
Ring Trial on Quantitative Assessment of Bile Acids Reveals a Method- and Analyte-Specific Accuracy and Reproducibility

 ,
,  ,
,  , , , , ,
, , , , , 
Abstract
:
1. Introduction
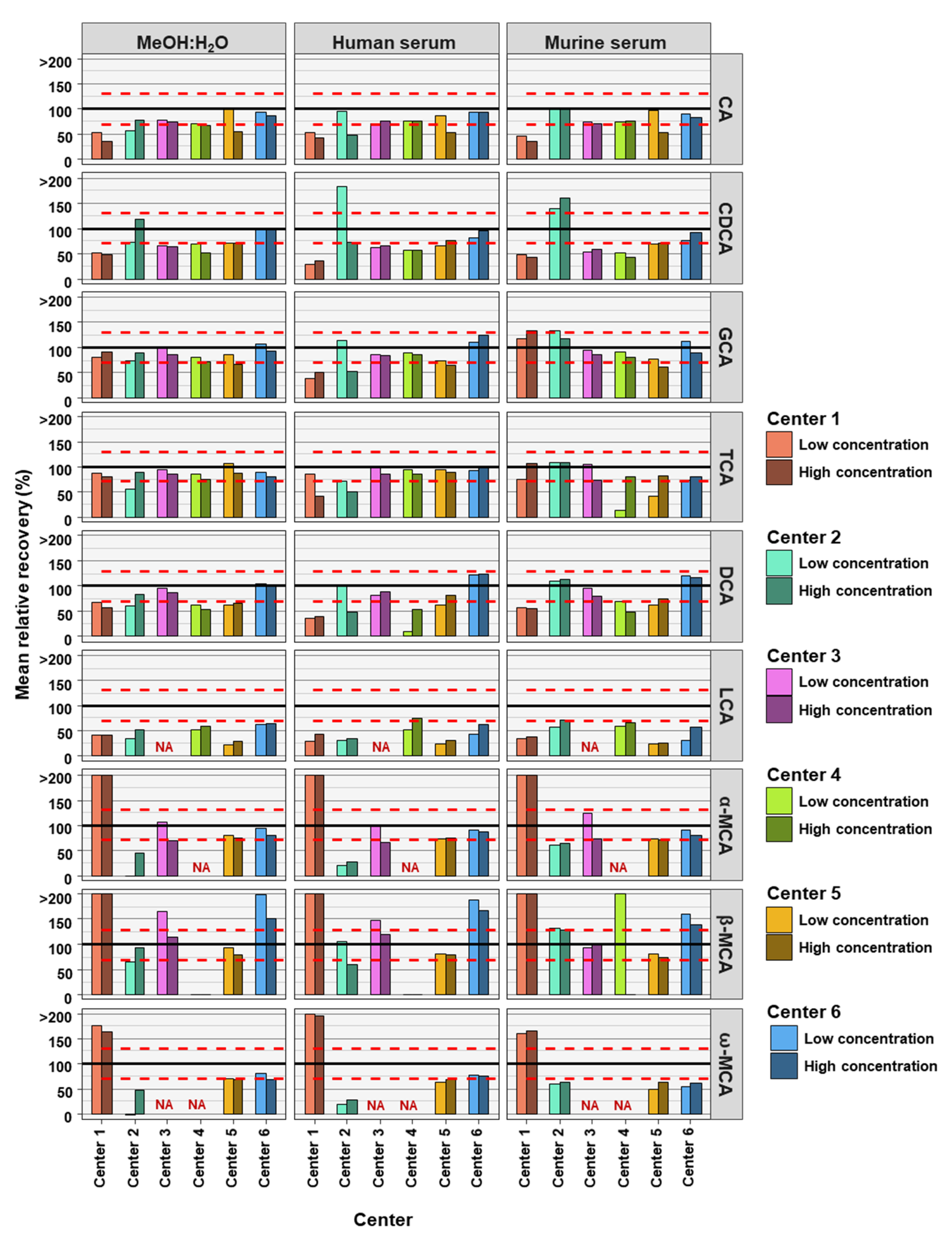
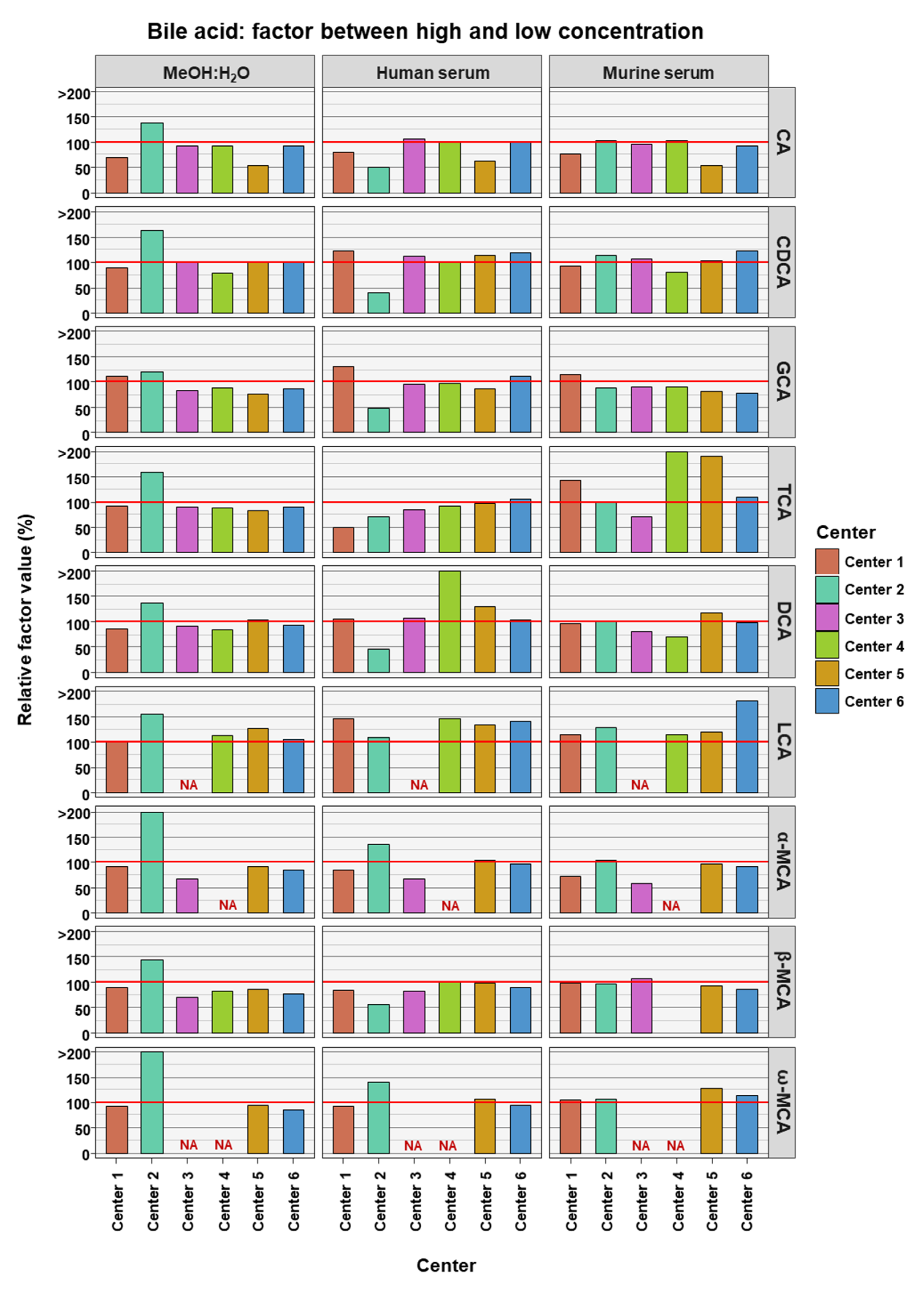
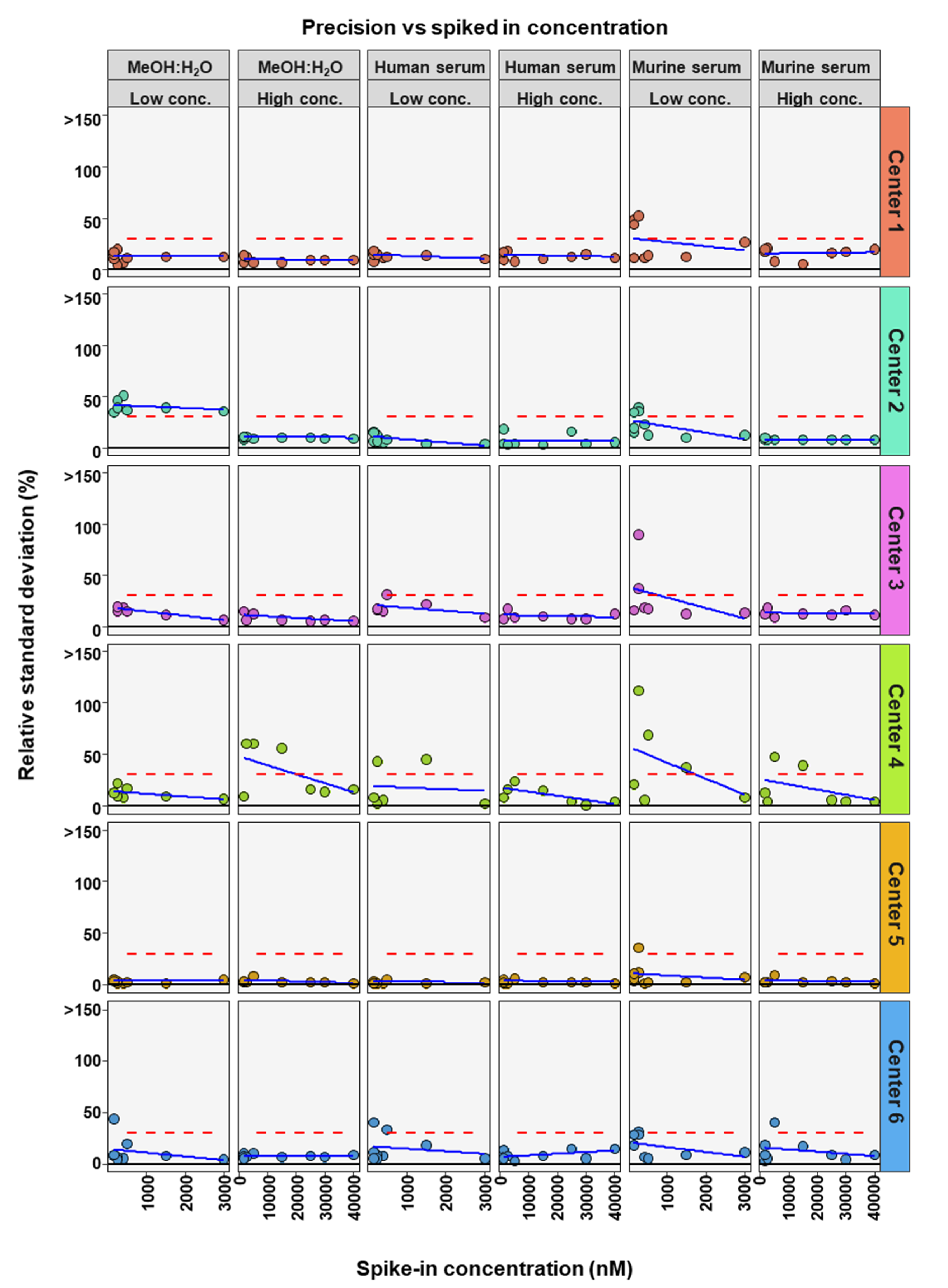
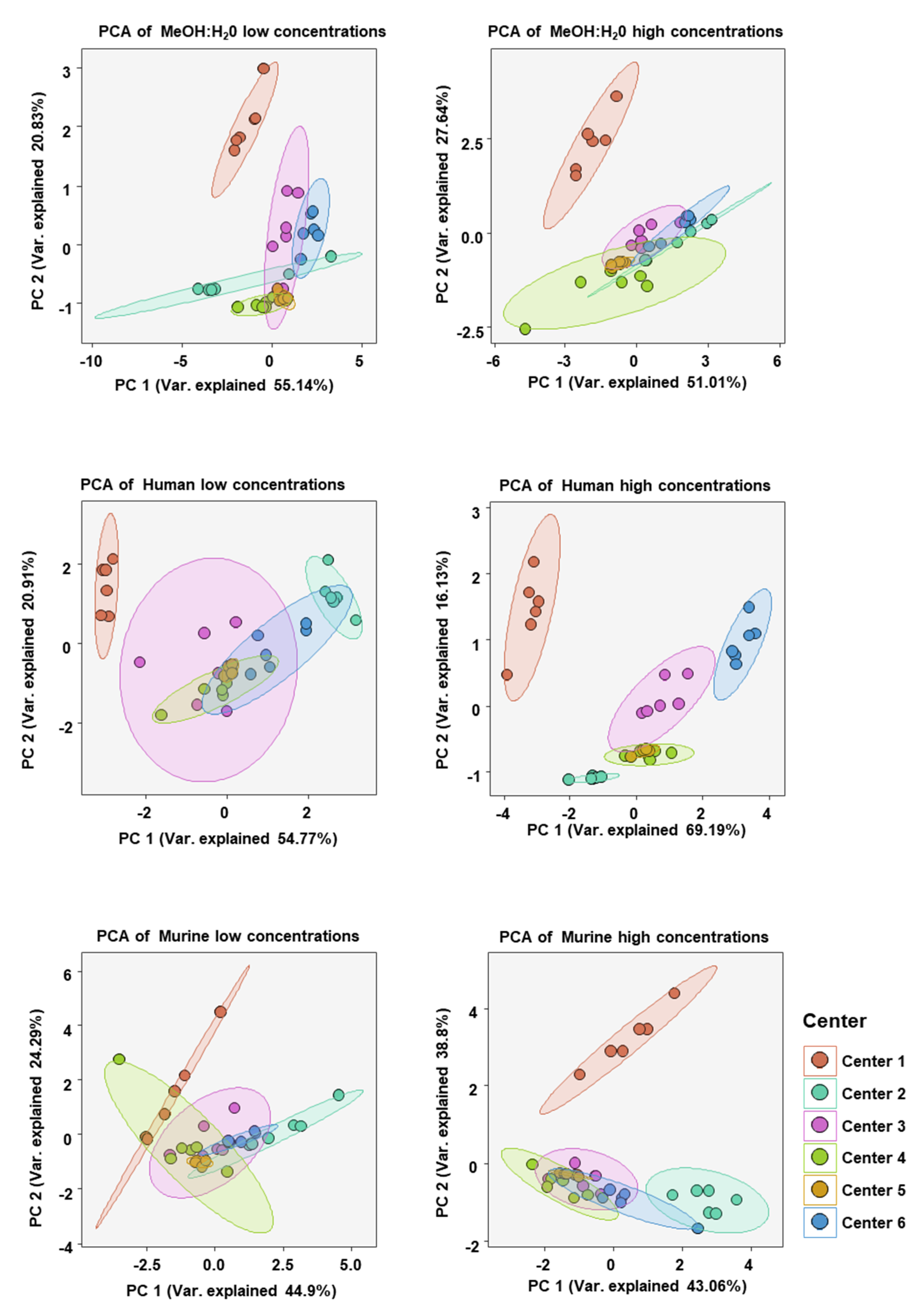
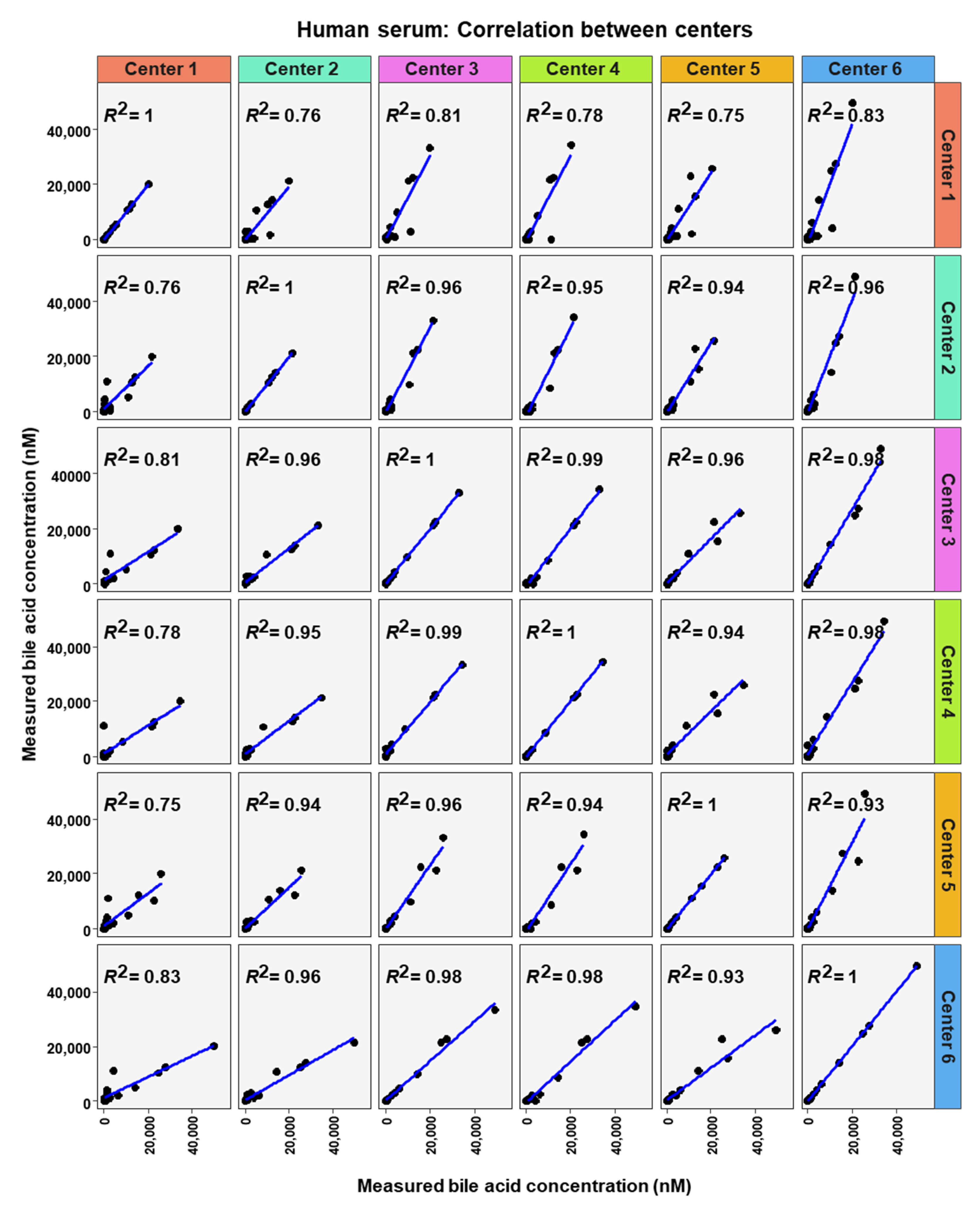
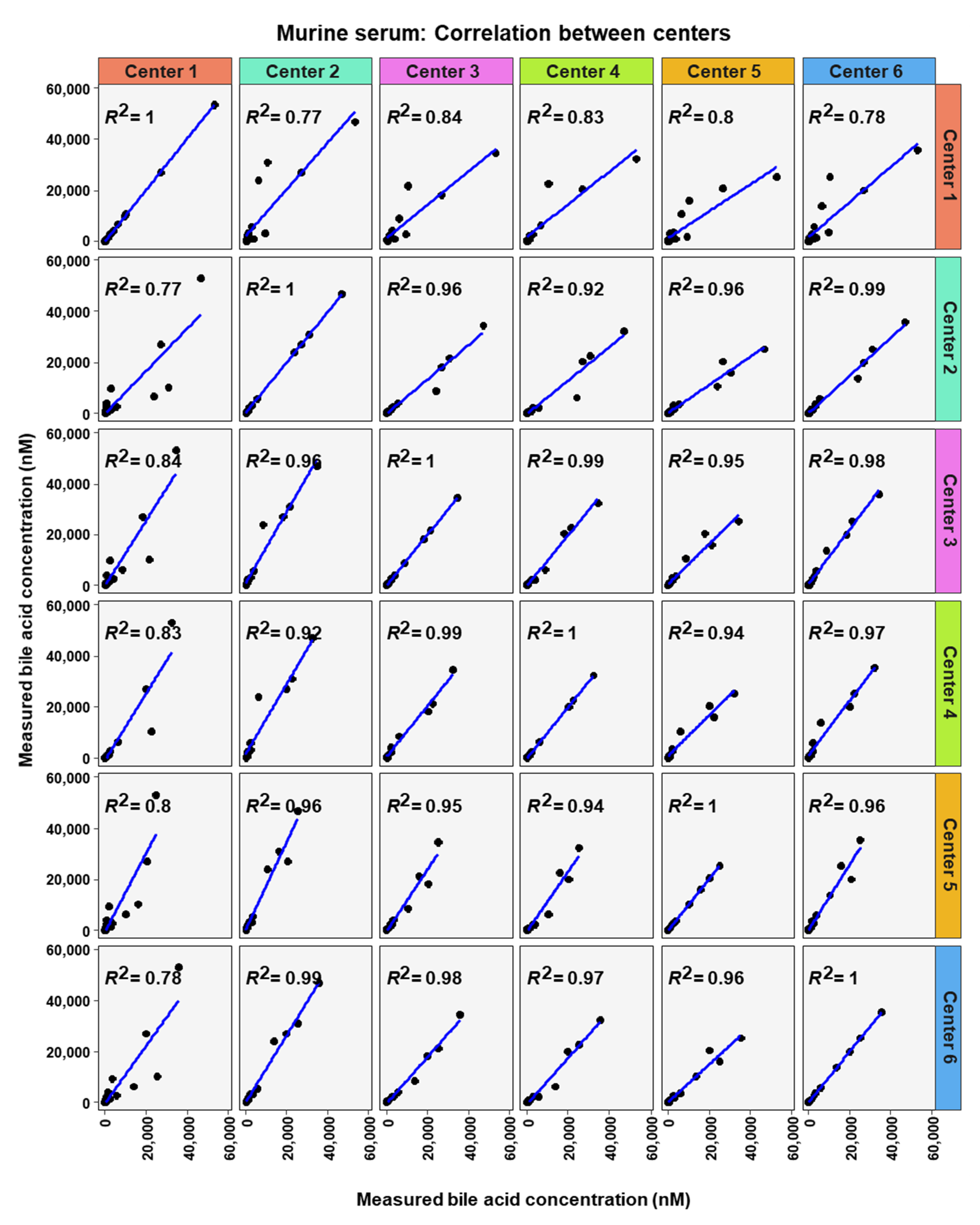
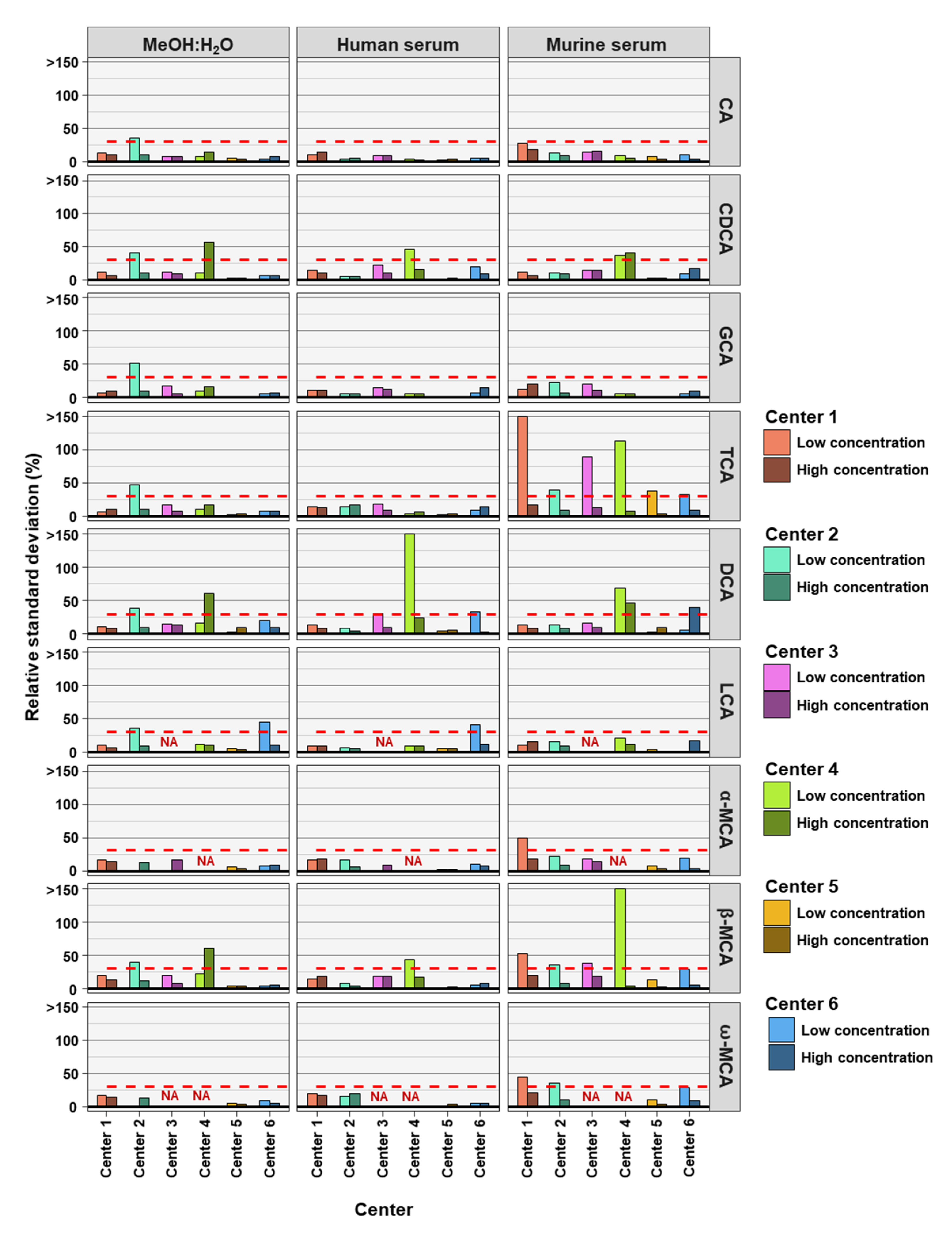
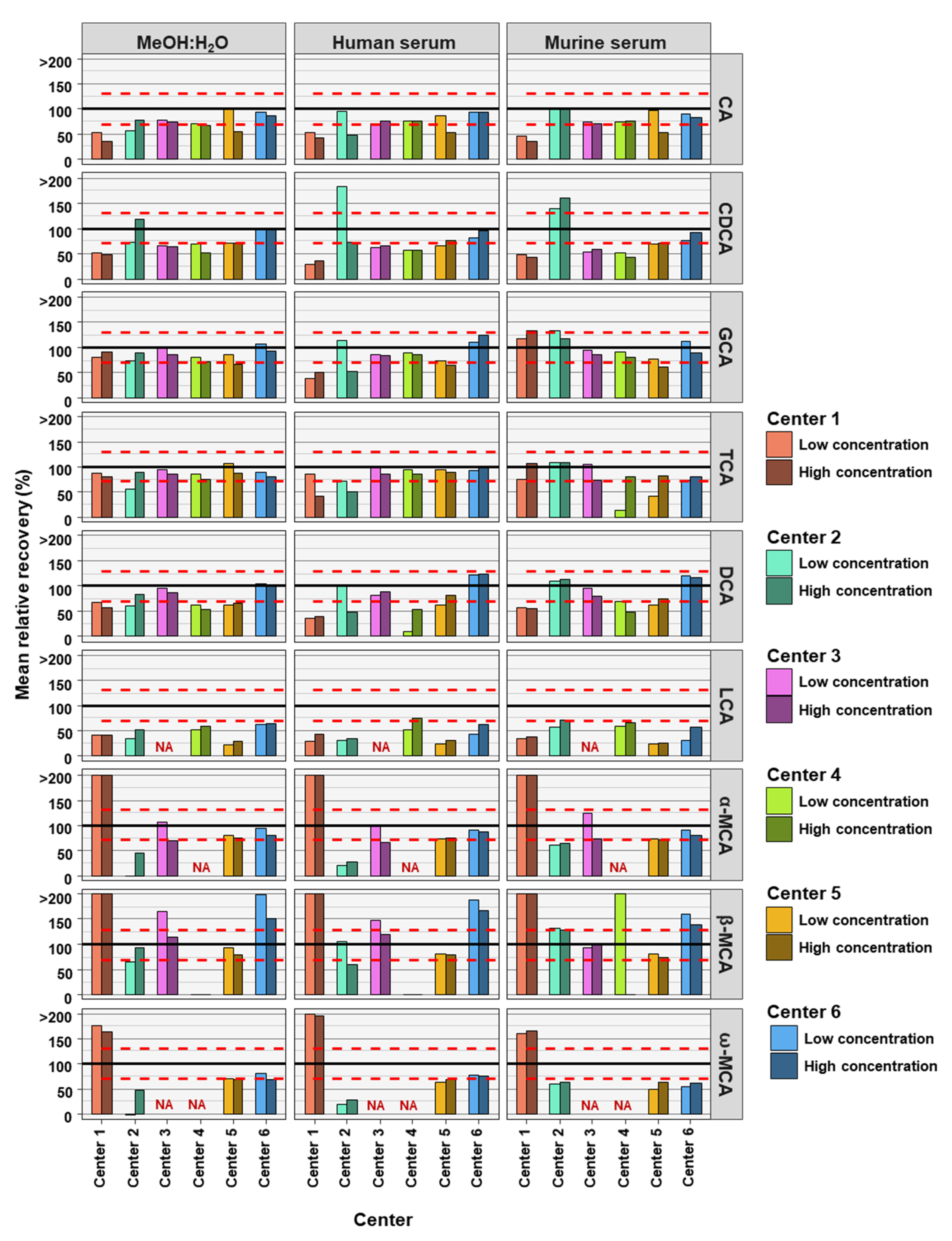
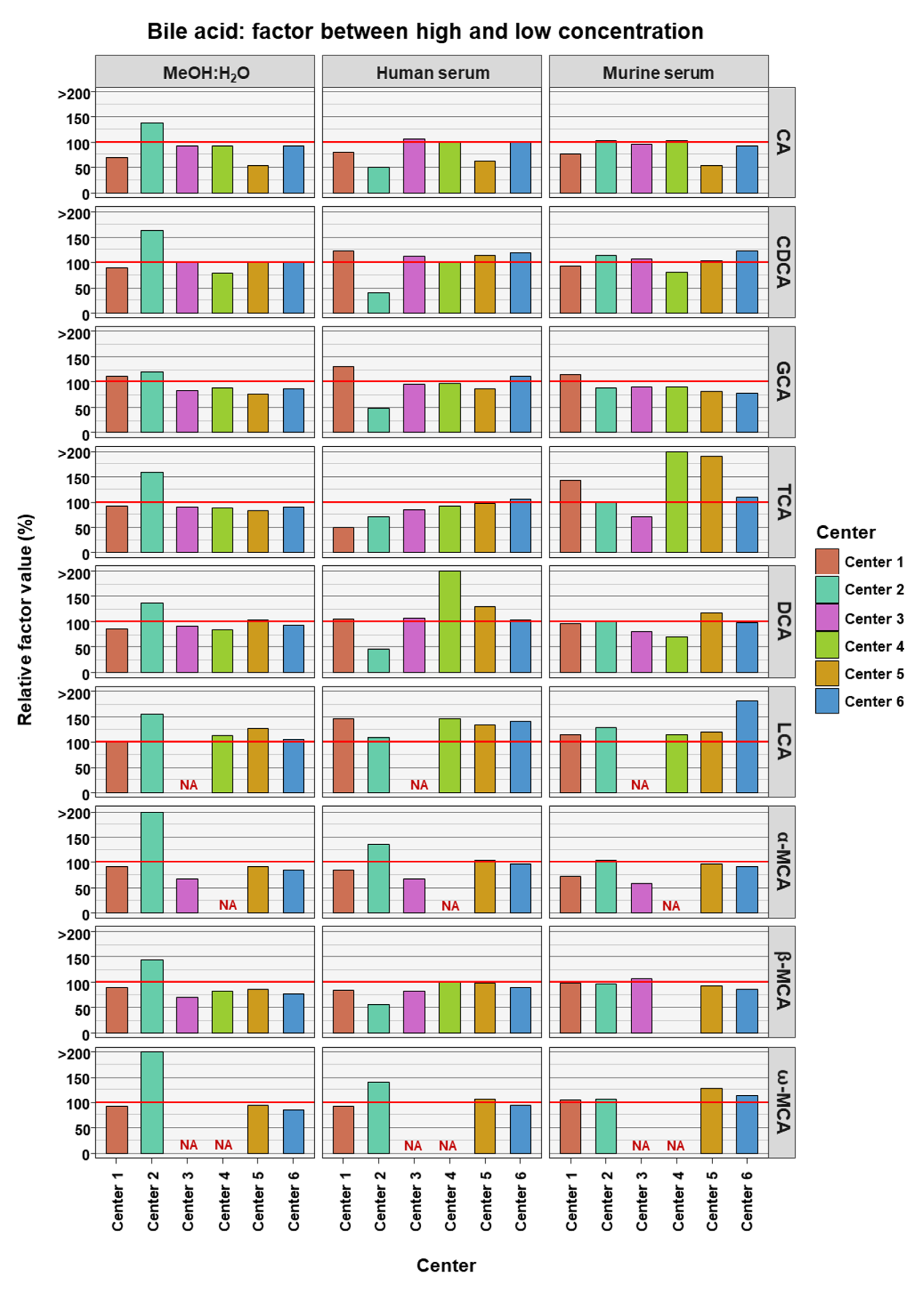
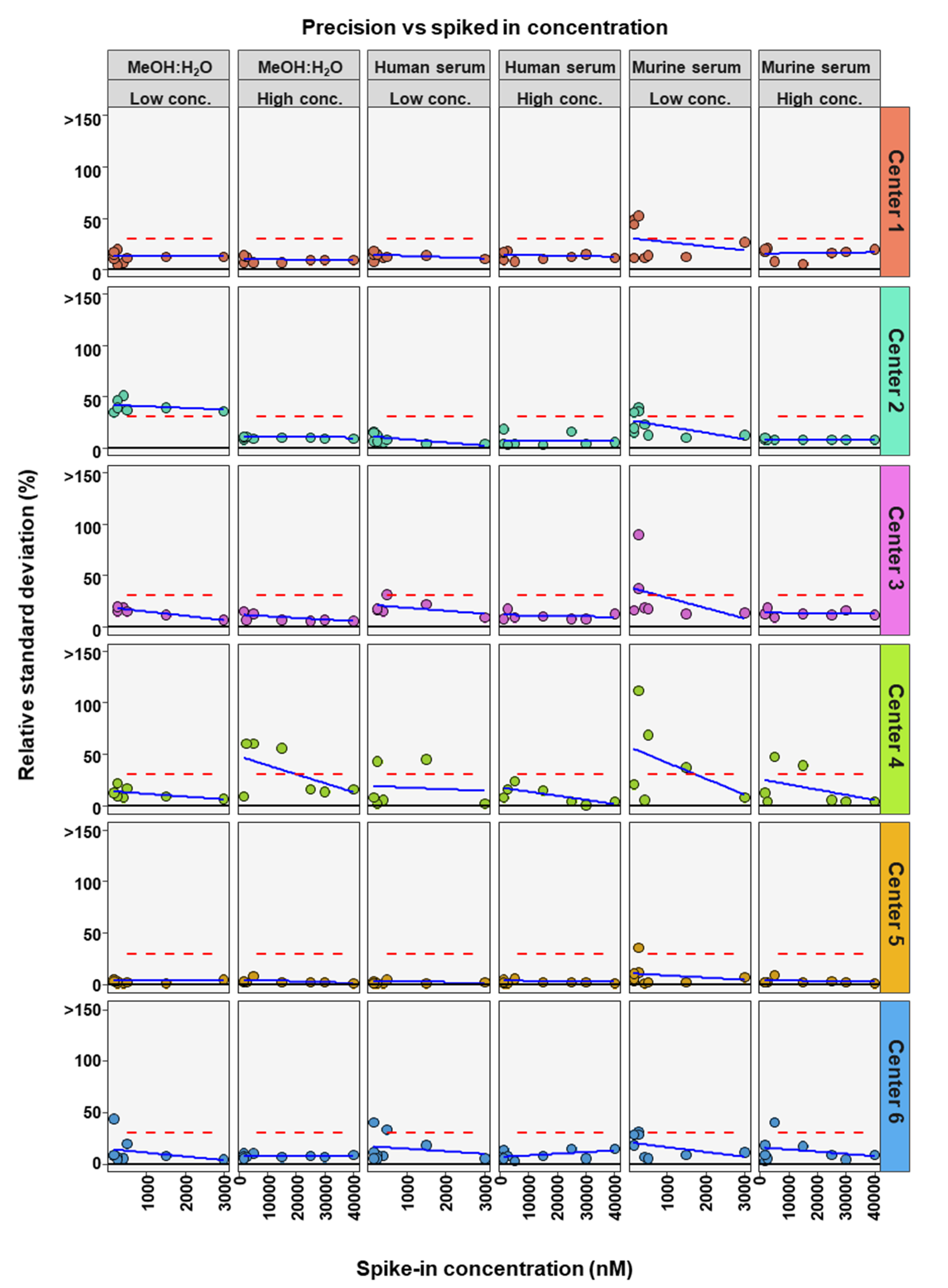
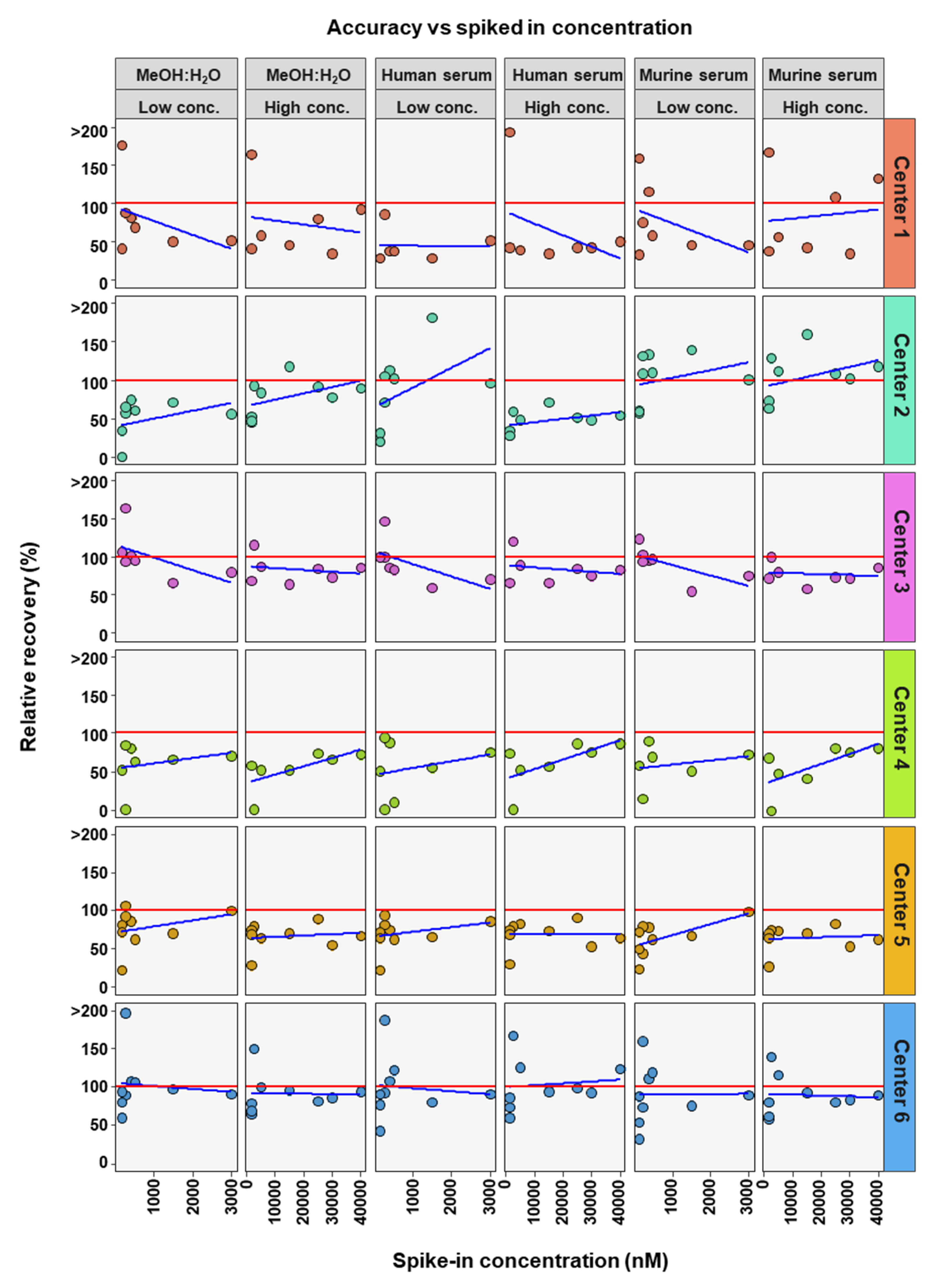
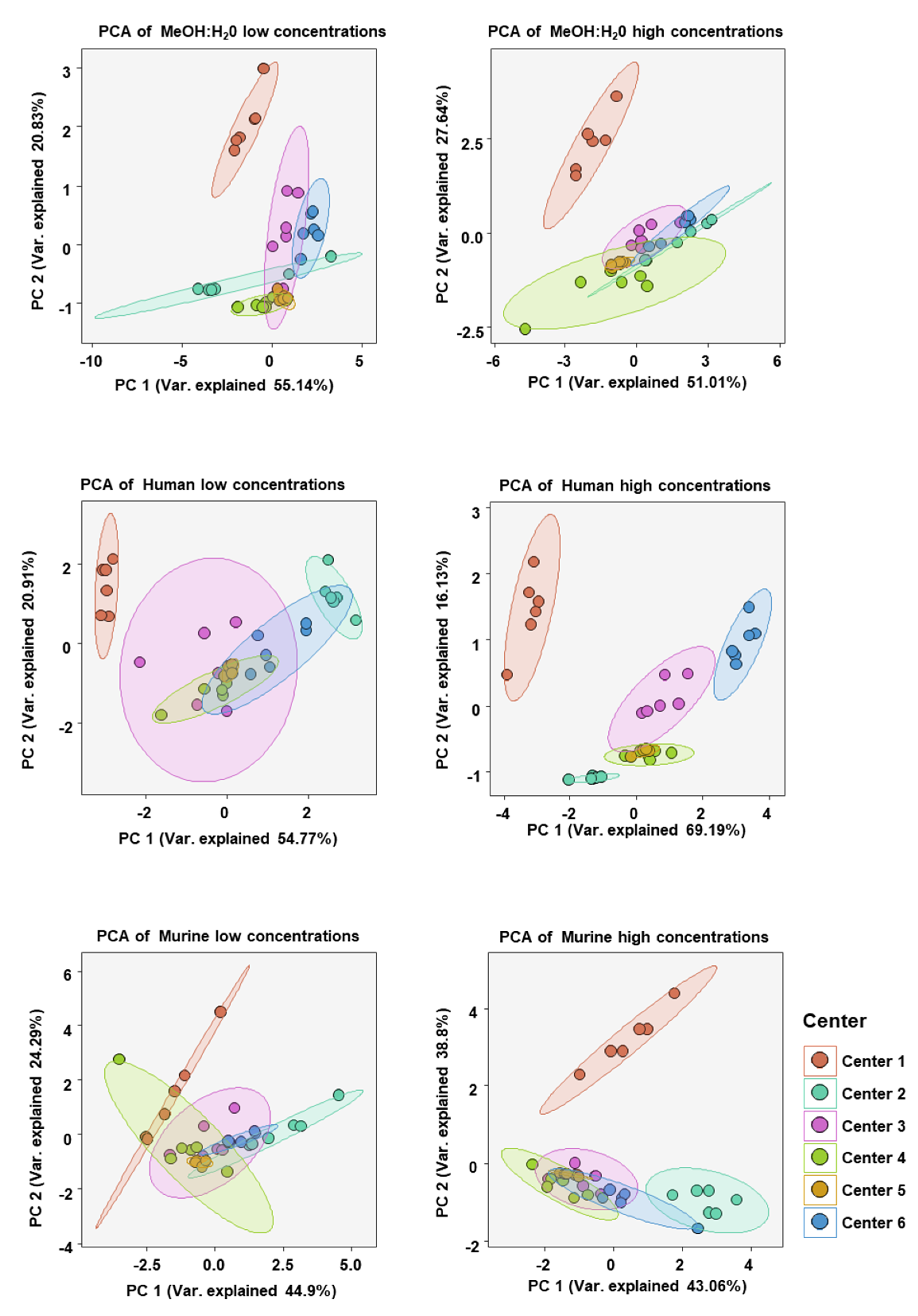
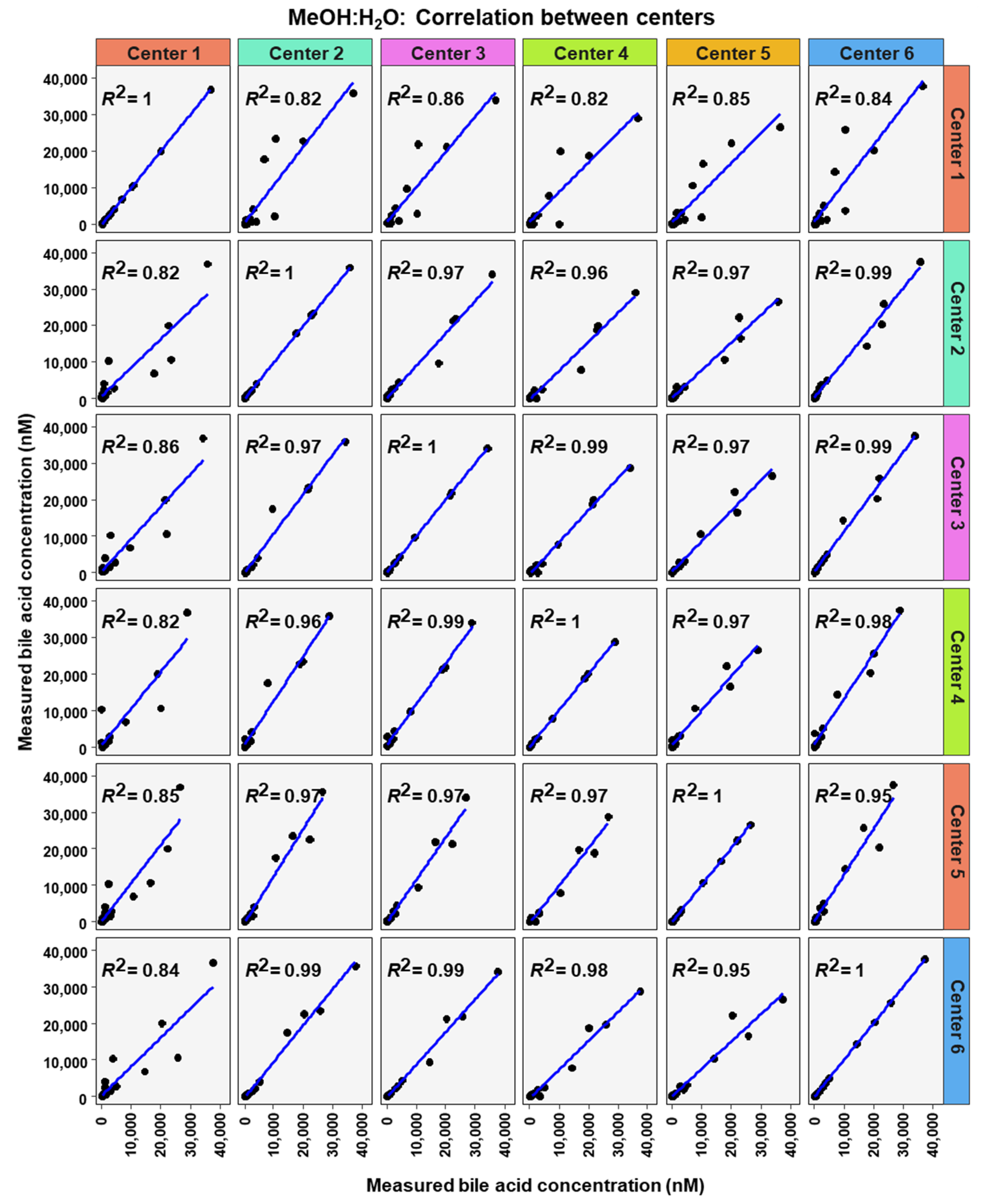
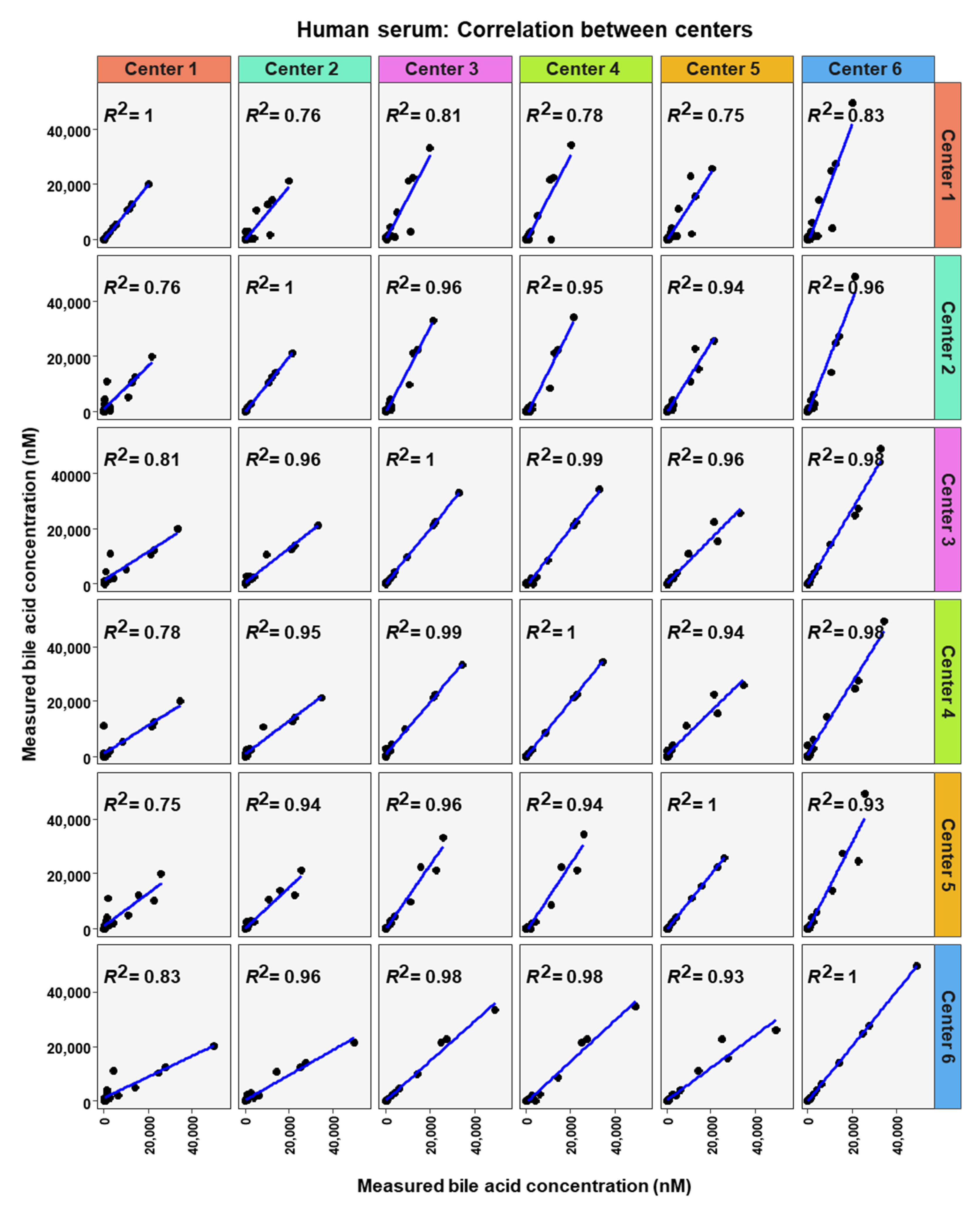
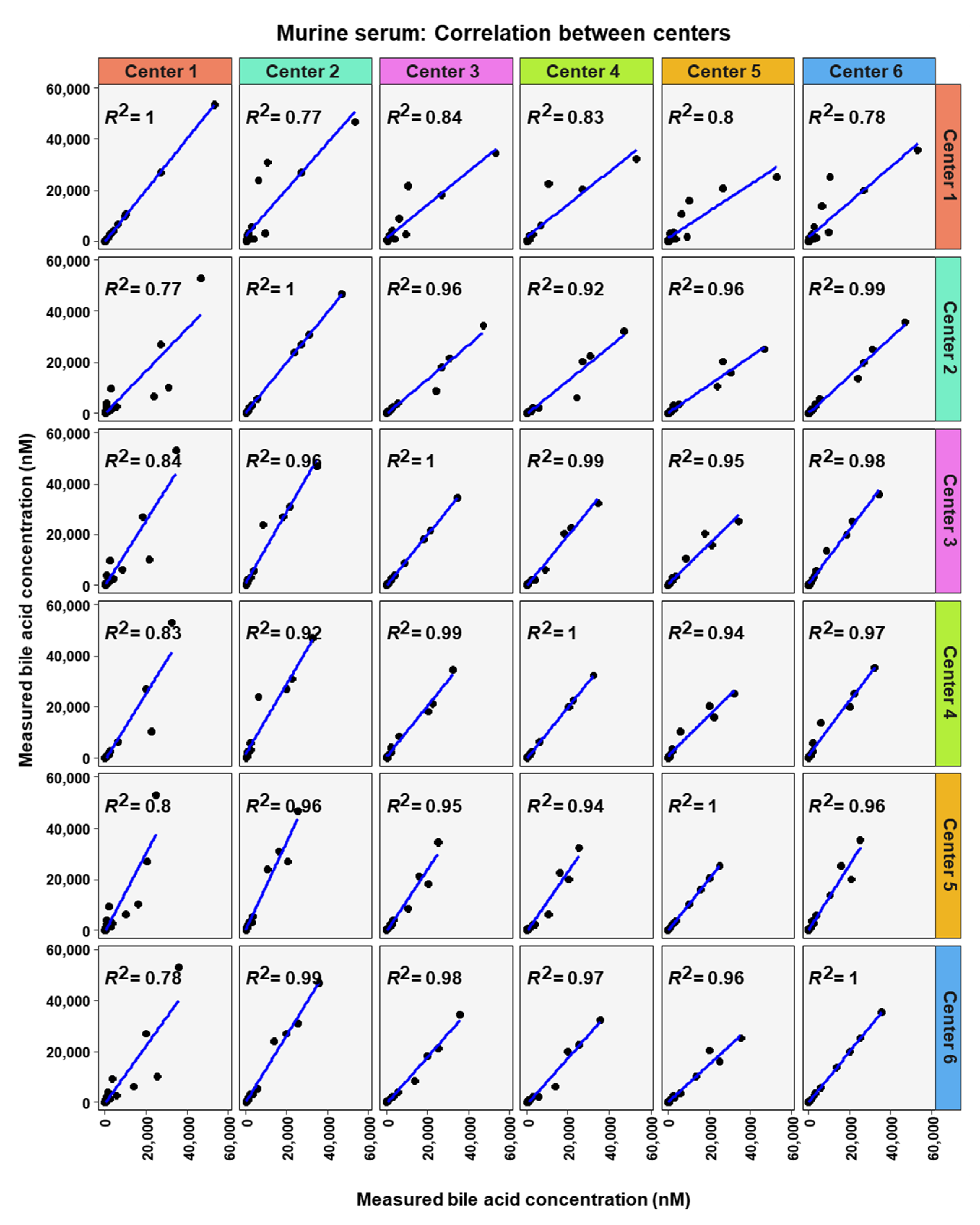
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Preparation for the Ring Trial
4.2. Center 1: Sample Preparation, LC-MS/MS, and Raw Data Analysis
4.2.1. Background
4.2.2. Materials, Calibration Standards and Quality Control
4.2.3. Sample Preparation
4.2.4. Equipment
4.2.5. Data Analysis
4.3. Center 2: Sample Preparation, LC-MS/MS, and Raw Data Analysis
4.3.1. Background
4.3.2. Calibration Standards and Quality Control
4.3.3. Sample Preparation
4.3.4. Equipment
4.3.5. Data Analysis
4.4. Center 3: Sample Preparation, LC-MS/MS, and Raw Data Analysis
4.4.1. Background
4.4.2. Materials, Calibration Standards and Quality Control
4.4.3. Sample Preparation
4.4.4. Equipment
4.4.5. Data Analysis
4.5. Center 4: Sample Preparation, LC-MS/MS, and Raw Data Analysis
4.5.1. Background
4.5.2. Materials, Calibration Standards and Quality Control
4.5.3. Sample Preparation
4.5.4. Equipment
4.5.5. Data Analysis
4.6. Center 5: Sample Preparation, LC-MS/MS, and Raw Data Analysis
4.6.1. Background
4.6.2. Materials, Calibration Standards and Quality Control
4.6.3. Sample Preparation
4.6.4. Equipment
4.6.5. Data Analysis
4.7. Center 6: Sample Preparation, LC-MS/MS, and Raw Data Analysis
4.7.1. Background
4.7.2. Materials, Calibration Standards and Quality Control
4.7.3. Sample Preparation
4.7.4. Equipment
4.7.5. Data Analysis
4.8. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, S.L.; Gahan, C.G.M.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.H.; O’Flaherty, S.; Barrangou, R.; Theriot, C.M. Bile salt hydrolases: Gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLoS Pathog. 2019, 15, e1007581. