Abstract
Hypertension poses a significant burden in the general population, being responsible for increasing cardiovascular morbidity and mortality, leading to adverse outcomes. Moreover, the association of hypertension with dyslipidaemia, obesity, and insulin resistance, also known as metabolic syndrome, further increases the overall cardiovascular risk of an individual. The complex pathophysiological overlap between the components of the metabolic syndrome may in part explain how novel antidiabetic drugs express pleiotropic effects. Taking into consideration that a significant proportion of patients do not achieve target blood pressure values or glucose levels, more efforts need to be undertaken to increase awareness among patients and physicians. Novel drugs, such as incretin-based therapies and renal glucose reuptake inhibitors, show promising results in decreasing cardiovascular events in patients with metabolic syndrome. The effects of sodium-glucose co-transporter-2 inhibitors are expressed at different levels, including renoprotection through glucosuria, natriuresis and decreased intraglomerular pressure, metabolic effects such as enhanced insulin sensitivity, cardiac protection through decreased myocardial oxidative stress and, to a lesser extent, decreased blood pressure values. These pleiotropic effects are also observed after treatment with glucagon-like peptide-1 receptor agonists, positively influencing the cardiovascular outcomes of patients with metabolic syndrome. The initial combination of the two classes may be the best choice in patients with type 2 diabetes mellitus and multiple cardiovascular risk factors because of their complementary mechanisms of action. In addition, the novel mineralocorticoid receptor antagonists show significant cardio-renal benefits, as well as anti-inflammatory and anti-fibrotic effects. Overall, the key to better control of hypertension in patients with metabolic syndrome is to consider targeting multiple pathogenic mechanisms, using a combination of the different therapeutic agents, as well as drastic lifestyle changes. This article will briefly summarize the association of hypertension with metabolic syndrome, as well as take into account the influence of antidiabetic drugs on blood pressure control.
1. Introduction
Hypertension, which possesses a significant prevalence in the general population, is one of the main constituents of metabolic syndrome. Hypertension is strongly associated with metabolic syndrome through the pathophysiology which involves obesity. Nevertheless, it represents the major risk factor responsible for elevating cardiovascular mortality and morbidity [1].
Hypertension is defined as repeated elevated office systolic blood pressure (SBP) values over 140 mmHg and/or diastolic BP (DBP) over 90 mmHg or average home BP over 135/85 mmHg [2,3].
Metabolic syndrome (MetS) has serious outcomes regarding the individual’s health, with increasing prevalence nowadays and a significant impact on healthcare systems. Its definition varied over time. MetS consists of several conditions, such as hypertension, elevated fasting glucose (over 100 mg/dL) or type 2 diabetes mellitus (T2DM), decreased high-density lipoprotein cholesterol levels (less than 40 mg/dL in men or 50 mg/dL in women), high triglycerides concentrations (over 150 mg/dL) and waist circumference over 40 inches (men) or 35 inches (women) [4].
MetS amplifies the risk for insulin resistance, cardiovascular and neurological complications [5]. The pathogenesis of MetS consists of multiple genetic and acquired mechanisms related to insulin resistance and chronic inflammation [6]. The burden of MetS is to be considered since it leads to adverse cardiovascular outcomes, which stand for the number one mortality cause worldwide [6].
Reaven, in 1988, discussed “syndrome X” as a metabolic syndrome represented by glucose intolerance, insulin resistance, dyslipidemia, hypertension and coronary artery disease [7]. However, earlier in 1973, the term “syndrome X” was first introduced by Kemp [8], representing symptoms of myocardial ischemia and electrocardiographic stress changes, but in the absence of atherosclerotic plaques of the coronary arteries [9].
Therefore, to distinguish between these two entities, Reaven’s “syndrome X” was associated with the term “metabolic” [8,9]. As we know, MetS includes central obesity, hypertension, insulin resistance and dyslipidemia. It is associated with an increased risk of developing complications such as diabetes and atherosclerotic cardiovascular disease [10]. Diabetes mellitus is a chronic metabolic disorder characterized by persistent high blood sugar levels [11]. It is classified into three types by etiology and clinical presentation, but type 2 diabetes accounts for around 90% of all cases [12]. Type 2 diabetes mellitus (T2DM) is an important problem that affects the entire planet and is constantly growing. In developed countries, diabetes is the main cause of cardiovascular diseases [12]. T2DM is characterized by insulin resistance that leads to insulin ineffectiveness. Initially, insulin production is increased to maintain glucose homeostasis, but over time it decreases Although people older than 45 years are most frequently affected, it is more and more common among young people due to multiple factors such as junk food, a sedentary lifestyle, and obesity [12]. Dysglycemia is a term used to describe impaired glucose regulation (or prediabetes) and diabetes mellitus [13]. A recent meta-analysis showed an inverse association between metabolic syndrome and intake of n-3 polyunsaturated fatty acids [14], which might explain why the increased intake of red meat decreases the incidence of dysglycemia [14]. Inadequate glucose metabolism (diabetes mellitus, impaired glucose tolerance, insulin resistance) or dyslipidemia are frequently associated with arterial hypertension [15,16,17]. In conclusion, hypertension is one of the most frequently diagnosed cardiovascular conditions, with well-known metabolic complications (hyperglycemia, diabetes mellitus, hyperlipidemia).
2. Pathogenesis of Hypertension in MetS
There are multiple mechanisms involved in the pathogenesis of MetS. Lifestyle, environmental, genetic, but also epigenetic [18] factors contribute to the development of MetS [6].
Insulin resistance can be found in most patients with impaired glucose tolerance or non-insulin dependent diabetes mellitus, but also in about a quarter of individuals with adequate weight and normal glucose tolerance [7]. Therefore, in these cases, the compensatory mechanism to keep glucose levels in optimal parameters is to increase insulin secretion in pancreatic cells, resulting in hyperinsulinemia [7].
Free fatty acids (FFA) play a role in the connection between insulin resistance, insulin levels and glucose tolerance. Plasma FFA can be lowered as a result of higher insulin concentrations. For that reason, hyperinsulinemia counteracts high FFA levels. When disruptions in these processes appear and hyperinsulinemia cannot be sustained, high FFA concentrations will amplify hepatic glucose production. Even slight elevations of hepatic glucose production can result in remarkable hyperglycemia [7].
There is a connection between hypertension, insulin resistance, hyperglycemia, and hyperinsulinemia [19,20,21,22,23,24,25,26,27]. Hyperinsulinemia is associated with elevated concentrations of plasmatic catecholamines, irrespective of plasmatic glucose levels [28,29]. Hypertension can also result from sodium and water metabolism through proximal renal tubular reabsorption, favoured by insulin [30].
Experimental studies indicated that hypertension could be induced in animal subjects while feeding them with food rich in fructose [7]. Due to accentuated sympathetic activity, hypertension can also be noted in sucrose-fed mice [31,32]. They conclude that blood pressure can increase as a response to dietary changes, leading to insulin resistance and hyperinsulinemia [7].
However, exercise can lower blood pressure values in people with initial hyperinsulinemia [28].
Resistance to insulin-stimulated glucose uptake is linked to hyperinsulinemia, glucose intolerance, hypertension, low HDL-cholesterol, and high triglycerides levels while also being able to influence coronary artery disease [7].
MetS is represented by a relationship between epigenetic, genetic, and environmental factors. The last two mentioned factors can lead to different expressions of orphan G protein-coupled receptors (GPCR) in MetS, such as GPR21 and GPR 82, raising the suspicion that they can represent a possible future therapeutic target gut metabolic syndrome [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33].
MicroRNA (miRNA) are small ribonucleotide acids, which regulate gene expression at the post-transcriptional level by connecting to the 3′UTRs region of messenger RNA, being able to promote or suppress translation [34]. It was observed that suppression of miR-33a can ameliorate the functional status of MetS patients since its activity consists of favoring atherosclerosis and regulation of glucose and cholesterol metabolism. Insulin resistance, statin use, and therefore low cholesterol levels can induce SREBP 1 and 2 genes, with the overexpression of miR-33a and miR-33b. This leads to low fatty acid beta-oxidation, decreased insulin signaling and low HDL-cholesterol levels, thus raising the risk for MetS [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. miR-221 and let-7 were found in high concentrations in females with MetS. Let-7 was correlated with hypertension and HDL-cholesterol levels [36].
Insulin resistance leads to hypertension through the loss of the vasodilatory effect of insulin and amplification of vasoconstriction due to FFA through reactive oxygen species [6,7,8,9,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37].
Metabolomics also plays a role in the pathogenesis of MetS. Choline, L-carnitine, and trimethylamine-N-oxide are associated with insulin resistance and unfavorable cardiometabolic events [6,38,39,40,41]. Alanine, glutamine, aspartate, asparagine, arginine, histidine methionine, cysteine, and lysine play a role in the evolution of insulin resistance [42]. Early markers for the possible occurrence of MetS can be phenylalanine, tryptophan, tyrosine, and phospholipids [43,44,45].
3. Links between Hypertension and Metabolic Syndrome
As we know, hypertension is an important element in MetS. The connections between these two entities are multiple, and complicated and they are not fully known until now.
Links that exist between hypertension and MetS include insulin resistance, central/visceral obesity, sympathetic overactivity, activated renin-angiotensin system, oxidative stress, increased inflammatory mediators, and obstructive sleep apnea [46]. In the following lines, we will discuss each separately to see how hypertension occurs in MetS.
MetS has insulin resistance as its main constituent. Studies have shown that insulin has an anti-natriuretic effect by stimulating sodium reabsorption, an effect that is not only preserved in insulin resistance but can even be increased [46]. Therefore, this can lead to hypertension within the metabolic syndrome. Likewise, other mechanisms by which insulin resistance contributes to the appearance of elevated blood pressure in the metabolic syndrome are the loss of the vasodilator effect of insulin, vasoconstriction caused by free fatty acids, and sympathetic hyperactivation [47].
Another thing worth mentioning about the link between insulin resistance and hypertension is the observation that drugs that improve insulin resistance and reduce hyperinsulinemia (such as metformin and sensitisers glitazones) also control hypertension very well. At the same time, some antihypertensives, such as angiotensin II converting enzyme inhibitors or angiotensin II receptor antagonists, also increase insulin sensitivity [48].
The second link is visceral or central obesity. Studies have shown that adipose tissue is an endocrine organ that secretes adipocytokines such as leptin, tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), angiotensinogen, and non-esterified fatty acids (NEFA), bioactive substances that have multiple roles in the body. An important effect of adipocytokines is the production of arterial hypertension. In addition, visceral obesity is the leading cause of MetS, which is why we can establish a link between it and hypertension [46].
Regarding sympathetic overactivity, the level of serum catecholamines and the activity of the sympathetic nervous system are increased in obese patients, especially for those with central obesity. People with obesity have an active renin-angiotensin system and a positive feedback relationship with the sympathetic nervous system, contributing to high blood pressure [38]. The plasma level of leptin is increased by insulin resistance, and leptin positively influences the activity of the nervous system, which leads us to think of hypertension associated with obesity [49].
There is a connection between endothelial dysfunction, which contributes to the appearance of high blood pressure, and insulin resistance, a fact highlighted by some epidemiological studies [38]. A prospective cohort study demonstrated that endothelial vasomotor dysfunction precedes and predicts the development of hypertension [50].
Another element involved in the occurrence of elevated blood pressure is the increase of inflammatory mediators [46]. Among the mediators involved in the pathophysiology of arterial hypertension within the MetS are tumour necrosis factor-α (TNF-α) and interleukin-6 (IL-6). The serum level of TNF-α was correlated with insulin resistance and systolic blood pressure. At the same time, IL-6 stimulates the sympathetic nervous system and induces an increase in the plasmatic level of angiotensinogen and angiotensin II, causing an increase in blood pressure [46].
