untargeted annotation. Larger cohorts are needed for confirmation. Furthermore, our cross-sectional design may underpower subgroup contrasts (TDT vs. NTDT) and cannot resolve causality. We did not quantify PLA2 activities, eicosanoids, FXR/TGR5 signaling, FGF19, or gut microbial composition; therefore, mechanistic interpretations involving these pathways should be viewed as exploratory. Findings should be viewed as hypothesis-generating and require targeted validation in larger, multi-center cohorts with independent replication.
4. Discussion
The present study utilized untargeted metabolomic analysis to identify a set of significantly up-regulated metabolites that cluster into two biologically coherent groups: (A) phosphatidylcholine (PC)–related species along the glycerophospholipid axis and (B) conjugated bile acids belonging to primary/secondary bile-acid biosynthesis. Together, these findings support a unifying model in β-thalassemia whereby oxidative membrane remodeling of erythrocytes and iron-related hepatobiliary signaling leave distinct, measurable footprints in the serum metabolome.
- A.
- Phosphatidylcholine biosynthesis and remodeling
Several phosphatidylcholines (PCs) and lysophosphatidylcholines (LysoPCs) were significantly elevated in β-thalassemia patients compared to healthy controls.
Phosphatidylcholines. PC(O-14:0/2:0) or 1-tetradecyl-2-acetyl-sn-glycero-3-phosphocholine PC(0:0/18:1(9E) or 2-oleoyl-sn-glycero-3-phosphocholine; PC(17:2(9Z,12Z)/19:0) or 1,2-diacyl-sn-glycero-3-phosphocholine; PC(20:4(5Z,8Z,11Z,14Z)/18:0); and PC(18:0/20:4(8Z,11Z,14Z,17Z)) are PCs (glycerophospholipids) in which a phosphorylcholine moiety occupies a glycerol substitution site which are intermediates in the metabolic pathway of PCs. PCs and sphingomyelin are the dominant phospholipids of the erythrocyte outer membrane layer, whereas phosphatidylethanolamine and phosphatidylserine (PS) reside largely on the inner layer. This transbilayer asymmetry—maintained by flippases/floppases/scramblases—is essential for RBC deformability and for evading macrophage recognition [21]. In β-thalassemia, excess α-globin chains, labile iron, and elevated intracellular Ca2+ promote lipid peroxidation and scramblase activation, causing PS externalization that accelerates splenic clearance and contributes to vascular/thrombotic complications [22]. Furthermore, phosphocholine and phosphatidylcholine catabolism play a key role in ATP production and terminal erythropoiesis, and the deregulation of this process may contribute to ineffective erythropoiesis in thalassemia [23].
Lysophosphatidylcholines (LysoPCs). LysoPC(18:1(11Z)) and LysoPC(18:3(9Z,12Z,15Z)) are monoacyl-glycerophospholipids bearing a phosphorylcholine headgroup. LPCs arise primarily from PC hydrolysis by lipoprotein-associated phospholipase A2 (Lp-PLA2) and from lecithin:cholesterol acyltransferase (LCAT) during cholesterol esterification. Beyond their structural role, LysoPCs function as bioactive lipids: they engage lysophospholipid-responsive pathways, generally upregulating genes involved in cholesterol biosynthesis while suppressing programs of hepatic fatty-acid oxidation, and at elevated concentrations they provoke endothelial dysfunction and oxidative stress. Excess LysoPCs production can reflect increased PLA2 flux, including Lp-PLA2 overexpression/activation [24]. Notably, β-thalassemia patients exhibit elevated circulating Lp-PLA2 activity [25], plausibly driven by monocyte/macrophage activation and heightened lipoprotein/erythrocyte oxidative stress. Together with our data, these observations support a mechanistic model in which erythroid and systemic redox imbalance expands the pool of oxidized PCs; PLA2 activities (including Lp-PLA2) cleave these substrates to generate LysoPCs that accumulate in the circulation and, in turn, amplify endothelial and inflammatory signaling. Because our untargeted approach does not quantify enzyme activities or downstream oxylipins, direct measurements of Lp-PLA2/sPLA2/cPLA2 activities and targeted eicosanoid profiling will be required to test this mechanism.