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Hu, X.J.; Singh, W.; Geng, W.; Tikhonova, I.G.; Lin, J. The complex structure of bile salt hydrolase from Lactobacillus salivarius reveals the structural basis of substrate specificity. Sci. Rep. 2019, 9, 12438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Seaton, S.C.; Ndousse-Fetter, S.; Adhikari, A.A.; DiBenedetto, N.; Mina, A.I.; Banks, A.S.; Bry, L.; Devlin, A.S. A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 2018, 7, e37182. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Urdaneta, V.; Casadesús, J. Interactions between Bacteria and Bile Salts in the Gastrointestinal and Hepatobiliary Tracts. Front. Med. 2017, 4, 163. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis: Thematic Review Series: Bile Acids. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. Nafld, Nash and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Kim, G.A.; Lee, H.C.; Choe, J.; Kim, M.J.; Lee, M.J.; Chang, H.S.; Bae, I.Y.; Kim, H.K.; An, J.; Shim, J.H.; et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2017, 68, 140–146. [Google Scholar] [CrossRef]
- van Best, N.; Rolle-Kampczyk, U.; Schaap, F.G.; Basic, M.; Olde Damink, S.W.M.; Bleich, A.; Savelkoul, P.H.M.; von Bergen, M.; Penders, J.; Hornef, M.W. Bile acids drive the newborn’s gut microbiota maturation. Nat. Commun. 2020, 11, 3692. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Su, L.; Zheng, P.C.; Bajaj, J.S.; Zhou, H. Bile Acids, Gut Microbiome and the Road to Fatty Liver Disease. Compr. Physiol. 2021, 12, 2719–2730. [Google Scholar] [CrossRef]
- Sun, L.; Cai, J.; Gonzalez, F.J. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 335–347. [Google Scholar] [CrossRef]
- Haange, S.-B.; Jehmlich, N.; Krügel, U.; Hintschich, C.; Wehrmann, D.; Hankir, M.; Seyfried, F.; Froment, J.; Hübschmann, T.; Müller, S.; et al. Gastric bypass surgery in a rat model alters the community structure and functional composition of the intestinal microbiota independently of weight loss. Microbiome 2020, 8, 13. [Google Scholar] [CrossRef]
- St John-Williams, L.; Mahmoudiandehkordi, S.; Arnold, M.; Massaro, T.; Blach, C.; Kastenmüller, G.; Louie, G.; Kueider-Paisley, A.; Han, X.; Baillie, R.; et al. Bile acids targeted metabolomics and medication classification data in the ADNI1 and ADNIGO/2 cohorts. Sci. Data 2019, 6, 212. [Google Scholar] [CrossRef] [Green Version]
- Behr, C.; Slopianka, M.; Haake, V.; Strauss, V.; Sperber, S.; Kamp, H.; Walk, T.; Beekmann, K.; Rietjens, I.; van Ravenzwaay, B. Analysis of metabolome changes in the bile acid pool in feces and plasma of antibiotic-treated rats. Toxicol. Appl. Pharmacol. 2019, 363, 79–87. [Google Scholar] [CrossRef]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef]