Sympathetic overactivity has also been associated with obstructive sleep apnea [46], another important element that can contribute to hypertension, and individuals with obstructive sleep apnea have a high prevalence of MetS [49]. In MetS, there is a sympathetic activation induced by the baroreflex dysfunction, which also characterizes obstructive sleep apnea [46]. How obstructive sleep apnea leads to sympathetic stimulation is the stimulation of arterial chemoreceptors through nocturnal hypoxia and hypercapnia [46]. Other mechanisms through which obstructive sleep apnea contributes to elevated blood pressure levels are insulin resistance, endothelial dysfunction, elevated angiotensin II and aldosterone levels, oxidative stress, hyperleptinemia, and inflammation [51,52]. In conclusion, these factors may induce vasoconstriction, decreased vasodilatation, sympathetic overactivity, and increased intravascular fluid, leading to the development of hypertension in MetS (Figure 1).
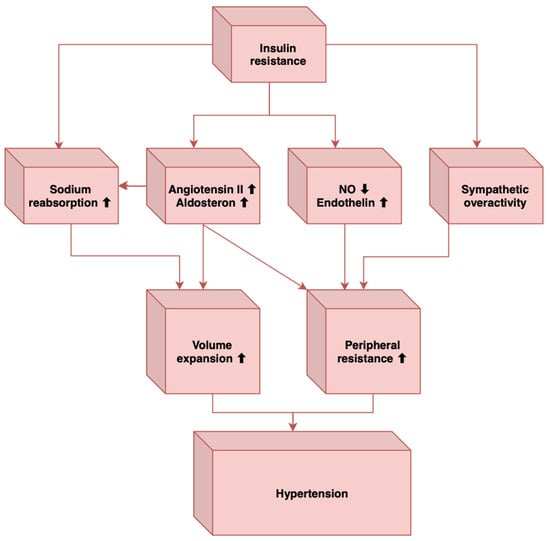
Figure 1.
The main mechanisms that link insulin resistance to hypertension (Adapted from [51]).
4. Dysmetabolic Hypertension—PROs and CONs
For many years, the constellation of hypertension, dyslipidaemia, obesity, and insulin resistance has been recognised as a distinct syndrome predisposing to the development of both type 2 diabetes mellitus and cardiovascular disease, among others [53]. On the one hand, high blood pressure, together with anthropometric and metabolic abnormalities, share common pathophysiological pathways, which partially explain why obesity and insulin resistance promotes the development of hypertension, on the other hand, how treatment of concurrent metabolic abnormalities can improve blood pressure control [54,55]. Although some controversy exists about whether MetS is indeed a syndrome or a mere association of unrelated phenotypes, the recognition of this syndrome has clinical and prognostic utility. It can help predict cardiovascular risk and guide treatment decisions, including lifestyle changes and pharmacologic treatment. The arguments in favor of the development of hypertension in patients with metabolic disturbances are controversial. Hypertension is known to be highly prevalent in patients with MetS [56]. Whether this is a causal relationship or a mere association represents a matter of discussion. Obese patients are prone to develop hypertension, but the reverse is not well established, as hypertension itself represents a poor predictor for the development of MetS [57,58]. Several mechanistic pathways that lead to a prohypertensive state have been considered in patients with MetS. Insulin resistance may promote hypertension through various processes [54]. Insulin and leptin are responsible for activating of the sympathetic nervous system, in response to over-nutrition states, which in turn increases BP values. In addition, insulin can influence the renin-angiotensin-aldosterone system, leading to an upregulation of angiotensin II type 1 receptor, potentiating peripheral vasoconstriction and plasma volume expansion [59]. These lead to a decreased response to natriuretic peptides and a rightward shift of the pressure–natriuresis curve. Furthermore, hypertensive patients with MetS are more likely to develop hypertension-mediated target organ damage than hypertensive patients alone, indicating that insulin resistance plays a central role. The components of the metabolic syndrome act synergistically to promote left ventricular hypertrophy, aortic stiffness, and microalbuminuria, which explains the increased morbidity and mortality associated with this syndrome [60].
Data from 2013 subjects from an Italian population study revealed that hypertension was present in more than 90% of individuals with MetS [61]. An increase in office blood pressure was the most frequent component found in patients with MetS, followed by hypertriglyceridemia and low plasma levels of HDL cholesterol. However, among the MetS components, only high-normal and hypertensive BP values, as well as impaired fasting glucose levels, were found to be associated with cardiovascular and all-cause mortality, while the other three components did not no prognostic association, nor did they demonstrate an increase in risk when added to the previous two risk factors. This suggests that the contribution of the different components of MetS is unbalanced, supporting the idea that metabolic syndrome is rather a heterogenous syndrome with no pathophysiological overlap [61].
On the other hand, other epidemiological data are not in accordance with this concept, suggesting that MetS is associated with an increased risk of cardiovascular events. In a study including 1750 hypertensive patients without cardiovascular disease, a significant proportion (34%) had MetS, which occurred particularly in older patients and those with longstanding hypertension. Patients with MetS had an increased risk of developing cardiovascular events than those without (hazard ratio, 3.23 vs. 1.76 per 100 patient-years; p value < 0.001). The association was observed even after multivariable adjustment (hazard ratio, 1.73; 95% confidence interval, 1.25 to 2.38; p value < 0.001), suggesting that MetS is an independent predictor of both cardiac and cerebrovascular events [62].
In another large French cohort study that included nearly 40,000 men and 20,000 women without cardiovascular disease, the prevalence of hypertension was 45% in men and 39% in women, while 19.3% of hypertensive men and 14.8% of hypertensive women had metabolic syndrome. The presence of MetS was associated with a minor increase in the risk of all-cause mortality among normotensive patients (hazard ratio, 1.09; 95% confidence interval, 0.68–1.75), while the risk among hypertensive patients was increased by 40% (hazard ratio, 1.40; 95% confidence interval, 1.13–1.74). However, no statistical significance was found after comparing the two groups. As a result, the impact of metabolic syndrome on all-cause mortality was similar between hypertensive and normotensive patients during a mean follow-up of 4.7 years. This indicates that MetS is associated with all-cause mortality, regardless of blood pressure values [63].
However, based on the existing data, we need to clearly establish whether the metabolic syndrome is indeed a clinical syndrome or a mere association of variables. The available data do not provide sufficient evidence regarding the clinical significance of MetS in hypertensive patients. Hypertension has been recognized as a major and independent risk factor for adverse cardiovascular events. It is acknowledged that hypertension tends to cluster with other metabolic abnormalities, but more efforts are needed in order to understand the pathophysiological overlap between them. This will ultimately help refine the notion of dysmetabolic hypertension.
5. Current Antidiabetic Drugs and the Influence of Hypertension
Globally, the prevalence of hypertension, systolic blood pressure (SBP) ≥140 mmHg, or diastolic blood pressure (DBP) ≥90 mmHg among patients with diabetes and obesity is 2–3 times higher than among those without this condition (over 60% in type 2 diabetes). In addition, a positive, linear relationship is observed between body mass index and the risk of hypertension [64].
Patients with type 2 diabetes (T2DM) and MetS frequently associate cardiovascular risk factors (CVRFs). Therefore, it has been shown that more than 70% of patients with diabetes also have hypertension or dyslipidemia.
The data provided by National Health and Nutritional Examination Survey (NHANES) indicates that 73.6% of adults with diabetes from the USA associate hypertension [65].
Apart from this, in Romania, the PREDATORR study showed that the prevalence of hypertension in diabetic patients was 61.7% [66].
Worldwide, the prevalence of dyslipidemia in diabetic patients varies between 72 and 85% [67]. Accordingly, the PREDATORR study revealed that 83.7% of patients with prediabetes and diabetes are also associated with dyslipidemia [66].
Atherogenic mechanisms operating in diabetic patients are multiple; insulin resistance and atherosclerosis are related to many factors. Novel drugs, incretin-based therapies (Glucagon-like peptide 1 (GLP-1) receptor agonist, dual-acting GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) receptor agonist, dipeptidyl peptidase 4 (DPP-4) inhibitors) and renal glucose reuptake inhibitors (sodium-glucose co-transporter-2 inhibitors, SGLT2-i) through pleiotropic properties lead to cardio-reno-metabolic benefits.
Many patients with MetS do not reach the target for CVD prevention (In EUROASPIRE IV) [68]. Multifactorial therapeutic management of factors that increase cardiovascular risk, obesity, blood glucose control, treatment of hypertension, and atherogenic dyslipidemia would induce a 75% decrease in cardiovascular events.
In the Steno-2 study, which included patients with T2DM and a very high risk of cardiovascular disease, an intensive multitargeted intervention including lifestyle modification and treatment for diabetes, hypertension, dyslipidemia, and microalbuminuria, along r with secondary prevention of cardiovascular disease with antiplatelet therapy with aspirin, decreases the risk of cardiovascular and microvascular outcomes by about 50% [69].
For the first time, in 2019, joint guidelines from the European Society of Cardiology and EASD in Guidelines on Diabetes, Prediabetes and Cardiovascular Diseases recommended an SGLT-2 inhibitor (empagliflozin, canagliflozin, and dapagliflozin) or GLP-1 RAs (liraglutide, semaglutide, and dulaglutide) as first-line therapy in patients with newly T2DM with prevalent cardiovascular disease (CVD) or very high/high cardiovascular (CV) risk, such as those with target-organ damage or several CVRFs [70]. The mechanisms by which the innovative therapy decreases cardiovascular mortality and brings cardiorenal benefits can be partly explained by influencing the cardiovascular risk factors associated with type 2 diabetes: obesity, hypertension, and dyslipidemia.
Although it was initially illustrated that among all antidiabetic medications with effect on hypertension (DPP4 inhibitors, GLP-1RAs, and SGLT2-i), the most significant decrease was observed in patients treated with GLP-1 RAs [71], a recent meta-analysis indicated that SGLT2-i have a more considerable effect [72].
5.1. Sodium-Glucose Co-Transporter-2 (SGLT2) Inhibitors
SGLT2-i reduces the reabsorption of filtered glucose, decreases the renal threshold for glucose, and promotes urinary glucose excretion and natriuresis [73]. SGLT2 co-transporters are found in the brush border of the renal tubular cells in the first segments of the proximal tubules (S1 and S2) and are responsible for the reabsorption of 90 to 97% of filtered glucose. The blood glucose-lowering effect is intermediate/high in conditions of adequate renal function, diminishing with its deterioration. The overall cardiovascular benefits of SGLT2-i do not seem to be related to BP and cholesterol reduction [74] and may be noticed even in patients without diabetes [75].
5.2. Features of Hypotensive Effects of SGLT2-i
The hypotensive effect of SGLT2-i is a class effect, and the decrease is modest, with an average of 3.6/1.7 mmHg [72], obtaining a similar outcome with a small dose of hydrochlorothiazide [76].
Both systolic and diastolic values are favourably influenced. The decrease in systolic blood pressure observed in most studies was between −3.5 to −4.4 mmHg, while the reduction of diastolic blood pressure was between −1.3 to −1.9 mmHg [77].
A more significant decrease in daytime values of blood pressure (possibly related to the variability of sympathetic tone) was noticed [78].
Compared to the GLP-1RAs, the decrease in blood pressure and plasmatic volume observed in SGLT2-i is not associated with an increased ventricular frequency, which can be explained by the reduction in both cardiac preload and afterload, with maintaining the cardiac output and decreasing the sympathetic nervous system activation.