Arachidonic acid (AA). We detected enrichment of arachidonic-acid–linked pathways, with AA-related features among the top discriminants. AA is a PUFA esterified predominantly in phosphatidylethanolamine, phosphatidylcholine, and phosphatidylinositol pools of cellular membranes. Upon activation, PLA2 hydrolyzes the sn-2 ester bond to liberate free AA, which is then converted by cyclooxygenases (COX-1/2), lipoxygenases (5-/12-/15-LOX), and cytochrome P450 epoxygenases into eicosanoids—potent autacoids governing inflammation, vascular tone, platelet function, and leukocyte trafficking [26].
In our cohort, the up-regulation of arachidonic-acid methyl ester and related signals indicates accelerated PUFA turnover and a shift toward pro-inflammatory oxylipin biosynthesis, consistent with elevated oxidative stress and sterile inflammation. These findings align with prior untargeted metabolomics in β-thalassemia that reported broad perturbations in lipid and energy pathways and a chronic pro-inflammatory milieu [9]. In thalassemia patients, chronic hemolysis and iron-driven reactive oxygen species (ROS) production may both increase PLA2-dependent AA liberation and promote non-enzymatic PUFA peroxidation, yielding non-classic eicosanoids, such as isoprostanes, that further amplify vascular inflammation [25,27]. Although we did not measure individual eicosanoids or PLA2 activities, the combined signatures—elevated LysoPCs, perturbation of PUFA-rich PCs, and pathway enrichment—are compatible with increased AA turnover. Confirmation will require targeted oxylipin MS and enzymatic assays.
- B.
- Bile acid biosynthesis
We observed up-regulation of conjugated primary bile acids—glycocholic acid (GCA) and glycolchenodeoxyglycocholic acid (GCDCA),—of the C27 intermediate (25R)-3α,7α-dihydroxy-5β-cholestan-27-oyl taurine alongside the secondary species glycoursodeoxycholic acid (GUDCA). Primary bile acids are synthesized from cholesterol in hepatocytes and conjugated to glycine/taurine before biliary secretion; most are reabsorbed in the ileum, but a fraction undergoes microbial deconjugation and 7α-dehydroxylation/epimerization to form secondary bile acids, e.g., ursodeoxycholic acid (UDCA), GUDCA, and deoxycholic acid (DCA) [28]. The elevation of bilirubin and primary bile acids in our patients suggests coupled effects of hemolysis-driven heme turnover and hepatic stress. The increased levels of bile acids may have several systemic effects, such as deterioration of hemolysis [29].
Functionally, bile acids can influence erythrocyte stability and systemic metabolism via the nuclear receptor FXR (farnesoid X receptor) and TGR5/GPBAR1 (Takeda G protein-coupled receptor 5/G protein-coupled bile acid receptor 1) receptor—pathways that regulate FGF19 (fibroblast growth factor 19) signaling [13]. We did not assess FXR/TGR5 activity, FGF19, or hepatic transcriptomics in this study; therefore, any link to receptor signaling should be considered hypothesis-generating. Prior work has reported that iron overload can suppress hepatocellular FXR and perturb bile-acid homeostasis [13]; our detection of a taurine-conjugated C27 intermediate is compatible with, but does not establish, such dysregulation in our cohort.
The presence of GUDCA could reflect microbiome-dependent epimerization of chenodeoxycholic acid followed by hepatic reconjugation [16]; however, we did not profile the gut microbiome, and hepatic and biliary handling may also contribute. Accordingly, any inference about microbiome involvement remains speculative and warrants integrated microbiome–metabolome studies.