- Xin, Z.; Zhai, Z.; Long, H.; Zhang, F.; Ni, X.; Deng, J.; Yi, L.; Deng, B. Metabolic Profiling by UPLC–Orbitrap–MS/MS of Liver from C57BL/6 Mice with DSS-Induced Inflammatory Bowel Disease. Mediat. Inflamm. 2020, 2020, 6020247. [Google Scholar] [CrossRef]
- Troise, A.D.; Ferracane, R.; Palermo, M.; Fogliano, V. Targeted metabolite profile of food bioactive compounds by Orbitrap high resolution mass spectrometry: The “FancyTiles” approach. Food Res. Int. 2014, 63, 139–146. [Google Scholar] [CrossRef]
- Pham, H.T.; Arnhard, K.; Asad, Y.J.; Deng, L.; Felder, T.K.; St John-Williams, L.; Kaever, V.; Leadley, M.; Mitro, N.; Muccio, S.; et al. Inter-Laboratory Robustness of Next-Generation Bile Acid Study in Mice and Humans: International Ring Trial Involving 12 Laboratories. J. Appl. Lab. Med. 2016, 1, 129–142. [Google Scholar] [CrossRef] [Green Version]
- García-Cañaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J. Lipid Res. 2012, 53, 2231–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amplatz, B.; Zöhrer, E.; Haas, C.; Schäffer, M.; Stojakovic, T.; Jahnel, J.; Fauler, G. Bile acid preparation and comprehensive analysis by high performance liquid chromatography-high-resolution mass spectrometry. Clin. Chim. Acta 2017, 464, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Reiter, S.; Dunkel, A.; Metwaly, A.; Panes, J.; Salas, A.; Haller, D.; Hofmann, T. Development of a Highly Sensitive Ultra-High-Performance Liquid Chromatography Coupled to Electrospray Ionization Tandem Mass Spectrometry Quantitation Method for Fecal Bile Acids and Application on Crohn’s Disease Studies. J. Agric. Food Chem. 2021, 69, 5238–5251. [Google Scholar] [CrossRef]
- Tagliacozzi, D.; Mozzi, A.F.; Casetta, B.; Bertucci, P.; Bernardini, S.; Di Ilio, C.; Urbani, A.; Federici, G. Quantitative analysis of bile acids in human plasma by liquid chromatography-electrospray tandem mass spectrometry: A simple and rapid one-step method. Clin. Chem. Lab. Med. 2003, 41, 1633–1641. [Google Scholar] [CrossRef]
- Zhou, J.; Yin, Y. Strategies for large-scale targeted metabolomics quantification by liquid chromatography-mass spectrometry. Analyst 2016, 141, 6362–6373. [Google Scholar] [CrossRef]