The decreasing effect of SGLT2-i on BP is ascertained immediately after administration, while the hypotensive effects of GLP-1 RAs were found only in chronic administration.
Lowering the BP is persistent and is independent of the dose of SGLT2-i, renal function, or glycemic control [77].
By primarily decreasing extracellular fluid volume, arterial hypotension is rarer than in patients treated with loop diuretics. However, in specific categories of patients (especially the elderly or those with associated or pre-existing diuretic medication), monitoring the BP more closely and, eventually, decreasing the dose of diuretic administered simultaneously is recommended.
In the EMPA-REG BP trial, treatment with empagliflozin 10 mg or 25 mg once daily for 12 weeks was associated with a significant reduction in 24-h systolic and diastolic blood pressures vs. placebo; adjusted mean (95% confidence interval) differences vs. placebo in 24-h SBP were −3.44 mmHg (95% CI: −4.78; −2.09) and −4.16 mmHg (95% CI: −5.50; −2.83), respectively; for 24-h DBP was −1.36 mmHg (95% CI: −2.15; −0.56) and −1.72 mmHg (95% CI: −2.51;−0.93), respectively [79].
In DECLARE-TIMI 58, dapagliflozin vs. placebo, at 48 months, the least squares (LS) mean ± SE was lower for SBP (132.96 ± 0.37 vs. 135.32 ± 0.37 mmHg) [80].
In a systematic review and network meta-analysis, including 424 trials (276 336 patients) compared with placebo, the weighted mean differences (WMD) for SGLT2-i on SBP levels were −2.89 mmHg (95% CI: −3.37; −2.4) [81]. The most significant placebo-subtracted reductions were evident with canagliflozin (WMD −3.38 mmHg (95% CI: −4.27; −2.49)) and empagliflozin (WMD −3.29 mmHg (95% CI: −4.16; −2.41) [66]; however, for ertugliflozin, WMD was −2.66 mmHg (95% CI: −3.86; −1.47) and for dapagliflozin −2.34 mmHg (95% CI: −3.08; −1.59) [81].
Regarding DBP, empagliflozin had the most significant lowering effect compared with placebo (WMD −1.68 mmHg (95% CI: −2.09; −1.27)) ([66]). Treatment with dapagliflozin was associated with reductions in DBP by −1.45 mmHg (95% CI: −1.87; −1.03) and canagliflozin by −1.42 mmHg (95% CI: −1.78; −1.05) [81].
Similar results were published in a previous systematic review and meta-analysis of 43 RCTs (22,528 patients) (Table 1) [77].

Table 1.
Effects of SGLT2-i on blood pressure [77].
In another systematic review and meta-analysis, treatment with SGLT2-i was related to significant reductions in the daytime and nighttime systolic and diastolic BP [82].
Treatment with SGLT2-i can decrease BP in patients with heart failure (HF). In patients with HF, a meta-analysis including 16 RCTs, therapy with an SGLT2-i, determined a statistically significant reduction in SBP from a baseline of −1.68 mmHg (95% CI −2.70; −0.66; p = 0.001) vs. control [83]. Because the cardio-reno-vascular benefits are not only associated with glycemic control (Figure 2), SGLT2-i has been added to the management of HF [84,85,86].
The most significant reduction in SBP was observed for luseogliflozin, −3.42 mmHg (95% CI: −7.40; 0.56), compared to empagliflozin, −2.30 mmHg (95% CI: −3.92; −0.67), canagliflozin −1.19 mmHg (95% CI: −4.19; 1.80), and dapagliflozin −1.02 mmHg (95% CI: −3.90, 1.86), respectively [83].
In this meta-analysis of patients with HF, there was no statistically significant difference in DBP between the SGLT2-i and control groups [83].
Several studies conducted on Japanese patients with T2DM relieved significant reductions in ambulatory BP, using empagliflozin (SACRA study) [87], canagliflozin (SHIFT-J) [88], luseogliflozin (LUSCAR study) [89], and dapagliflozin (Y-AIDA study) [90].
5.3. Possible Mechanisms Involved in Lowering Blood Pressure Using SGLT2-i
Osmotic diuresis (110–470 mL/day) and natriuresis are responsible for the initial, early effect of decreasing BP that may be important, of −4.7 mmHg for dapagliflozin [91,92] (Figure 2).

Figure 2.
Pleiotropic effects of SGLT2-i (adapted from [91,92,95,97]) (BP, blood pressure; SGLT2-i, sodium-glucose co-transporter-2 inhibitors; PCT, proximal convoluted tubule; GLUT2, Glucose transporter 2, K, potassium; Na, sodium, ATP, adenosine triphoshat; FFA, fatty free acids).
Figure 2.
Pleiotropic effects of SGLT2-i (adapted from [91,92,95,97]) (BP, blood pressure; SGLT2-i, sodium-glucose co-transporter-2 inhibitors; PCT, proximal convoluted tubule; GLUT2, Glucose transporter 2, K, potassium; Na, sodium, ATP, adenosine triphoshat; FFA, fatty free acids).

Decreasing the sympathetic nervous system activity may be responsible for the persistence of the hypotensive effect of SGLT2-i (Figure 2) [93].
For decreasing of body weight, it should be mentioned that, in some studies, the effect of lowering the BP was independent of the weight loss. The glucosuric effect of SGLT2-i is associated with weight loss (Figure 2). In a recent meta-analysis, treatment with SGLT2-i, compared with placebo, generated a significant mean weight reduction of −1.79 kg (95% CI: −1.93; −1.66, p < 0.001) [94]. In patients with HF, SGLT2-i reduced body weight by −1.36 kg; (95% CI: −1.68; −1.03; p < 0.001) [83].
SGLT2-i suppresses the renal renin-angiotensin system by increasing the sodium concentration at the level of the juxtaglomerular apparatus [95] and decreasing of vascular rigidity by direct effect [96] or anti-inflammatory properties (Figure 2) [97].
Other mechanisms suggested to be involved in lowering BP are oxidative stress and enhancement of endothelial dysfunction (Figure 2) [98,99]. These combined actions result in a significant decrease in BP.
The effect of SGLT2-i on blood pressure remains unchanged regardless of the dose of SGLT2-i [99], independent of renal function and glycemic control [100,101].
Side effects of SGLT2-i include mycotic genital infections (vaginitis in women, balanitis in men), urinary infections, volume depletion, arterial hypotension, and dizziness (especially in the elderly or in combination with diuretics, angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers), Fournier’s gangrene of the genital organs; distal lower limb amputations and fractures (for canagliflozin) [70,74,77,80].
A rare complication, especially in patients with severely impaired insulin secretory function, is diabetic ketoacidosis [70,74,77,80].
The kidney, metabolic, and cardiovascular effects explain possible mechanisms involved in lowering blood pressure using SGLT2-i. Kidney effects include the rise of glucosuria, natriuresis, uricosuria and diuresis, reduced intraglomerular pressure, and hyperfiltration.
Metabolic effects can be explained by increasing insulin secretion and glucagon to insulin ratio and reducing glucose toxicity; on lipids, metabolism reducing visceral adiposity, epicardiac fat and inflammation; consecutive increased muscle FFA uptake. Heart effects, incompletely elucidated, include reduced cardiac pre- and afterload, but also improvement in cardiac efficacity, reduced myocardial oxidative stress and inflammation consecutive with decreased epicardiac fat.
At the level of the blood vessel, improved endothelial function, decreased oxidative stress, arterial stiffness and peripheral vascular resistance.
6. GLP-1 Receptor Agonist (GLP-1RAs)
GLP-1 (glucagon-like peptide-1) is an incretin hormone produced by differential posttranslational processing of the proglucagon protein by the enteroendocrine L cells” [102].
GLP-1 receptor agonists are drugs administered as subcutaneous injections (the exception is semaglutide, for which there are also formulations for oral administration). These can be with short-acting (exenatide twice daily, lixisenatide once daily, and oral semaglutide once daily) and long-acting agents (liraglutide—once daily; albiglutide, dulaglutide, exenatide, and semaglutide administrated once weekly). The glycemic effects of GLP-1 are mainly mediated by binding to its selective heptahelical G-protein–coupled receptor GLP-1R and the formation of cAMP via Gs signaling [103].
Glucose control is attained through several mechanisms of action: augmentation of glucose-dependent insulin secretion, suppressed glucagon secretion, reduced appetite, slowed gastric emptying, and concomitant reduction of food intake.
Moreover, GLP-1RAs also exert beneficial roles in multiple organ systems in which the GLP-1 receptors exist, including the cardiovascular system. In clinical trials, the identified effect of GLP-1RAs on BP differed from a slight increase, as suggested in a study with dulaglutide ([104]), to neutrality or a substantial BP-lowering impact [105,106,107].
6.1. BP Changes Observed in Clinical Trials with GLP-1 RAs
The administration of GLP-1 in intravenous infusion does not cause a decrease in BP [108]. Contrariwise, in a small study, including ten overweight men, intravenous administration of exenatide acutely increases heart rate (HR) by 6.8 beats/min (95% CI: 1.7; 11.9, p < 0.05) and increases SBP by 9.8 mmHg (95% CI: 3.5;16.1, p < 0.01) [109].
In chronic administration, a moderate decrease in SBP was observed [110]. In a meta-analysis including 32 trials, GLP-1 agonists decreased systolic blood pressure by −1.79 mmHg (95% CI: −2.94; −0.64) and −2.39 mmHg (95% CI: −3.35; −1.42) compared to placebo and active control, respectively ([93]). There was no statistically significant reduction in diastolic blood pressure −0.54 mmHg (95% CI: −1.15; 0.07) vs. placebo and −0.50 mmHg (95% CI: −1.24; 0.24) vs. active control) [111].
The most important BBP lowering effect was found with semaglutide 1 mg administered weekly vs. exenatide ten mcg with daily administration (−4.6 mmHg vs. −2.2 mmHg) without significant difference in DBP [112].
Dulaglutide 1.5 mg significantly reduced mean 24-h SBP at 16 and 26 weeks vs. placebo (least squares mean difference −2.8 mmHg (95% CI: −4.6; −1, p ≤ 0.001) at 16 wk and −2.7 mmHg (95% CI: −4.5; −0.8, p ≤ 0.002 at 26 wk ([95]). Dulaglutide 1.5 mg increase heart rate by 2.8 bpm (95% CI: 1.5, 4.2) [113].
Overall, GLP-1 agonists increased the HR by 1.86 beats/min (bpm) (95% CI: 0.85; 2.87) vs. placebo and 1.90 bpm (95% CI: 1.30; 2.50) vs. active control [111,114].
Simultaneously with the decrease in BP values, an increase in ventricular frequency was also reported [110]. It may be explained by the enhancement of the sympathetic nervous system (SNS) [115] related to the inhibition of the autonomic nervous system, especially the parasympathetic nervous system [116], with a reduced total peripheral resistance [117]. The sinoatrial node stimulation contributes to GLP-1RA–related increases in heart rate [116].
In a systematic review and network meta-analysis, including 424 trials (276 336 patients) compared with placebo, the weighted mean differences (WMD) for GLP-1RAs on SBP levels varied from 2.93 to 2.34 mmHg [81]—with exenatide ER and dulaglutide being the least efficient (Table 2) [81]. In terms of DBP, decline ranged from 1.02 mmHg for exenatide twice-daily to −0.53 mmHg for semaglutide PO; dulaglutide, exenatide ER, and lixisenatide had a neutral effect [81].
Table 2.