5. Conclusions
Untargeted UHPLC–Orbitrap MS metabolomics of β-thalassemia serum samples revealed a dual metabolic signature: (i) disrupted glycerophospholipid metabolism with elevated PC/LysoPC species and (ii) altered bile acid biosynthesis marked by increased conjugated bile acids and a taurine-linked C27 intermediate. These findings suggest that oxidative stress from iron overload and hemolysis drives membrane remodeling and inflammation, while hepatobiliary and gut-derived signals reshape the bile acid pool. Notably, these metabolic alterations were consistent across TD and NTD patients. The results nominate potential biomarker candidates (LysoPCs, AA-enriched PCs, conjugated bile acids) and generate hypotheses regarding pathways (oxidative stress, PLA2, bile-acid signaling) for future validation. Larger longitudinal studies integrating metabolomics with clinical outcomes are needed for biomarker validation and clinical translation.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/metabo16030170/s1. Section S1: Instrumental Conditions [30]; Section S2: Feature Annotation; Section S3: Statistical Analyses.
Author Contributions
Conceptualization, A.M. and V.S.; methodology, T.P., D.P., E.G. and V.S.; software, E.G., D.P. and V.S.; formal analysis, D.P., E.G. and V.S.; data curation, A.M., T.P., E.H. and E.K.; writing—original draft preparation, A.M., E.H. and V.S.; writing—review and editing, A.M., E.H., E.G. and V.S.; supervision, A.M. and V.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of University Hospital of Ioannina, Greece (512/28-1-2026), approval date: 29 September 2015.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Dataset available on request from the authors.
Acknowledgments
During the preparation of this manuscript, the authors used Claude Sonnet 4.6 for the preparation of the graphical abstract. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AA | Arachidonic acid |
| ATP | Adenosine triphosphate |
| BA | Bile acid (s) |
| COX | Cyclooxygenase |
| DCA | Deoxycholic acid |
| DFO | Deferoxamine |
| DFP | Deferiprone |
| DFX | Deferasirox |
| ESI | Electrospray ion source |
| FC | Fold change |
| FDR | False discovery rate |
| FGF19 | Fibroblast growth factor 19 |
| FTMS | Fourier transform mass spectrometry |
| FWHM | Full width at half maximum |
| FXR | Farnesoid X receptor |
| GC | Gas chromatography |
| GCA | Glycocholic acid |
| GCDCA | Glycolchenodeoxyglycocholic acid |
| GPBAR1 | G protein-coupled bile acid receptor 1 |
| GUDCA | Glycoursodeoxycholic acid |
| HRMS | High-resolution mass spectrometry |
| IQR | Interquartile range |
| KNN | k-nearest neighbor |
| LCAT | Lecithin:cholesterol acyltransferase |
| LIC | Liver iron concentration |
| LOOCV | Leave-one-out cross-validation |
| LOX | Lipoxygenases |
| Lp-PLA2 | Lipoprotein-associated phospholipase A2 |
| LTQ | Linear trap quadrupole |
| LysoPC | Lysophosphatidylcholine |
| MRI | Magnetic resonance imaging |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| NTDT | Non-transfusion-dependent thalassemia |
| OPLS-DA | Orthogonal projection to latent structures–discriminant analysis |
| PC | Phosphatidylcholine |
| PCA | Principal Component Analysis |
| PLA | Phospholipase |
| PLS-DA | Partial least square–discriminant analysis |
| PS | Phosphatidylserine |
| PUFA | Polyunsaturated fatty acid |
| QC | Quality control |
| RBC | Red blood cell |
| ROS | Reactive oxygen species |