- Yin, P.; Lehmann, R.; Xu, G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal. Bioanal. Chem. 2015, 407, 4879–4892. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, M.; Angeli, P.; Claria, J.; Moreau, R.; Gines, P.; Jalan, R.; Caraceni, P.; Fernandez, J.; Gerbes, A.L.; O’Brien, A.J.; et al. Albumin in decompensated cirrhosis: New concepts and perspectives. Gut 2020, 69, 1127–1138. [Google Scholar] [CrossRef] [Green Version]
- Ceryak, S.; Bouscarel, B.; Fromm, H. Comparative binding of bile acids to serum lipoproteins and albumin. J. Lipid Res. 1993, 34, 1661–1674. [Google Scholar] [CrossRef]
- Steiner, C.; von Eckardstein, A.; Rentsch, K.M. Quantification of the 15 major human bile acids and their precursor 7α-hydroxy-4-cholesten-3-one in serum by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2010, 878, 2870–2880. [Google Scholar] [CrossRef] [Green Version]
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129. [Google Scholar] [CrossRef]
- Ay, Ü.; Leníček, M.; Classen, A.; Olde Damink, S.W.M.; Bolm, C.; Schaap, F.G. New Kids on the Block: Bile Salt Conjugates of Microbial Origin. Metabolites 2022, 12, 176. [Google Scholar] [CrossRef]
- Lucas, L.N.; Barrett, K.; Kerby, R.L.; Zhang, Q.; Cattaneo, L.E.; Stevenson, D.; Rey, F.E.; Amador-Noguez, D.; Manichanh, C. Dominant Bacterial Phyla from the Human Gut Show Widespread Ability To Transform and Conjugate Bile Acids. mSystems 2021, 6, e00805–e00821. [Google Scholar] [CrossRef]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Frank, O.; Kreissl, J.K.; Daschner, A.; Hofmann, T. Accurate determination of reference materials and natural isolates by means of quantitative (1)h NMR spectroscopy. J. Agric. Food Chem. 2014, 62, 2506–2515. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bile Acid | High Concentration (nM) | Low Concentration (nM) | Factor (High Conc./Low Conc.) | Bile Acid Class |
|---|---|---|---|---|
| CA | 30,000 | 3000 | 10 | primary |
| CDCA | 15,000 | 1500 | 10 | primary |
| GCA | 40,000 | 400 | 100 | conjugated primary |
| TCA | 25,000 | 250 | 100 | conjugated primary |
| DCA | 5000 | 500 | 10 | secondary |
| LCA | 1500 | 150 | 10 | secondary |
| α-MCA | 1500 | 150 | 10 | murine primary |
| β-MCA | 2500 | 250 | 10 | murine primary |
| ω-MCA | 1500 | 150 | 10 | murine secondary |
| Center | Matrix | Conc. Level | # BAs in Method | # BAs in RSD Limits | Bile Acid RSD in Limits | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CA | CDCA | GCA | TCA | DCA | LCA | α-MCA | β-MCA | ω-MCA | |||||
| Center 1 | MeOH:H2O | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Low | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Human serum | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Low | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Murine serum | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Low | 9 | 5 | Yes | Yes | Yes | - | Yes | Yes | - | - | - | ||