Effects of GLP-1RAs on blood pressure (adapted after [81]).
6.2. Mechanisms Possibly Involved in Influencing BP Values
Data from several multicenter, long-term cardiovascular outcome trials (CVOTs) with GLP-1 RAs indicated cardiovascular benefit in patients with T2DM with CVD or at very high/high risk (Lixisenatide ELIXA [118], Liraglutide LEADER [119], Semaglutide SUSTAIN 6 [120], Exenatide EXSCEL [121], Albiglutide Harmony Outcomes [122], Dulaglutide REWIND [123], and Oral semaglutide PIONEER 6 [124].
In this study, treatment with GLP-1 RAs revealed a slight but significantly reduced BP. The cardiovascular effects of GLP-1 RAs might be facilitated by improving cardiovascular risk factors such as HBA1c, BP, and body weight, reducing anti-inflammatory markers (hs-CRP) (Figure 3). In addition, it decreases vascular resistance partially by increasing nitric oxide production at the endothelium level [125] and reducing endothelial dysfunction by inhibiting oxidative stress and inflammation [126] (Figure 3).
GLP-1 RAs act at the level of the myocardium through specific receptors and inhibit cardiomyocyte apoptosis [127]. In experimental models, GLP-1 improves myocardial function and cardiac output, improves insulin sensitivity, and myocardial glucose utilisation. In animal models of myocardial ischemia, GLP-1 administration reduced infarct size, and in human subjects with acute myocardial infarction and severe systolic dysfunction after successful primary angioplasty, GLP-1 administration improved regional and global left ventricular function [128].
GLP-1 RAs increase natriuresis and diuresis reducing BP [129], in part, to inhibition of the proximal tubule Na+/H+ exchanger isoform 3 in the proximal renal tubule [130,131].
At the CNS level, GLP-1 RAs increase sympathetic activity and decrease vagal activity [116].
The renin-angiotensin-aldosterone system (RAAS) is modulated at the renal level [132]. In twelve healthy male subjects, synthetic human GLP-1 infusion decreased the circulating concentration of ANG II by 19% (95% CI: −28; −8%, p = 0.003) without significant changes for renin or aldosterone or other components of the renin-angiotensin-aldosterone system [132]. Liraglutide decreased angiotensin II (ANG II) level by 21% (p = 0.02), but there were no effects on other renin-angiotensin system components, atrial natriuretic peptides (ANPs), metanephrine or excretion of catecholamines [133].
Dulaglutide was not associated with significant changes in serum aldosterone, plasma renin activity, plasma metanephrines, normetanephrine, or N-terminal pro-brain natriuretic peptide [113].
In addition, GLP1-RAs may improve endothelial cell function, have anti-proliferative effects on smooth muscle cells, limit activation and recruitment of macrophages in atherosclerotic plaques, decrease inflammatory cytokines, and increase endogenous antioxidant defences [134].
Weight loss is important for treating hypertension, being associated with BP reduction [135]. In a study with dulaglutide, no correlation was found between weight loss and BP reduction [113]. The most common side effects of GLP-1RAs are nausea, vomiting, and diarrhoea, which decrease over time [136,137,138]. Data from clinical studies and medical practice attest that GLP-1 Ras do not predispose to acute pancreatitis, pancreatic adenocarcinoma or medullary thyroid carcinoma [136,137,138].
At the kidney level, GLP-1 RAs increase natriuresis and diuresis, lowering BP and decreasing renal inflammation and oxidative stress. On the blood vessel, decrease vascular resistance partially by raising nitric oxide production at the endothelium level and reducing endothelial dysfunction by inhibiting oxidative stress and inflammation. GLP-1 RAs at the level of the myocardium inhibit cardiomyocyte apoptosis, improve myocardial function and cardiac output, improve myocardial glucose utilization, and reduce inflammation.

Figure 3.
Pleiotropic effects of GLP-1 RA (adapted from [136,137,138]) (BP, blood pressure; FFA, fatty free acids; LDL, low-density lipoprotein; GFR, glomerular filtration rate; NO, nitric oxide).
Figure 3.
Pleiotropic effects of GLP-1 RA (adapted from [136,137,138]) (BP, blood pressure; FFA, fatty free acids; LDL, low-density lipoprotein; GFR, glomerular filtration rate; NO, nitric oxide).

The combination of GLP-1 RAs and SGLT2-is is of interest because of complementary mechanisms of action: GLP-1 RAs enrich insulin secretion, slow gastric emptying, and lower body weight, and SGLT2-i facilitate urinary glucose excretion and decrease body weight. Agents from both classes have been demonstrated to reduce CV risk. Thus, combined treatment including a GLP-1 RAs and an SGLT2-1, with or without metformin, should be the best choice to start therapy for T2DM with CVRFs (obesity, hypertension, dyslipidemia) because, in addition to reducing glucose levels, body fat will also be decreased as well BP and cardiovascular risk. With these modifications, there is the possibility for a decline in cardiac outcomes, cardiac and total mortality, and a slowdown of the decrease in renal function.
6.3. Tirzepatide, a Dual GIP (Glucose-Dependent Insulinotropic Polypeptide)/GLP-1 (Glucagon-like Peptide-1) Receptor Co-Agonist
In the SURMOUNT-1 trial [139], a significant decrease in 24 h ambulatory BP was found, with a potential beneficial effect in reducing cardiovascular risk in overweight or obese people. The reduction in blood pressure values was associated with an insignificant increase in ventricular frequency.
Thus, SBP decreased from the initial value by 5.6, 8.8, and 6.2 mmHg in patients who received doses of 5, 10, and 15 mg of tirzepatide weekly, while in the control group, SBP increased by 1.8 mmHg. DBP values in the three treatment groups decreased on average by 1.5, 2.4, and 0.0 mmHg, while they increased by 0.5 mmHg in the placebo-controlled group. This clinically significant BP reduction observed in the first 24 weeks of treatment is superior to the BP-lowering effect observed in studies with GLP-1 Ras [139].
The ventricular rate decreased on average by 1.8 bpm in the control group and increased by 0.3, 0.5, and 3.6 bpm, respectively, in the group treated with tirzepatide, similar to the effect of GLP-1 Ras [139].
In this study, the curve of decrease in blood pressure values was steep in the first 24 weeks of treatment, so, later, it remained on the plateau.
It should be mentioned, however, that the blood pressure values of the patients enrolled in the SURMOUNT-1 study were normal, one of the exclusion criteria being a BP value >160 mmHg [139].
The hypotensive effect is expected to be more consistent in hypertensive patients.
By acting on cardiovascular risk factors (significant weight loss, lowering BP, and improving the metabolic profile through the glycemic and lipid components), tirzepatide can reduce cardiovascular risk [139].
6.4. DPP-4 Inhibitors
Few studies have analyzed the effects of DPP-4 inhibitors on BP in T2D. Some studies indicate that sitagliptin may decrease SBP [140,141,142], although this result has not been observed in all studies [143]. Linagliptin, 5 mg once daily vs. placebo, was associated with small decreases in SBP and DBP [144]. Acute administration of vildagliptin reduced SBP and DBP and raised HR without affecting superior mesenteric artery blood flow [145]. In chronic administration, vildagliptin is associated with a decrease in SBP (−2.6 ± 0.3, p < 0.001) and DBP (−1.64 ± 0.8, p < 0.001) [146].
7. New Antihypertensive Drugs, New Perspectives
It is important to attain and maintain an optimal BP target (130/80 mmHg) ([129]) to reduce the risk of the progression of chronic micro- and macrovascular complications. Individualization of treatment targets is based on clinical characteristics (high or very high cardiovascular risk, chronic kidney diseases (CKD), stroke, older age, frailty) and potential adverse effects of antihypertensive treatment (syncope, orthostatic hypotension, kidney injury) [147].
Treatment for hypertension in patients with diabetes should include any of the antihypertensive pharmacotherapy drug classes with demonstrated to reduce cardiovascular risk: angiotensin-converting (ACE) inhibitors, angiotensin receptor blockers (ARB), thiazide-like diuretics (chlorthalidone and indapamide), or dihydropyridine calcium channel antagonists, and the mineralocorticoid receptor antagonists finerenonă [147,148].
Nontraditional BP-lowering agents such as SGLT2-i and GLP-1 ARs can be used, but monotherapy may be inadequate to control BP [148].
In patients with resistant hypertension, the addition of a mineralocorticoid receptor antagonist (MRA) may be considered. Recent studies assume that sacubitril/valsartan could be used in the treatment of patients with resistant hypertension, with or without additional MRA therapy [149].
Nonsteroidal Mineralocorticoid Receptor Antagonist
Finerenone is a new, selective, nonsteroidal MR antagonist with a more selective activity than spironolactone and eplerenone. Finerenone blocks MR-mediated sodium reabsorption and mineralocorticoid receptor overactivation [150]. The benefits are related to anti-inflammatory and anti-fibrotic effects observed in preclinical models [150].
Three extensive studies have been published on finerenone: FIDELIO-DKD [151], FIGARO-DKD [152], and FIDELITY [153].
In FIDELIO-DKD, finerenone showed a significant reduction in the primary kidney composite outcome, lowering the risk for CKD progression (hazard ratio, 0.82; 95% CI 0.73; 0.93, p = 0.001) among patients with predominantly stage 3–4 CKD with severely increased albuminuria and T2DM; a lower risk of cardiovascular event was observed in the finerenone group [151].
In FIGARO-DKD study, finerenone significantly improved cardiovascular composite outcomes vs. placebo, in patients with T2DM and CKD (moderately elevated albuminuria with stage 2–4 CKD or severely elevated albuminuria and stage 1–2 CKD) [152].
The FIDELIO-DKD and FIGARO-DKD trials show that finerenone has a modest impact on SBP in patients with DKD. The mean SBP decline was −2.1 mmHg at 12 months in FIDELIO-DKD [133], and −2.85 mmHg at 12 months in FIGARO-DKD [134]. In FIDELITY, the benefit on SBP was modest; in the finerenone group, was noticed the decrease of mean SBP with −3.2 mmHg at four months, −2.5 mmHg at 12 months, and −1.8 mmHg at 48 months [153].
Finerenone is recommended for patients with T2DM, an eGFR 25 mL/min/1.73 m2, normal serum potassium, and urinary albumin-to-creatinine ratio >30 mg/dL in addition to an ACE inhibitor an ARB at the maximum tolerated dose [130] or for persistent albuminuria in addition to a renin-angiotensin system inhibitor and SGLT2-i or in people with T2DM and CKD who cannot take an SGLT2-i [148].
In the FIDELITY study, at baseline, some of the included patients received treatment with SGLT2-i (6.7%) or GLP-1 RAs (7.2%). The cardiorenal benefits are maintained regardless of whether there is an SGLT2-i or GLP-1 RAs in the treatment [153].
Esaxerenone, another novel non-steroidal MRA, was authorised to treat hypertension and diabetic CKD [154].
In monotherapy, esaxerenone was associated with decreased BP during the study period −18.5/−8.8 mmHg; add-on to a RAS inhibitor, and the decline was −17.8/−8.1 mmHg [119].
In the ESAX-DN study, in patients with T2DM with microalbuminuria, esaxerenone showed a raised probability of normalising a higher urinary albumin-to-creatinine ratio and declining the progression of albuminuria [155].