| SIMCA-P | Soft independent modeling of class analogy (Pro) |
| T2* | Myocardial T2* (cardiac MRI) |
| TDT | Transfusion-dependent thalassemia |
| TGR5/GP | Takeda G protein-coupled receptor 5/G protein-coupled bile acid receptor 1 |
| UDCA | Ursodeoxycholic acid |
| UHPLC | Ultrahigh-performance liquid chromatography |
| USA | United States of America |
| UV | Unit variance |
| VIP | Variable importance in projection |
References
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. Beta-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef]
- Kattamis, A.; Kwiatkowski, J.L.; Aydinok, Y. Thalassaemia. Lancet 2022, 399, 2310–2324. [Google Scholar] [CrossRef]
- Chaliasos, N.; Challa, A.; Hatzimichael, E.; Koutsouka, F.; Bourantas, D.K.; Vlahos, A.P.; Siamopoulou, A.; Bourantas, K.L.; Makis, A. Serum adipocytokine and vascular inflammation marker levels in Beta-thalassaemia major patients. Acta Haematol. 2010, 124, 191–196. [Google Scholar] [CrossRef]
- Makis, A.; Voskaridou, E.; Papassotiriou, I.; Hatzimichael, E. Novel Therapeutic Advances in beta-Thalassemia. Biology 2021, 10, 546. [Google Scholar] [CrossRef]
- Trivedi, D.K.; Hollywood, K.A.; Goodacre, R. Metabolomics for the masses: The future of metabolomics in a personalized world. New Horiz. Transl. Med. 2017, 3, 294–305. [Google Scholar] [CrossRef]
- Katsantoni, E. Omics Studies in Hemoglobinopathies. Mol. Diagn. Ther. 2019, 23, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Capolongo, G.; Zacchia, M.; Beneduci, A.; Costantini, S.; Cinque, P.; Spasiano, A.; De Luca, G.; Di Pietro, M.E.; Ricchi, P.; Trepiccione, F.; et al. Urinary Metabolic Profile of Patients with Transfusion-Dependent beta-Thalassemia Major Undergoing Deferasirox Therapy. Kidney Blood Press. Res. 2020, 45, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Sayani, F.; Zhang, Y.; Abdolmohammadi, N.; Lauf, A.M.; Evans, P.; Porter, J.B.; Weljie, A.M. Metabolomic biomarkers as predictors of iron load and oxidative damage in iron-overloaded thalassemia patients. In Proceedings of the 54th Annual Meeting of the American Society of Hematology, Atlanta, GA, USA, 8–11 December 2012. [Google Scholar]
- Musharraf, S.G.; Iqbal, A.; Ansari, S.H.; Parveen, S.; Khan, I.A.; Siddiqui, A.J. beta-Thalassemia Patients Revealed a Significant Change of Untargeted Metabolites in Comparison to Healthy Individuals. Sci. Rep. 2017, 7, 42249. [Google Scholar] [CrossRef]
- Iqbal, A.; Ansari, S.H.; Parveen, S.; Khan, I.A.; Siddiqui, A.J.; Musharraf, S.G. Hydroxyurea Treated beta-Thalassemia Children Demonstrate a Shift in Metabolism Towards Healthy Pattern. Sci. Rep. 2018, 8, 15152. [Google Scholar] [CrossRef]
- Monni, G.; Murgia, F.; Corda, V.; Peddes, C.; Iuculano, A.; Tronci, L.; Balsamo, A.; Atzori, L. Metabolomic Investigation of beta-Thalassemia in Chorionic Villi Samples. J. Clin. Med. 2019, 8, 798. [Google Scholar] [CrossRef]
- Lisk, C.; Cendali, F.; Setua, S.; Thangaraju, K.; Pak, D.I.; Swindle, D.; Dzieciatkowska, M.; Gamboni, F.; Hassell, K.; Nuss, R.; et al. Metabolic and Proteomic Divergence Is Present in Circulating Monocytes and Tissue-Resident Macrophages from Berkeley Sickle Cell Anemia and beta-Thalassemia Mice. J. Proteome Res. 2023, 22, 2925–2935. [Google Scholar] [CrossRef]