| Center 2 | MeOH:H2O | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Low | 9 | 1 | - | - | - | - | - | Yes | Yes | - | - | ||
| Human serum | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Low | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Murine serum | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Low | 9 | 6 | Yes | Yes | Yes | - | Yes | Yes | Yes | - | - | ||
| Center 3 | MeOH:H2O | High | 7 | 7 | Yes | Yes | Yes | Yes | Yes | NA | Yes | Yes | NA |
| Low | 7 | 6 | Yes | Yes | Yes | Yes | Yes | NA | - | Yes | NA | ||
| Human serum | High | 7 | 7 | Yes | Yes | Yes | Yes | Yes | NA | Yes | Yes | NA | |
| Low | 7 | 5 | Yes | Yes | Yes | Yes | - | NA | - | Yes | NA | ||
| Murine serum | High | 7 | 7 | Yes | Yes | Yes | Yes | Yes | NA | Yes | Yes | NA | |
| Low | 7 | 5 | Yes | Yes | Yes | - | Yes | NA | Yes | - | NA | ||
| Center 4 | MeOH:H2O | High | 7 | 4 | Yes | - | Yes | Yes | - | Yes | NA | - | NA |
| Low | 7 | 7 | Yes | Yes | Yes | Yes | Yes | Yes | NA | Yes | NA | ||
| Human serum | High | 7 | 7 | Yes | Yes | Yes | Yes | Yes | Yes | NA | Yes | NA | |
| Low | 7 | 4 | Yes | - | Yes | Yes | - | Yes | NA | - | NA | ||
| Murine serum | High | 7 | 5 | Yes | - | Yes | Yes | - | Yes | NA | Yes | NA | |
| Low | 7 | 3 | Yes | - | Yes | - | - | Yes | NA | - | NA | ||
| Center 5 | MeOH:H2O | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Low | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Human serum | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Low | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Murine serum | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Low | 9 | 8 | Yes | Yes | Yes | - | Yes | Yes | Yes | Yes | Yes | ||
| Center 6 | MeOH:H2O | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Low | 9 | 8 | Yes | Yes | Yes | Yes | Yes | - | Yes | Yes | Yes | ||
| Human serum | High | 9 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Low | 9 | 7 | Yes | Yes | Yes | Yes | - | - | Yes | Yes | Yes | ||
| Murine serum | High | 9 | 8 | Yes | Yes | Yes | Yes | - | Yes | Yes | Yes | Yes | |
| Low | 9 | 7 | Yes | Yes | Yes | - | Yes | - | Yes | Yes | Yes | ||
| Center | Matrix | Conc. Level | # BAs in Method | # BAs in Rel. Recovery Limits | Bile Acid in Relative Recovery Limits | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CA | CDCA | GCA | TCA | DCA | LCA | α-MCA | β-MCA | ω-MCA | |||||