In conclusion, there is much evidence from several CVOTs that indicate cardio-reno-metabolic benefits using an SGLT2-i and GLP-1 RAs in patients with metabolic syndrome at very high/high CV risk or with atherosclerotic cardiovascular diseases. SGLT2-i (empagliflozin, canagliflozin, dapagliflozin) yielded a more marked BP decline (SBP/DBP –2.46/–1.46 mmHg) without heart rate differences [70]. The BP-lowering effects of these drugs must be considered when managing BP. Treatment with these innovative molecules should be started as early as possible in type 2 diabetes. Furthermore, finerenone decreases the risk of cardiovascular outcomes and kidney disease progression in a broad spectrum of patients with CKD and T2DM [153].
8. Conclusions
Given that MetS is a constellation of abnormalities such as abdominal obesity, dyslipidemia, hypertension, and hyperglycemia, its treatment includes medication capable of targeting each of these elements. Thus, metabolic diseases are treated with anti-obesity, lipid-lowering, anti-hypertensive and anti-diabetic drugs.
Firstly, lifestyle changes are very important in patients with MetS. Losing weight can increase insulin sensitivity, reducing the risk of type 2 diabetes and can lower blood pressure. This can be achieved through diet and regular exercise. However, a potential solution for treating metabolic diseases would be the development of drugs with multiple actions. An ideal drug for MetS therapy involves decreased weight, blood pressure, inflammation, plasma lipids, and blood glucose levels.
Secondly, better adherence, rethinking the initial antihypertensive pharmacotherapy, using the triple or quadruple fixed combination that includes small doses of different therapeutic agents, thus targeting multiple mechanisms involved in the pathogenesis of hypertension, is also important.
The data of the latest studies have led to a paradigm shift regarding the management of cardiometabolic disorders. The patient-centred approach and new therapeutic classes improve the glycemic balance and reduce numerous cardiovascular risk factors. Early identification and treatment of cardiometabolic factors and conditions associated with metabolic syndrome will be related to a favourable impact on mortality and morbidity. The last years’ research brings hope in obtaining better blood pressure control, in parallel with cardio-reno-metabolic protection.
Author Contributions
Conceptualization, S.S., E.R., C.A.S. and M.J.; methodology, S.S., A.C.R., B.A.; software, D.M., E.R. and A.M.V.; validation, S.S., E.R., R.I., M.J. and C.A.S., formal analysis, S.S., C.A.S.; and investigation E.R., D.M., R.I. and C.A.S.; resources, S.S.; data curation, R.I., M.J.; writing—original draft preparation, S.S., E.R., C.A.S. and M.J.; writing—review and editing, C.A.S., B.A.; D.M., A.M.V. and A.C.R.; visualization, S.S.; supervision, M.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Franklin, S.S. Hypertension in the Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2006, 4, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Bergler-Klein, J. What’s New in the ESC 2018 Guidelines for Arterial Hypertension: The Ten Most Important Messages. Wien. Klin. Wochenschr. 2019, 131, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the Management of Arterial Hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Swarup, S.; Goyal, A.; Grigorova, Y.; Zeltser, R. Metabolic Syndrome; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Bovolini, A.; Garcia, J.; Andrade, M.A.; Duarte, J.A. Metabolic Syndrome Pathophysiology and Predisposing Factors. Int. J. Sports Med. 2021, 42, 199–214. [Google Scholar] [CrossRef]
- Fahed, C.; Aoun, L.; Zerdan, M.B.; Allam, S.; Zerdan, M.B.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef]
- Reaven, G.M. Banting Lecture 1988. Role of Insulin Resistance in Human Disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Kemp, H.G. Left Ventricular Function in Patients with the Anginal Syndrome and Normal Coronary Arteriograms. Am. J. Cardiol. 1973, 32, 375–376. [Google Scholar] [CrossRef]
- Cheng, T.O. Cardiac Syndrome X versus Metabolic Syndrome X. Int. J. Cardiol. 2007, 119, 137–138. [Google Scholar] [CrossRef]
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic syndrome: Pathophysiology, management, and modulation by natural compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/books/NBK513253 (accessed on 21 December 2022).
- Regufe, V.M.G.; Pinto, C.M.C.B.; Perez, P.M.V.H.C. Metabolic syndrome in type 2 diabetic patients: A review of current evidence. Porto Biomed. J. 2020, 5, e101. [Google Scholar] [CrossRef]
- Jambi, H.; Enani, S.; Malibary, M.; Bahijri, S.; Eldakhakhny, B.; Al-Ahmadi, J.; Al Raddadi, R.; Ajabnoor, G.; Boraie, A.; Tuomilehto, J. The Association Between Dietary Habits and Other Lifestyle Indicators and Dysglycemia in Saudi Adults Free of Previous Diagnosis of Diabetes. Nutr. Metab. Insights 2020, 15, 1178638820965258. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Park, K. Omega-3 and omega-6 polyunsaturated fatty acids and metabolic syndrome: A systemic review and meta-analysis. Clin. Nutr. 2020, 39, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Onat, A.; Hergenc, G.; Sari, I.; Turkmen, S.; Can, G.; Sansoy, V. Dyslipidemic hypertension: Distinctive features and cardiovascular risk in a prospective population-based study. Am. J. Hypertens. 2005, 18, 409–416. [Google Scholar] [CrossRef]
- Garcia-Puig, J.; Ruilope, L.M.; Luque, M.; Fernandez, J.; Ortega, R.; Dal-Re, R. Glucose metabolism in patients with essential hypertension. Am. J. Med. 2006, 119, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, F.; He, C.; Zhu, Y.; Tan, W. Elevated prevalence of abdominal glucose metabolism in patients with primary aldosteronism: A meta-analysis. Ir. J. Med. Sci. 2014, 183, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Fathi Dizaji, B. The Investigations of Genetic Determinants of the Metabolic Syndrome. Diabetes Metab. Syndr. 2018, 12, 783–789. [Google Scholar] [CrossRef]
- Stamler, J.; Rhomberg, P.; Schoenberger, J.A.; Shekelle, R.B.; Dyer, A.; Shekelle, S.; Stamler, R.; Wannamaker, J. Multivariate Analysis of the Relationship of Seven Variables to Blood Pressure: Findings of the Chicago Heart Association Detection Project in Industry, 1967–1972. J. Chronic. Dis. 1975, 28, 527–548. [Google Scholar] [CrossRef]
- Florey, C.V.; Uppal, S.; Lowy, C. Relation between Blood Pressure, Weight, and Plasma Sugar and Serum Insulin Levels in Schoolchildren Aged 9–12 Years in Westland, Holland. Br. Med. J. 1976, 1, 1368–1371. [Google Scholar] [CrossRef]
- Jarrett, R.J.; Keen, H.; Mccartney, M.; Fuller, J.H.; Hamilton, P.J.S.; Reid, D.D.; Rose, G. Glucose Tolerance and Blood Pressure in Two Population Samples: Their Relation to Diabetes Mellitus and Hypertension. Int. J. Epidemiol. 1978, 7, 15–24. [Google Scholar] [CrossRef]
- Persky, V.; Dyer, A.; Stamler, J.; Shekelle, R.B.; Schoenberger, J.; Wannamaker, J.; Upton, M. The Relationship between Post-Load Plasma Glucose and Blood Pressure at Different Resting Heart Rates. J. Chronic. Dis. 1979, 32, 263–268. [Google Scholar] [CrossRef]
- Voors, A.W.; Radhakrishnamurthy, B.; Srinivasan, S.R.; Webber, L.S.; Berenson, G.S. Plasma Glucose Level Related to Blood Pressure in 272 Children, Ages 7-15 Years, Sampled from a Total Biracial Population. Am. J. Epidemiol. 1981, 113, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.P.; Estigarribia, J.A.; Darga, L.L.; Reaven, G.M. Insulin and Blood Pressure in Obesity. Hypertension 1985, 7, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.; Gödicke, W.; Voigt, S.; Hajdu, I.; Weiss, M. Postprandial Hyperinsulinemia in Patients with Mild Essential Hypertension. Hypertension 1985, 7, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Modan, M.; Halkin, H.; Almog, S.; Lusky, A.; Eshkol, A.; Shefi, M.; Shitrit, A.; Fuchs, Z. Hyperinsulinemia. A Link between Hypertension Obesity and Glucose Intolerance. J. Clin. Investig. 1985, 75, 809–817. [Google Scholar]
- Manicardi, V.; Camellini, L.; Bellodi, G.; Coscelli, C.; Ferrannini, E. Evidence for an Association of High Blood Pressure and Hyperinsulinemia in Obese Man. J. Clin. Endocrinol. Metab. 1986, 62, 1302–1304. [Google Scholar] [CrossRef]
- Rowe, J.W.; Young, J.B.; Minaker, K.L.; Stevens, A.L.; Pallotta, J.; Landsberg, L. Effect of Insulin and Glucose Infusions on Sympathetic Nervous System Activity in Normal Man. Diabetes 1981, 30, 219–225. [Google Scholar] [CrossRef]
- Christensen, N.J.; Gundersen, H.J.G.; Hegedüs, L.; Jacobsen, F.; Mogensen, C.E.; Østerby, R.; Vittinghus, E. Acute Effects of Insulin on Plasma Noradrenaline and the Cardiovascular System. Metabolism 1980, 29 (Suppl. 1), 1138–1145. [Google Scholar] [CrossRef]
- Baum, M. Insulin Stimulates Volume Absorption in the Rabbit Proximal Convoluted Tubule. J. Clin. Investig. 1987, 79, 1104–1109. [Google Scholar] [CrossRef]
- Landsberg, L.; Young, J.B. Diet and the Sympathetic Nervous System: Relationship to Hypertension. Int. J. Obes. 1981, 5, 79–91. [Google Scholar]
- Hwang, I.S.; Ho, H.; Hoffman, B.B.; Reaven, G.M. Fructose-Induced Insulin Resistance and Hypertension in Rats. Hypertension 1987, 10, 512–516. [Google Scholar] [CrossRef]
- Romero-Nava, R.; García, N.; Aguayo-Cerón, K.A.; Sánchez Muñoz, F.; Huang, F.; Hong, E.; Villafaña, S. Modifications in GPR21 and GPR82 Genes Expression as a Consequence of Metabolic Syndrome Etiology. J. Recept. Signal Transduct. Res. 2021, 41, 38–44. [Google Scholar] [CrossRef]
- Ionescu, R.F.; Enache, R.M.; Cretoiu, S.M.; Cretoiu, D. The Interplay Between Gut Microbiota and MiRNAs in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9, 856901. [Google Scholar] [CrossRef] [PubMed]