- Rana, N.K.; Lisk, C.; Cendali, F.; Lucero, M.J.; Grier, A.; Setua, S.; Thangaraju, K.; Khan, A.; Reisz, J.A.; Dzieciatkowska, M.; et al. Metabolic and Proteomic Divergence is Present in Spleens and Livers from Berkeley Sickle Cell Anemia and beta-Thalassemia Mice. J. Proteome Res. 2025, 24, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Theocharaki, K.; Anastasiadi, A.T.; Delicou, S.; Tzounakas, V.L.; Barla, I.; Rouvela, S.; Kazolia, E.; Tzafa, G.; Mpekoulis, G.; Gousdovas, T.; et al. Cellular and biochemical heterogeneity contributes to the phenotypic diversity of transfusion-dependent I(2)-thalassemia. Blood Adv. 2025, 9, 2091–2107. [Google Scholar] [CrossRef]
- Guo, X.; Lin, S.; Zhang, X.; Li, M.; Wang, Z.; Peng, Y.; He, X.; Liu, J. Integrated metabolomic and microbiome analysis identifies Cupriavidus metallidurans as a potential therapeutic target for beta-thalassemia. Ann. Hematol. 2024, 103, 5169–5179. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, C.; Han, L.; Xu, T.; Saeed, K.; Han, J.; Liu, J.; Klaassen, C.D.; Gonzalez, F.J.; Lu, Y.; et al. Suppressed farnesoid X receptor by iron overload in mice and humans potentiates iron-induced hepatotoxicity. Hepatology 2022, 76, 387–403. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. Guidelines for the Management of Non-Transfusion-Dependent β-Thalassaemia, 3rd ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2023. [Google Scholar]
- Taher, A.T.; Farmakis, D.; Porter, J.B.; Cappellini, M.D.; Musallam, K.M. Guidelines for the Management of Transfusion-Dependent β-Thalassaemia (TDT), 5th ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2025. [Google Scholar]
- MetaboAnalyst. Available online: https://www.metaboanalyst.ca/MetaboAnalyst/ModuleView.xhtml (accessed on 22 November 2025).
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef]
- Yashar, V.B.B.; Barenholz, Y.; Hy-Am, E.; Rachmilewitz, E.A.; Eldor, A. Phosphatidylserine in the outer leaflet of red blood cells from beta-thalassemia patients may explain the chronic hypercoagulable state and thrombotic episodes. Am. J. Hematol. 1993, 44, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.J.; Lin, Y.C.; Lin, C.Y.; Pishesha, N.; Lewis, C.A.; Freinkman, E.; Farquharson, C.; Millan, J.L.; Lodish, H. Enhanced phosphocholine metabolism is essential for terminal erythropoiesis. Blood 2018, 131, 2955–2966. [Google Scholar] [CrossRef]
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef]
- Tselepis, A.D.; Hahalis, G.; Tellis, C.C.; Papavasiliou, E.C.; Mylona, P.T.; Kourakli, A.; Alexopoulos, D.C. Plasma levels of lipoprotein-associated phospholipase A(2) are increased in patients with beta-thalassemia. J. Lipid Res. 2010, 51, 3331–3341. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Sun, J.; Zhang, W.; Guo, Z.; Ma, Q. Arachidonic acid metabolism in health and disease. MedComm 2023, 4, e363. [Google Scholar] [CrossRef]
- Matayatsuk, C.; Lee, C.Y.; Kalpravidh, R.W.; Sirankapracha, P.; Wilairat, P.; Fucharoen, S.; Halliwell, B. Elevated F2-isoprostanes in thalassemic patients. Free Radic. Biol. Med. 2007, 43, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Pozdeev, V.I.; Gatidis, S.; Qadri, S.M.; Haussinger, D.; Kubitz, R.; Herebian, D.; Mayatepek, E.; Lang, F.; Lang, K.S.; et al. Bile Acid-Induced Suicidal Erythrocyte Death. Cell. Physiol. Biochem. 2016, 38, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Ji, F.; Chen, Y.; Cai, Z. StatTarget: A streamlined tool for signal drift correction and interpretations of quantitative mass spectrometry-based omics data. Anal. Chim. Acta 2018, 1036, 66–72. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.