| Center 1 | MeOH:H2O | High | 9 | 2 | - | - | Yes | Yes | - | - | - | - | - |
| Low | 9 | 2 | - | - | Yes | Yes | - | - | - | - | - | ||
| Human serum | High | 9 | 0 | - | - | - | - | - | - | - | - | - | |
| Low | 9 | 1 | - | - | - | Yes | - | - | - | - | - | ||
| Murine serum | High | 9 | 1 | - | - | - | Yes | - | - | - | - | - | |
| Low | 9 | 2 | - | - | Yes | Yes | - | - | - | - | - | ||
| Center 2 | MeOH:H2O | High | 9 | 5 | Yes | - | Yes | Yes | Yes | - | - | Yes | - |
| Low | 9 | 2 | - | Yes | Yes | - | - | - | - | - | - | ||
| Human serum | High | 9 | 1 | - | Yes | - | - | - | - | - | - | - | |
| Low | 9 | 5 | Yes | - | Yes | Yes | Yes | - | - | Yes | - | ||
| Murine serum | High | 9 | 4 | Yes | - | Yes | Yes | Yes | - | - | - | - | |
| Low | 9 | 3 | Yes | - | - | Yes | Yes | - | - | - | - | ||
| Center 3 | MeOH:H2O | High | 7 | 5 | Yes | - | Yes | Yes | Yes | NA | - | Yes | NA |
| Low | 7 | 5 | Yes | - | Yes | Yes | Yes | NA | Yes | - | NA | ||
| Human serum | High | 7 | 5 | Yes | - | Yes | Yes | Yes | NA | - | Yes | NA | |
| Low | 7 | 5 | Yes | - | Yes | Yes | Yes | NA | Yes | - | NA | ||
| Murine serum | High | 7 | 6 | Yes | - | Yes | Yes | Yes | NA | Yes | Yes | NA | |
| Low | 7 | 6 | Yes | - | Yes | Yes | Yes | NA | Yes | Yes | NA | ||
| Center 4 | MeOH:H2O | High | 7 | 2 | - | - | Yes | Yes | - | - | NA | - | NA |
| Low | 7 | 3 | Yes | - | Yes | Yes | - | - | NA | - | NA | ||
| Human serum | High | 7 | 4 | Yes | - | Yes | Yes | - | Yes | NA | - | NA | |
| Low | 7 | 3 | Yes | - | Yes | Yes | - | - | NA | - | NA | ||
| Murine serum | High | 7 | 3 | Yes | - | Yes | Yes | - | - | NA | - | NA | |
| Low | 7 | 2 | Yes | - | Yes | - | - | - | NA | - | NA | ||
| Center 5 | MeOH:H2O | High | 9 | 4 | - | Yes | - | Yes | - | - | Yes | Yes | - |
| Low | 9 | 6 | Yes | - | Yes | Yes | - | - | Yes | Yes | Yes | ||
| Human serum | High | 9 | 5 | - | Yes | - | Yes | Yes | - | Yes | Yes | - | |
| Low | 9 | 5 | Yes | - | Yes | Yes | - | - | Yes | Yes | - | ||
| Murine serum | High | 9 | 3 | - | - | - | Yes | Yes | - | - | Yes | - | |
| Low | 9 | 4 | Yes | - | Yes | - | - | - | Yes | Yes | - | ||
| Center 6 | MeOH:H2O | High | 9 | 6 | Yes | Yes | Yes | Yes | Yes | - | Yes | - | - |
| Low | 9 | 7 | Yes | Yes | Yes | Yes | Yes | - | Yes | - | Yes | ||
| Human serum | High | 9 | 7 | Yes | Yes | Yes | Yes | Yes | - | Yes | - | Yes | |
| Low | 9 | 7 | Yes | Yes | Yes | Yes | Yes | - | Yes | - | Yes | ||
| Murine serum | High | 9 | 6 | Yes | Yes | Yes | Yes | Yes | - | Yes | - | - | |