- Gharipour, M.; Sadeghi, M. Pivotal Role of MicroRNA-33 in Metabolic Syndrome: A Systematic Review. ARYA Atheroscler. 2013, 9, 372. [Google Scholar] [PubMed]
- Wang, Y.T.; Tsai, P.C.; Liao, Y.C.; Hsu, C.Y.; Juo, S.H.H. Circulating MicroRNAs Have a Sex-Specific Association with Metabolic Syndrome. J. Biomed. Sci. 2013, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Mohanty, P.; Dhindsa, S.; Syed, T.; Ghanim, H.; Aliada, A.; Dandona, P. Elevation of Free Fatty Acids Induces Inflammation and Impairs Vascular Reactivity in Healthy Subjects. Diabetes 2003, 52, 2882–2887. [Google Scholar] [CrossRef] [PubMed]
- Lent-Schochet, D.; McLaughlin, M.; Ramakrishnan, N.; Jialal, I. Exploratory Metabolomics of Metabolic Syndrome: A Status Report. World J. Diabetes 2019, 10, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.; Tomiyama, H.; Yambe, M.; Koji, Y.; Motobe, K.; Shiina, K.; Yamamoto, Y.; Yamashina, A. Elevated Serum Levels of Alanine Aminotransferase and Gamma Glutamyltransferase Are Markers of Inflammation and Oxidative Stress Independent of the Metabolic Syndrome. Atherosclerosis 2006, 189, 198–205. [Google Scholar] [CrossRef]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Hazen, S.L. Microbiome, Trimethylamine N-Oxide, and Cardiometabolic Disease. Transl. Res. 2017, 179, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.M.T.; Vogelzangs, N.; Mook-Kanamori, D.O.; Brahimaj, A.; Nano, J.; van der Heijden, A.A.W.A.; van Dijk, K.W.; Slieker, R.C.; Steyerberg, E.W.; Ikram, A.; et al. Blood Metabolomic Measures Associate With Present and Future Glycemic Control in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 4569–4579. [Google Scholar] [CrossRef]
- Adams, S.H. Emerging Perspectives on Essential Amino Acid Metabolism in Obesity and the Insulin-Resistant State. Adv. Nutr. 2011, 2, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zheng, X.; Ma, X.; Bao, Y.; Ni, Y.; Hu, C.; Rajani, C.; Huang, F.; Zhao, A.; Jiia, W.; et al. Tryptophan Predicts the Risk for Future Type 2 Diabetes. PLoS ONE 2016, 11, e0162192. [Google Scholar] [CrossRef]
- Oxenkrug, G.; van der Hart, M.; Summergrad, P. Elevated Anthranilic Acid Plasma Concentrations in Type 1 but Not Type 2 Diabetes Mellitus. Integr. Mol. Med. 2015, 2, 365–368. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yanai, H.; Tomono, Y.; Ito, K.; Furutani, N.; Yoshida, H.; Tada, N. The Underlying Mechanisms for Development of Hypertension in the Metabolic Syndrome. Nutr. J. 2008, 7, 10. [Google Scholar] [CrossRef]
- Manrique, C.; Lastra, G.; Sowers, J.R. New Insights into Insulin Action and Resistance in the Vasculature. Ann. N. Y. Acad. Sci. 2014, 1311, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Mendizábal, Y.; Llorens, S.; Nava, E. Hypertension in Metabolic Syndrome: Vascular Pathophysiology. Int. J. Hypertens. 2013, 2013, 230868. [Google Scholar] [CrossRef]
- Alvarez, G.E.; Beske, S.D.; Ballard, T.P.; Davy, K.P. Sympathetic Neural Activation in Visceral Obesity. Circulation 2002, 106, 2533–2536. [Google Scholar] [CrossRef]
- de Jongh, R.T.; Serné, E.H.; Ijzerman, R.G.; de Vries, G.; Stehouwer, C.D.A. Impaired Microvascular Function in Obesity: Implications for Obesity-Associated Microangiopathy, Hypertension, and Insulin Resistance. Circulation 2004, 109, 2529–2535. [Google Scholar] [CrossRef]
- Kirpichnikov, D.; Sowers, J.R. Diabetes Mellitus and Diabetes-Associated Vascular Disease. Trends Endocrinol. Metab. 2001, 12, 225–230. [Google Scholar] [CrossRef]
- Umeda, M.; Kanda, T.; Murakami, M. Effects of Angiotensin II Receptor Antagonists on Insulin Resistance Syndrome and Leptin in Sucrose-Fed Spontaneously Hypertensive Rats. Hypertens. Res. 2003, 26, 485–492. [Google Scholar] [CrossRef]
- Simmons, R.K.; Alberti, K.G.; Gale, E.A.; Colagiuri, S.; Tuomilehto, J.; Qiao, Q.; Ramachandran, A.; Tajima, N.; Brajkovich Mirchov, I.; Ben-Nakhi, A.; et al. The metabolic syndrome: Useful concept or clinical tool? Report of a WHO expert consultation. Diabetologia 2010, 53, 600–605. [Google Scholar] [CrossRef]
- Marcunsi, C.; Izzo, R.; di Gioia, G.; Losi, M.A.; Barbato, E.; Morisco, C. Insulin resistance the hinge between hypertension and type 2 diabetes. High Blood Press. Cardiovasc. Prev. 2020, 27, 515–526. [Google Scholar]
- Leggio, M.; Lombardi, M.; Caldarone, E.; Severi, P.; D’Emidio, S.; Armeni, M.; Bravi, V.; Bendini, M.G.; Mazza, A. The relationship between obesity and hypertension: An updated comprehensive overview on vicious twins. Hypertens. Res. 2017, 40, 947–963. [Google Scholar] [CrossRef]
- Katsimardou, A.; Imprialos, K.; Stavropoulos, K.; Sachinidis, A.; Doumas, M.; Athyros, V. Hypertension in metabolic syndrome: Novel insights. Curr. Hypertens. Rev. 2019, 16, 12–18. [Google Scholar]
- Bramlage, P.; Pittrow, D.; Wittchen, H.U.; Kirch, W.; Boehler, S.; Lehnert, H.; Hoefler, M.; Unger, T.; Sharma, A.M. Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am. J. Hypertens. 2004, 17, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Garrison, R.J.; Kannel, W.B.; Stokes, J.; Castelli, W.P. Incidence and precursors of hypertension in young adults: The Framingham offspring study. Prev. Med. 1987, 16, 235–251. [Google Scholar] [CrossRef]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Mulè, G.; Calcaterra, I.; Nardi, E.; Cerasola, G.; Cottone, S. Metabolic Syndrome in Hypertensive Patients: An Unholy Alliance. World J. Cardiol. 2014, 6, 890. [Google Scholar] [CrossRef]
- Mancia, G.; Cannon, C.P.; Tikkanen, I.; Zeller, C.; Ley, L.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Impact of Empagliflozin on Blood Pressure in Patients with Type 2 Diabetes Mellitus and Hypertension by Background Antihypertensive Medication. Hypertension 2016, 68, 1355–1364. [Google Scholar] [CrossRef]
- Schillaci, G.; Pirro, M.; Vaudo, G.; Gemelli, F.; Marchesi, S.; Porcellati, C.; Mannarino, E. Prognostic Value of the Metabolic Syndrome in Essential Hypertension. J. Am. Coll. Cardiol. 2004, 43, 1817–1822. [Google Scholar] [CrossRef]
- Pannier, B.; Thomas, F.; Bean, K.; Jégo, B.; Benetos, A.; Guize, L. The Metabolic Syndrome: Similar Deleterious Impact on All-Cause Mortality in Hypertensive and Normotensive Subjects. J. Hypertens. 2008, 26, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Mouton, A.J.; da Silva, A.A.; Wang, Z.; Li, X.; do Carmo, J.M. Obesity, Kidney Dysfunction, and Inflammation: Interactions in Hypertension. Cardiovasc. Res. 2021, 117, 1859–1876. [Google Scholar] [CrossRef] [PubMed]
- National Diabetes Statistics Report. Centers for Disease Control and Prevention. In National Diabetes Statistics Report, 2020; Centers for Disease Control and Prevention: Atlanta, GA, USA.; Dept of Health and Human Services: Washington, DC, USA, 2020. [Google Scholar]
- Mota, M.; Popa, S.G.; Mota, E.; Mitrea, A.; Catrinoiu, D.; Cheta, D.M.; Guja, C.; Hancu, N.; Ionescu-Tirgoviste, C.; Lichiardopol, R.; et al. Prevalence of Diabetes Mellitus and Prediabetes in the Adult Romanian Population: PREDATORR Study. J. Diabetes 2016, 8, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B. Pathophysiology of Diabetic Dyslipidaemia: Where Are We? Diabetologia 2015, 58, 886–899. [Google Scholar] [CrossRef]
- Kotseva, K.; Wood, D.; de Bacquer, D.; de Backer, G.; Rydé, L.; Jennings, C.; Gyberg, V.; Amouyel, P.; Bruthans, J.; Castro Conde, A. European Society of Cardiology® Original Scientific Paper EUROASPIRE IV: A European Society of Cardiology Survey on the Lifestyle, Risk Factor and Therapeutic Management of Coronary Patients from 24 European Countries. Eur. J. Prev. Cardiol. 2016, 23, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Gæde, P.; Vedel, P.; Larsen, N.; Jensen, G.V.H.; Parving, H.-H.; Pedersen, O. Multifactorial Intervention and Cardiovascular Disease in Patients with Type 2 Diabetes. N. Engl. J. Med. 2003, 348, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef]
- Khangura, D.; Kurukulasuriya, L.R.; Sowers, J.R. Treatment of Hypertension in Diabetes: A Contemporary Approach with a Focus on Improving Cardiovascular Outcomes. Expert. Rev. Endocrinol. Metab. 2016, 11, 41–50. [Google Scholar] [CrossRef]
- Liakos, C.I.; Papadopoulos, D.P.; Sanidas, E.A.; Markou, M.I.; Hatziagelaki, E.E.; Grassos, C.A.; Velliou, M.L.; Barbetseas, J.D. Blood Pressure-Lowering Effect of Newer Antihyperglycemic Agents (SGLT-2 Inhibitors, GLP-1 Receptor Agonists, and DPP-4 Inhibitors). Am. J. Cardiovasc. Drugs 2021, 21, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Padda, I.S.; Mahtani, A.U.; Parmar, M. Sodium-Glucose Transport Protein 2 (SGLT2) Inhibitors; StatPearls: Tampa, FL, USA, 2022. [Google Scholar]
- Fitchett, D.; Inzucchi, S.E.; Zinman, B.; Wanner, C.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Ofstad, A.P.; Salsali, A.; George, J.T.; et al. Mediators of the Improvement in Heart Failure Outcomes with Empagliflozin in the EMPA-REG OUTCOME Trial. ESC Heart Fail 2021, 8, 4517. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 Inhibitors and Mechanisms of Cardiovascular Benefit: A State-of-the-Art Review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Georgianos, P.I.; Agarwal, R. Ambulatory Blood Pressure Reduction With SGLT-2 Inhibitors: Dose-Response Meta-Analysis and Comparative Evaluation With Low-Dose Hydrochlorothiazide. Diabetes Care Am. Diabtes Adssociation. 2019, 42, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Mazidi, M.; Rezaie, P.; Gao, H.K.; Kengne, A.P. Effect of Sodium-Glucose Cotransport-2 Inhibitors on Blood Pressure in People With Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of 43 Randomized Control Trials With 22,528 Patients. J. Am. Heart Assoc. 2017, 6, e004007. [Google Scholar] [CrossRef]