| Low | 9 | 6 | Yes | Yes | Yes | Yes | Yes | - | Yes | - | - | ||
| Center | Column Type | Mass Spectrometer | Flow Rate (mL/min) | Analysis Time (min) | Total Turn-Around Time (min) | Sample Volume Required for BA Extraction (µL) | Sample Volume Injected (µL) | Software | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Center 1 | ACQUITY UPLC BEH Shield RP18 column (particle size: 1.7 µm, dimensions: 100 × 2.1 mm) | Xevo TQ-S (Waters) | 0.7 | 13 | 19 | 25 | 5 | Target Lynx v4.1 (Waters) | García-Cañaveras et al. [23] |
| Center 2 | Kinetex C18 reversed phase column (particle size: 2.6 μm, dimensions: 100 × 3.0 mm) | Q Exactive hybrid quadrupole-Orbitrap (Thermo Fisher Scientific) | 0.5 | 23 | 25 | 10 | 10 | Xcalibur 2.3 (Thermo Fisher Scientific) | Amplatz et al. [24] |
| Center 3 | Kinetex C18 reversed phase column (particle size: 1.7 μm, dimensions: 100 × 2.1 mm) and a SecurityGuard ULTRA Cartridges UHPLC C18 2.1 mm column | QTrap 5500 (Sciex) | 0.4 | 23 | 25 | 30 | 1 | Multiquant 3.0.3 (Sciex) | Reiter et al. [25] |
| Center 4 | ACQUITY UPLC BEH Shield RP18 column (particle size: 1.7 µm, dimensions: 50 × 2.1 mm) | Xevo TQ-S (Waters) | 0.4 | 18.6 | 20 | 50 | 5 | Target Lynx v4.1 (Waters) | Tagliacozzi et al. [26] |
| Center 5 | Kinetex C18 reversed phase column (particle size: 1.7 μm, dimensions: 100 × 2.1 mm) | QTrap 5500 (Sciex) | 0.4 | 18.6 | 20 | 50 | 5 | Multiquant 3.0.3 (Sciex) | García-Cañaveras et al. [23] |
| Center 6 | ACQUITY UPLC System (UHPLC Column from Biocrates P.-No 91220052120868) | QTrap 5500 (Sciex) | 0.5 | 3.5 | 5 | 10 | 5 | Analyst 1.7.1 (Sciex) | Phamet al. [22] |
| Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) | |
|---|---|---|---|
| Analysis | 0 | 0.7 | 85 |
| 9 | 0.7 | 60 | |
| 10 | 0.7 | 20 | |
| 13 | 0.7 | 0 | |
| Column Regeneration | 16 | 0.7 | 0 |
| 16.1 | 0.7 | 85 | |
| 19 | 0.7 | 85 |
| Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) | |
|---|---|---|---|
| Analysis | 0 | 0.4 | 75 |
| 2 | 0.4 | 75 | |
| 3.5 | 0.4 | 73 | |
| 5.5 | 0.4 | 65 | |
| 10 | 0.4 | 65 | |
| 11 | 0.4 | 57 | |
| 12 | 0.4 | 57 | |
| 14 | 0.4 | 42 | |
| 17 | 0.4 | 42 | |
| 17.5 | 0.4 | 35 | |
| 18 | 0.4 | 20 | |
| 19 | 0.4 | 0 | |
| 20 | 0.4 | 0 | |
| 23 | 0.4 | 75 | |
| Column Regeneration | 25 | 0.4 | 75 |
| Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) | |
|---|---|---|---|
| Analysis | 0 | 0.4 | 75 |
| 2 | 0.4 | 75 | |
| 3.5 | 0.4 | 73 | |
| 5.5 | 0.4 | 65 | |
| 10 | 0.4 | 65 | |
| 11 | 0.4 | 57 | |
| 12 | 0.4 | 57 | |
| 14 | 0.4 | 42 | |
| 17 | 0.4 | 42 | |