- Baker, W.L.; Buckley, L.F.; Kelly, M.S.; Bucheit, J.D.; Parod, E.D.; Brown, R.; Carbone, S.; Abbate, A.; Dixon, D.L. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on 24-Hour Ambulatory Blood Pressure: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2017, 6, e005686. [Google Scholar] [CrossRef] [PubMed]
- Tikkanen, I.; Narko, K.; Zeller, C.; Green, A.; Salsali, A.; Broedl, U.C.; Woerle, H.J.; EMPA-REG BP Investigators (2015). Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care 2014, 38, 420–428. [Google Scholar] [CrossRef]
- Cahn, A.; Raz, I.; Leiter, L.A.; Mosenzon, O.; Murphy, S.A.; Goodrich, E.L.; Yanuv, I.; Rozenberg, A.; Bhatt, D.L.; McGuire, D.K.; et al. Cardiovascular, Renal, and Metabolic Outcomes of Dapagliflozin Versus Placebo in a Primary Cardiovascular Prevention Cohort: Analyses From DECLARE-TIMI 58. Diabetes Care 2021, 44, 1159–1167. [Google Scholar] [CrossRef]
- Tsapas, A.; Karagiannis, T.; Kakotrichi, P.; Avgerinos, I.; Mantsiou, C.; Tousinas, G.; Manolopoulos, A.; Liakos, A.; Malandris, K.; Matthews, D.R.; et al. Comparative Efficacy of Glucose-Lowering Medications on Body Weight and Blood Pressure in Patients with Type 2 Diabetes: A Systematic Review and Network Meta-Analysis. Diabetes Obes. Metab. 2021, 23, 2116–2124. [Google Scholar] [CrossRef]
- . Tsapas, A.; Avgerinos, I.; Karagiannis, T.; Malandris, K.; Manolopoulos, A.; Andreadis, P.; Liakos, A.; Matthews, D.R.; Bekiari, E. Comparative Effectiveness of Glucose-Lowering Drugs for Type 2 Diabetes: A Systemic Review and Network Meta-analysis. Ann. Intern. Med. 2020, 18, 278–286. [Google Scholar] [CrossRef]
- Li, M.; Yi, T.; Fan, F.; Qiu, L.; Wang, Z.; Weng, H.; Ma, W.; Zhang, Y.; Huo, Y. Effect of Sodium-Glucose Cotransporter-2 Inhibitors on Blood Pressure in Patients with Heart Failure: A Systematic Review and Meta-Analysis. Cardiovasc. Diabetol. 2022, 21, 1–12. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar]
- Brito, D.; Bettencourt, P.; Carvalho, D.; Ferreira, J.; Fontes-Carvalho, R.; Franco, F.; Moura, B.; Silva-Cardoso, J.C.; de Melo, R.T.; Fonseca, C. Sodium-Glucose Co-transporter 2 Inhibitors in the Failing Heart: A Growing Potential. Cardiovasc. Drugs Ther. 2020, 34, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Muscoli, S.; Barillà, F.; Tajmir, R.; Meloni, M.; Della Morte, D.; Bellia, A.; Di Daniele, N.; Lauro, D.; Andreadi, A. The New Role of SGLT2 Inhibitors in the Management of Heart Failure: Current Evidence and Future Perspective. Pharmaceutics 2022, 14, 1730. [Google Scholar] [CrossRef]
- Kario, K.; Okada, K.; Kato, M.; Nishizawa, M.; Yoshida, T.; Asano, T.; Uchiyama, K.; Niijima, Y.; Katsuya, T.; Urata, H.; et al. Twenty-Four-Hour Blood Pressure–Lowering Effect of a Sodium-Glucose Cotransporter 2 Inhibitor in Patients With Diabetes and Uncontrolled Nocturnal Hypertension. Circulation 2019, 139, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Kario, K.; Hoshide, S.; Okawara, Y.; Tomitani, N.; Yamauchi, K.; Ohbayashi, H.; Itabashi, N.; Matsumoto, Y.; Kanegae, H. Effect of Canagliflozin on Nocturnal Home Blood Pressure in Japanese Patients with Type 2 Diabetes Mellitus: The SHIFT-J Study. J. Clin. Hypertens. 2018, 20, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Kario, K.; Okada, K.; Murata, M.; Suzuki, D.; Yamagiwa, K.; Abe, Y.; Usui, I.; Tsuchiya, N.; Iwashita, C.; Harada, N.; et al. Effects of Luseogliflozin on Arterial Properties in Patients with Type 2 Diabetes Mellitus: The Multicenter, Exploratory LUSCAR Study. J. Clin. Hypertens. 2020, 22, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Kinguchi, S.; Wakui, H.; Ito, Y.; Kondo, Y.; Azushima, K.; Osada, U.; Yamakawa, T.; Iwamoto, T.; Yutoh, J.; Misumi, T.; et al. Improved Home BP Profile with Dapagliflozin Is Associated with Amelioration of Albuminuria in Japanese Patients with Diabetic Nephropathy: The Yokohama Add-on Inhibitory Efficacy of Dapagliflozin on Albuminuria in Japanese Patients with Type 2 Diabetes Study (Y-AIDA Study). Cardiovasc. Diabetol. 2019, 18, 110. [Google Scholar]
- Sarzani, R.; Giulietti, F.; di Pentima, C.; Spannella, F. Sodium-Glucose Co-Transporter-2 Inhibitors: Peculiar “Hybrid” Diuretics That Protect from Target Organ Damage and Cardiovascular Events. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1622–1632. [Google Scholar] [CrossRef]
- Scheen, A.J. Pharmacodynamics, Efficacy and Safety of Sodium-Glucose Co-Transporter Type 2 (SGLT2) Inhibitors for the Treatment of Type 2 Diabetes Mellitus. Drugs 2015, 75, 33–59. [Google Scholar] [CrossRef]
- Wan, N.; Rahman, A.; Hitomi, H.; Nishiyama, A. The Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Sympathetic Nervous Activity. Front. Endocrinol. 2018, 9, 421. [Google Scholar] [CrossRef]
- Cheong, A.J.Y.; Teo, Y.N.; Teo, Y.H.; Syn, N.L.; Ong, H.T.; Ting, A.Z.H.; Chia, A.Z.Q.; Chong, E.Y.; Chan, M.Y.; Lee, C.H.; et al. Inhibitors on Weight and Body Mass: A Meta-Analysis of 116 Randomized-Controlled Trials. Obesity 2022, 30, 117–128. [Google Scholar] [CrossRef]
- Shin, S.J.; Chung, S.; Kim, S.J.; Lee, E.M.; Yoo, Y.H.; Kim, J.W.; Ahn, Y.B.; Kim, E.S.; Moon, S.D.; Kim, M.J.; et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0165703. [Google Scholar] [CrossRef] [PubMed]
- Chilton, R.; Tikkanen, I.; Cannon, C.P.; Crowe, S.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Effects of Empagliflozin on Blood Pressure and Markers of Arterial Stiffness and Vascular Resistance in Patients with Type 2 Diabetes. Diabetes Obes. Metab. 2015, 17, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.A.; Tan, L. Empagliflozin and canagliflozin attenuate inflammatory cytokines interferon-λ, tumor necrosis factor-α, interleukin-6: Possible mechanism of decreasing cardiovascular risk in diabetes mellitus. J. Am. Coll. Cardiol. 2018, 71, A1830. [Google Scholar] [CrossRef]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of SGLT2 Selective Inhibitor Ipragliflozin on Hyperglycemia, Hyperlipidemia, Hepatic Steatosis, Oxidative Stress, Inflammation, and Obesity in Type 2 Diabetic Mice. Eur. J. Pharmacol. 2013, 715, 246–255. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia. 2017, 60, 215–225. [Google Scholar] [CrossRef]
- Jones, B. Chronic Kidney Disease: Empagliflozin-One Step Closer to Glycaemic Control in Patients with Type II Diabetes and CKD? Nat. Rev. Nephrol. 2014, 10, 181. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Fioretto, P.; Tang, W.; List, J.F. Long-Term Study of Patients with Type 2 Diabetes and Moderate Renal Impairment Shows That Dapagliflozin Reduces Weight and Blood Pressure but Does Not Improve Glycemic Control. Kidney Int. 2014, 85, 962–971. [Google Scholar] [CrossRef]
- Rouille, Y.; Martin, S.; Steiner, D.F. Differential Processing of Proglucagon by the Subtilisin-like Prohormone Convertases PC2 and PC3 to Generate Either Glucagon or Glucagon-like Peptide. J. Biol. Chem. 1995, 270, 26488–26496. [Google Scholar] [CrossRef]
- Thorens, B. Expression Cloning of the Pancreatic Beta Cell Receptor for the Gluco-Incretin Hormone Glucagon-like Peptide 1. Proc. Natl. Acad. Sci. USA 1992, 89, 8641–8645. [Google Scholar] [CrossRef]
- Odawara, M.; Miyagawa, J.; Iwamoto, N.; Takita, Y.; Imaoka, T.; Takamura, T. Once-Weekly Glucagon-like Peptide-1 Receptor Agonist Dulaglutide Significantly Decreases Glycated Haemoglobin Compared with Once-Daily Liraglutide in Japanese Patients with Type 2 Diabetes: 52 Weeks of Treatment in a Randomized Phase III Study. Diabetes Obes. Metab. 2016, 18, 249–257. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. Drug Ther. Bull. 2016, 54, 101. [Google Scholar]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.M.B.; Todd, J.F.; Ghatei, M.A.; Bloom, S.R. Subcutaneous Glucagon-like Peptide-1 (7-36) Amide Is Insulinotropic and Can Cause Hypoglycaemia in Fasted Healthy Subjects. Clin. Sci. 1998, 95, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.M.; Muskiet, M.H.A.; Tonneijck, L.; Hoekstra, T.; Kramer, M.H.H.; Diamant, M.; van Raalte, D.H. Exenatide Acutely Increases Heart Rate in Parallel with Augmented Sympathetic Nervous System Activation in Healthy Overweight Males. Br. J. Clin. Pharmacol. 2016, 81, 613–620. [Google Scholar] [CrossRef]
- Dalsgaard, N.B.; Vilsbøll, T.; Knop, F.K.; Filip Knop, C.K. Effects of Glucagon-like Peptide-1 Receptor Agonists on Cardiovascular Risk Factors: A Narrative Review of Head-to-Head Comparisons. Diabetes Obes. Met. 2018, 20, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.E.; Holt, T.A.; Rees, K.; Randeva, H.S.; O’Hare, J.P. Effects of Exenatide and Liraglutide on Heart Rate, Blood Pressure and Body Weight: Systematic Review and Meta-Analysis. BMJ. Open 2013, 24, e001986. [Google Scholar] [CrossRef]
- Ahmann, A.J.; Capehorn, M.; Charpentier, G.; Dotta, F.; Henkel, E.; Lingvay, I.; Holst, A.G.; Annett, M.P.; Aroda, V.R. Efficacy and Safety of Once-Weekly Semaglutide Versus Exenatide ER in Subjects With Type 2 Diabetes (SUSTAIN 3): A 56-Week, Open-Label, Randomized Clinical Trial. Diabetes Care 2018, 41, 258–266. [Google Scholar] [CrossRef]
- Ferdinand, K.C.; White, W.B.; Calhoun, D.A.; Lonn, E.M.; Sager, P.T.; Brunelle, R.; Jiang, H.H.; Threlkeld, R.J.; Robertson, K.E.; Geiger, M.J. Effects of the Once-Weekly Glucagon-like Peptide-1 Receptor Agonist Dulaglutide on Ambulatory Blood Pressure and Heart Rate in Patients with Type 2 Diabetes Mellitus. Hypertension 2014, 64, 731–737. [Google Scholar] [CrossRef]
- Sun, F.; Wu, S.; Guo, S.; Yu, K.; Yang, Z.; Li, L.; Zhang, Y.; Quan, X.; Ji, L.; Zhan, S. Impact of GLP-1 receptor agonists on blood pressure, heart rate and hypertension among patients with type 2 diabetes: A systematic review and network meta-analysis. Diabetes. Res. Clin. Pract. 2015, 110, 26–37. [Google Scholar] [CrossRef]
- Meier, J.J.; Rosenstock, J.; Hincelin-Méry, A.; Roy-Duval, C.; Delfolie, A.; Coester, H.V.; Menge, B.A.; Forst, T.; Kapitza, C. Contrasting Effects of Lixisenatide and Liraglutide on Postprandial Glycemic Control, Gastric Emptying, and Safety Parameters in Patients With Type 2 Diabetes on Optimized Insulin Glargine With or Without Metformin: A Randomized, Open-Label Trial. Diabetes Care 2015, 38, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Kawabe, A.; Matsumura, M.; Aso, Y.; Yasu, T.; Banba, N.; Nakamoto, T. Effects of GLP-1 Receptor Agonists on Heart Rate and the Autonomic Nervous System Using Holter Electrocardiography and Power Spectrum Analysis of Heart Rate Variability. Diabetes Care 2016, 39, e22–e23. [Google Scholar] [CrossRef] [PubMed]
- Mendis, B.; Simpson, E.; Macdonald, I.; Mansell, P. Investigation of the Haemodynamic Effects of Exenatide in Healthy Male Subjects. Br. J. Clin. Pharmacol. 2012, 74, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Claggett, B.; Diaz, R.; Dickstein, K.; Gerstein, H.C.; Køber, L.V.; Lawson, F.C.; Ping, L.; Wei, X.; Lewis, E.F.; et al. Lixisenatide in Patients with Type 2 Diabetes and Acute Coronary Syndrome. N. Engl. J. Med. 2015, 373, 2247–2257. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 28, 311–322. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R. Cardiovascular Safety and Benefits of Semaglutide in Patients With Type 2 Diabetes: Findings From SUSTAIN 6 and PIONEER 6. Front. Endocrinol. 2021, 29, 645566. [Google Scholar] [CrossRef]
- Holman, R.R.; Bethel, M.A.; George, J.; Sourij, H.; Doran, Z.; Keenan, J.; Khurmi, N.S.; Mentz, R.J.; Oulhaj, A.; Buse, J.B.; et al. Rationale and design of the EXenatide Study of Cardiovascular Event Lowering (EXSCEL) trial. Am. Heart J. 2016, 174, 103–110. [Google Scholar] [CrossRef]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes and Cardiovascular Disease (Harmony Outcomes): A Double-Blind, Randomised Placebo-Controlled Trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and Cardiovascular Outcomes in Type 2 Diabetes (REWIND): A Double-Blind, Randomised Placebo-Controlled Trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef]
- Dong, Z.; Chai, W.; Wang, W.; Zhao, L.; Fu, Z.; Cao, W.; Liu, Z. Protein Kinase A Mediates Glucagon-like Peptide 1-Induced Nitric Oxide Production and Muscle Microvascular Recruitment. Am. J. Physiol. Endocrinol. Metab. 2013, 304, 222–228. [Google Scholar] [CrossRef]
- Ceriello, A.; Novials, A.; Ortega, E.; Canivell, S.; la Sala, L.; Pujadas, G.; Esposito, K.; Giugliano, D.; Genovese, S. Glucagon-like Peptide 1 Reduces Endothelial Dysfunction, Inflammation, and Oxidative Stress Induced by Both Hyperglycemia and Hypoglycemia in Type 1 Diabetes. Diabetes Care 2013, 36, 2346–2350. [Google Scholar] [CrossRef]
- Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.S.; Drucker, D.J.; Husain, M. Cardioprotective and Vasodilatory Actions of Glucagon-Like Peptide 1 Receptor Are Mediated Through Both Glucagon-Like Peptide 1 Receptor–Dependent and –Independent Pathways. Circulation 2008, 117, 2340–2350. [Google Scholar] [CrossRef]
- Nikolaidis, L.A.; Mankad, S.; Sokos, G.G.; Miske, G.; Shah, A.; Elahi, D.; Shannon, R.P. Effects of Glucagon-like Peptide-1 in Patients with Acute Myocardial Infarction and Left Ventricular Dysfunction after Successful Reperfusion. Circulation 2004, 109, 962–965. [Google Scholar] [CrossRef]
- Dieter, B.P.; Alicic, R.Z.; Tuttle, K.R. Translational Physiology: GLP-1 Receptor Agonists in Diabetic Kidney Disease: From the Patient-Side to the Bench-Side. Am. J. Physiol. Renal Physiol. 2018, 315, F1519. [Google Scholar] [CrossRef]
- Martins, F.L.; Bailey, M.A.; Girardi, A.C.C. Endogenous Activation of Glucagon-Like Peptide-1 Receptor Contributes to Blood Pressure Control: Role of Proximal Tubule Na+/H+Exchanger Isoform 3, Renal Angiotensin II, and Insulin Sensitivity. Hypertension 2020, 76, 839–848. [Google Scholar] [CrossRef]
- Crajoinas, R.O.; Oricchio, F.T.; Pessoa, T.D.; Pacheco, B.P.M.; Lessa, L.M.A.; Malnic, G.; Girardi, A.C.C. Mechanisms Mediating the Diuretic and Natriuretic Actions of the Incretin Hormone Glucagon-like Peptide-1. Am. J. Physiol. Renal Physiol. 2011, 301, 355–363. [Google Scholar] [CrossRef]
- Skov, J.; Dejgaard, A.; Frøkiær, J.; Holst, J.J.; Jonassen, T.; Rittig, S.; Christiansen, J.S. Glucagon-Like Peptide-1 (GLP-1): Effect on Kidney Hemodynamics and Renin-Angiotensin-Aldosterone System in Healthy Men. J. Clin. Endocrinol. Metab. 2013, 98, E664–E671. [Google Scholar] [CrossRef]
- Skov, J.; Pedersen, M.; Holst, J.J.; Madsen, B.; Goetze, J.P.; Rittig, S.; Jonassen, T.; Frøkiær, J.; Dejgaard, A.; Christiansen, J.S. Short-Term Effects of Liraglutide on Kidney Function and Vasoactive Hormones in Type 2 Diabetes: A Randomized Clinical Trial. Diabetes Obes. Metab. 2016, 18, 581–589. [Google Scholar] [CrossRef]
- Erdogdu, Ö.; Nathanson, D.; Sjöholm, Å.; Nyström, T.; Zhang, Q. Exendin-4 Stimulates Proliferation of Human Coronary Artery Endothelial Cells through ENOS-, PKA- and PI3K/Akt-Dependent Pathways and Requires GLP-1 Receptor. Mol. Cell Endocrinol. 2010, 325, 26–35. [Google Scholar] [CrossRef]
- Neter, J.E.; Stam, B.E.; Kok, F.J.; Grobbee, D.E.; Geleijnse, J.M. Influence of Weight Reduction on Blood Pressure: A Meta-Analysis of Randomized Controlled Trials. Hypertension 2003, 42, 878–884. [Google Scholar] [CrossRef]
- Przezak, A.; Bielka, W.; Pawlik, A. Incretins in the Therapy of Diabetic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 12312. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Pharmacology, Physiology, and Mechanisms of Incretin Hormone Action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef]
- Ussher, J.R.; Drucker, D.J. Cardiovascular Biology of the Incretin System. Endocr. Rev. 2012, 33, 187–215. [Google Scholar] [CrossRef]
- Rosenstock, J.; Wysham, C.; Frías, J.P.; Kaneko, S.; Lee, C.J.; Fernández Landó, L.; Mao, H.; Cui, X.; Karanikas, C.A.; Thieu, V.T. Efficacy and Safety of a Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide in Patients with Type 2 Diabetes (SURPASS-1): A Double-Blind, Randomised, Phase 3 Trial. Lancet 2021, 398, 143–155. [Google Scholar] [CrossRef]
- Pratley, R.; Nauck, M.; Bailey, T.; Montanya, E.; Cuddihy, R.; Filetti, S.; Garber, A.; Thomsen, A.B.; Hartvig, H.; Davies, M. One Year of Liraglutide Treatment Offers Sustained and More Effective Glycaemic Control and Weight Reduction Compared with Sitagliptin, Both in Combination with Metformin, in Patients with Type 2 Diabetes: A Randomised, Parallel-Group, Open-Label Trial. Int. J. Clin. Pract. 2011, 65, 397. [Google Scholar] [CrossRef]
- Ogawa, S.; Ishiki, M.; Nako, K.; Okamura, M.; Senda, M.; Mori, T.; Ito, S. Sitagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Decreases Systolic Blood Pressure in Japanese Hypertensive Patients with Type 2 Diabetes. Tohoku J. Exp. Med. 2011, 223, 133–135. [Google Scholar] [CrossRef]
- Kubota, A.; Maeda, H.; Kanamori, A.; Matoba, K.; Jin, Y.; Minagawa, F.; Obana, M.; Iemitsu, K.; Ito, S.; Amemiya, H.; et al. Pleiotropic Effects of Sitagliptin in the Treatment of Type 2 Diabetes Mellitus Patients. J. Clin. Med. Res. 2012, 4, 309–313. [Google Scholar] [CrossRef]
- Koren, S.; Shemesh-Bar, L.; Tirosh, A.; Peleg, R.K.; Berman, S.; Hamad, R.A.; Vinker, S.; Golik, A.; Efrati, S. The Effect of Sitagliptin versus Glibenclamide on Arterial Stiffness, Blood Pressure, Lipids, and Inflammation in Type 2 Diabetes Mellitus Patients. Diabetes Technol. Ther. 2012, 14, 561–567. [Google Scholar] [CrossRef]
- von Eynatten, M.; Gong, Y.; Emser, A.; Woerle, H.J. Efficacy and Safety of Linagliptin in Type 2 Diabetes Subjects at High Risk for Renal and Cardiovascular Disease: A Pooled Analysis of Six Phase III Clinical Trials. Cardiovasc. Diabetol. 2013, 12, 60. [Google Scholar] [CrossRef]
- Wu, T.; Trahair, L.G.; Little, T.J.; Bound, M.J.; Zhang, X.; Wu, H.; Sun, Z.; Horowitz, M.; Rayner, C.K.; Jones, K.L. Effects of Vildagliptin and Metformin on Blood Pressure and Heart Rate Responses to Small Intestinal Glucose in Type 2 Diabetes. Diabetes Care 2017, 40, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Schweizer, A.; Foley, J.E. Blood pressure and fasting lipid changes after 24 weeks’ treatment with vildagliptin: A pooled analysis in >2,000 previously drug-naïve patients with type 2 diabetes mellitus. Vasc. Health Risk Manag. 2016, 12, 337–340. [Google Scholar] [PubMed]
- American Diabetes Association Professional Practice Committee. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S144–S174.
- Blonde, L.; Umpierrez, G.E.; Reddy, S.S.; McGill, J.B.; Berga, S.L.; Bush, M.; Chandrasekaran, S.; De Fronzo, R.A.; Einhorn, D.; Galindo, R.J.; et al. American Association of Clinical Endocrinology Clinical Practice Guideline: Developing a Diabetes Mellitus Comprehensive Care Plan—2022 Update. Endocr. Pract. 2022, 28, 923–1049. [Google Scholar] [PubMed]
- Tadic, M.; Cuspidi, C. Sacubitril/Valsartan in the Treatment of Resistant Hypertension: Raising Star or Illusion? J. Clin. Med. 2022, 11, 3081. [Google Scholar] [CrossRef]
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and Non-Steroidal Mineralocorticoid Receptor Antagonists in Cardiorenal Medicine. Eur. Heart J. 2021, 42, 152–161. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M.; et al. Cardiovascular and Kidney Outcomes with Finerenone in Patients with Type 2 Diabetes and Chronic Kidney Disease: The FIDELITY Pooled Analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef]
- Duggan, S. Esaxerenone: First Global Approval. Drugs 2019, 79, 477–481. [Google Scholar] [CrossRef]
- Ito, S.; Kashihara, N.; Shikata, K.; Nangaku, M.; Wada, T.; Okuda, Y.; Sawanobori, T. Esaxerenone (CS-3150) in Patients with Type 2 Diabetes and Microalbuminuria (ESAX-DN): Phase 3 Randomized Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1715–1727. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).