| 17.5 | 0.4 | 35 | |
| 18 | 0.4 | 20 | |
| 19 | 0.4 | 0 | |
| 20 | 0.4 | 0 | |
| 23 | 0.4 | 75 | |
| Column Regeneration | 25 | 0.4 | 75 |
| Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) | |
|---|---|---|---|
| Analysis | 0 | 0.4 | 80 |
| 1 | 0.4 | 81 | |
| 5 | 0.4 | 65 | |
| 14.5 | 0.4 | 5 | |
| 14.6 | 0.4 | 0 | |
| 18.5 | 0.4 | 0 | |
| 18.6 | 0.4 | 80 | |
| Column Regeneration | 20 | 0.4 | 80 |
| Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) | |
|---|---|---|---|
| Analysis | 0 | 0.4 | 80 |
| 1 | 0.4 | 80 | |
| 5 | 0.4 | 65 | |
| 14.5 | 0.4 | 5 | |
| 14.6 | 0.4 | 0 | |
| 18.5 | 0.4 | 0 | |
| 18.6 | 0.4 | 80 | |
| Column Regeneration | 20 | 0.4 | 80 |
| Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) | |
|---|---|---|---|
| Analysis | 0 | 0.5 | 65 |
| 0.25 | 0.5 | 65 | |
| 0.35 | 0.5 | 60 | |
| 1.9 | 0.5 | 55 | |
| 2.1 | 0.8 | 45 | |
| 3.3 | 1.0 | 35 | |
| 3.5 | 1.0 | 0 | |
| Column Regeneration | 4.0 | 1.0 | 0 |
| 4.1 | 0.9 | 65 | |
| 5 | 0.5 | 65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haange, S.-B.; Till, A.; Bergh, P.-O.; Fauler, G.; Gigl, M.; Löfgren-Sandblom, A.; Schaap, F.G.; Clavel, T.; Trautwein, C.; Fenske, W.; et al. Ring Trial on Quantitative Assessment of Bile Acids Reveals a Method- and Analyte-Specific Accuracy and Reproducibility. Metabolites 2022, 12, 583. https://doi.org/10.3390/metabo12070583
Haange S-B, Till A, Bergh P-O, Fauler G, Gigl M, Löfgren-Sandblom A, Schaap FG, Clavel T, Trautwein C, Fenske W, et al. Ring Trial on Quantitative Assessment of Bile Acids Reveals a Method- and Analyte-Specific Accuracy and Reproducibility. Metabolites. 2022; 12(7):583. https://doi.org/10.3390/metabo12070583
Chicago/Turabian StyleHaange, Sven-Bastiaan, Andreas Till, Per-Olof Bergh, Günter Fauler, Michael Gigl, Anita Löfgren-Sandblom, Frank G. Schaap, Thomas Clavel, Christian Trautwein, Wiebke Fenske, and et al. 2022. "Ring Trial on Quantitative Assessment of Bile Acids Reveals a Method- and Analyte-Specific Accuracy and Reproducibility" Metabolites 12, no. 7: 583. https://doi.org/10.3390/metabo12070583
APA StyleHaange, S.-B., Till, A., Bergh, P.-O., Fauler, G., Gigl, M., Löfgren-Sandblom, A., Schaap, F. G., Clavel, T., Trautwein, C., Fenske, W., Kleigrewe, K., Marschall, H.-U., Olde Damink, S. W. M., Moustafa, T., von Bergen, M., & Rolle-Kampczyk, U. (2022). Ring Trial on Quantitative Assessment of Bile Acids Reveals a Method- and Analyte-Specific Accuracy and Reproducibility. Metabolites, 12(7), 583. https://doi.org/10.3390/metabo12070583