
3,5-T2-an Endogenous Thyroid Hormone Metabolite as Promising Lead Substance in Anti-Steatotic Drug Development?


Abstract

1. Introduction
1.1. Initial Decades of Promising 3,5-T2 Research
1.2. Discovery of 3,5-T2 in the Serum and Subsequent Research
2. Direct Mitochondrial Actions of 3,5-T2
3. Mechanisms Postulated for 3,5-T2 Actions
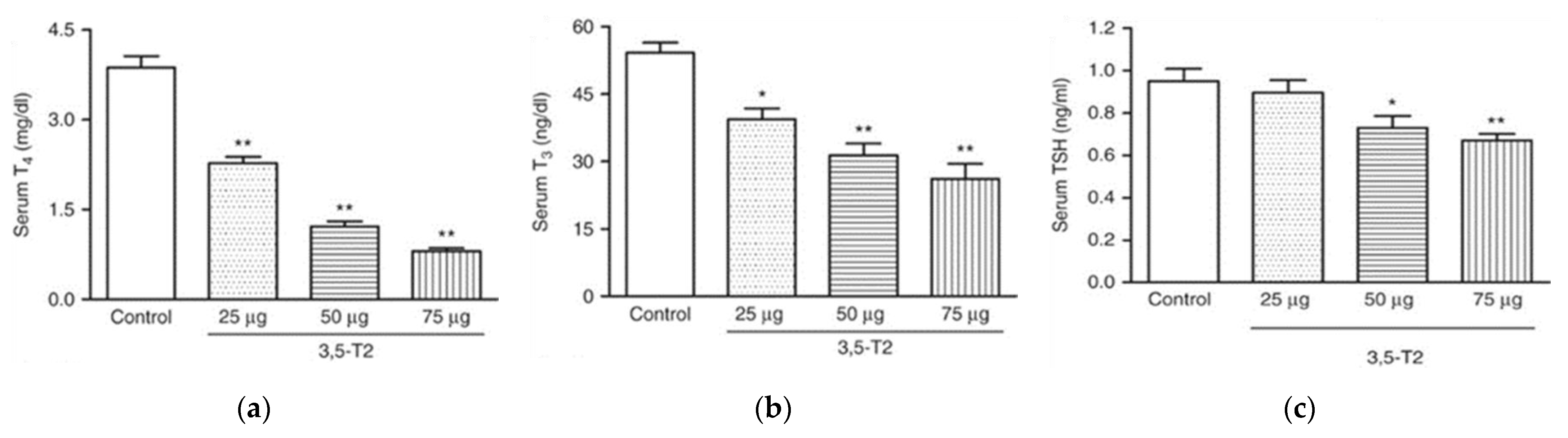
4. Effects of 3,5-T2 in Tissues of the HPT Axis
4.1. 3,5-T2 and the Pituitary
4.2. 3,5-T2 Effects May Depend on the Thyroid State
5. Hepatic Effects of 3,5-T2
5.1. Canonical 3,5-T2 Actions in the Liver of Rodents
5.2. Noncanonical 3,5-T2 Actions in the Liver of Rodents
5.3. Lipid Metabolism
5.4. Thyromimetic Effects of 3,5-T2 on Glucose Metabolism
5.5. Hepatic Accumulation, Selective Uptake and Inefficient Hepatic Elimination of 3,5-T2
5.6. Implications of High 3,5-T2 in Context of Low T3, T4, and Suppressed TSH for Hepatic Lipid Metabolism
5.7. Hepatic 3,5-T2 Effects Differ between Lean and HFD Obese Mice
6. 3,5-T2 and the Heart
7. Dose Issues of 3,5-T2
8. Limitations, Deficits, Problem Areas and Challenges in 3,5-T2 Research
9. Clinical Potential
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jonas, W.; Lietzow, J.; Wohlgemuth, F.; Hoefig, C.S.; Wiedmer, P.; Schweizer, U.; Köhrle, J.; Schürmann, A. 3,5-Diiodo-l-thyronine (3,5-T2) Exerts thyromimetic effects on hypothalamus-pituitary-thyroid axis, body composition, and energy metabolism in male diet- induced obese mice. Endocrinology 2015, 156, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J.; Lehmphul, I.; Pietzner, M.; Renko, K.; Rijntjes, E.; Richards, K.; Anselmo, J.; Danielsen, M.; Jonklaas, J. 3,5-T2—A Janus-Faced Thyroid Hormone Metabolite Exerts Both Canonical T3-Mimetic Endocrine and Intracrine Hepatic Action. Front. Endocrinol. 2020, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Magnus-Levy, A. Ueber den respiratorischen Gaswechsel unter Einfluss der Thyroidea sowie unter verschiedenen pathologischen Zuständen. Berl. Klin Wochschr 1895, 32, 650–652. [Google Scholar]
- Magnus-Levy, A. Gaswechsel und Fettumsatz bei Myxoedem und Schilddrüsenfütterung. Verhandl. D XIV Kongr. F Inn. Med. S 1896, 140, 137–142. [Google Scholar]
- Boothby, W.M.; Sandiford, I. Basal metabolism. Physiol. Rev. 1924, 4, 69–162. [Google Scholar] [CrossRef]
- Gaddum, J.H. Quantitative observations on thyroxine and allied substances: I. The use of tadpoles. J. Physiol. 1927, 64, 246–254. [Google Scholar] [CrossRef]
- Gaddum, J.H. Quantitative observations on thyroxine and allied substances. J. Physiol. 1930, 68, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.; Harington, C.; Lyon, D.M. The use of 3: 5-Diiodothyronine in the treatment of myxoedema. Lancet 1933, 222, 1081–1084. [Google Scholar] [CrossRef]
- Byrom, F. The nature of myxoedema. Clin. Sci. 1934, 1, 1933–1934. [Google Scholar]
- Karmisholt, J.; Andersen, S.; Laurberg, P. Weight loss after therapy of hypothyroidism is mainly caused by excretion of excess body water associated with myxoedema. J. Clin. Endocrinol. Metab. 2011, 96, E99–E103. [Google Scholar] [CrossRef]
- Gross, J.; Pitt-Rivers, R. 3:5:3′-triiodothyronine. 1. Isolation from thyroid gland and synthesis. Biochem. J. 1953, 53, 645. [Google Scholar] [CrossRef] [PubMed]
- Harington, C.R.; Barger, G. Chemistry of Thyroxine: Constitution and Synthesis of Thyroxine. Biochem. J. 1927, 21, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Gemill, C.L. Effects of thyroxine, 3,5-diiodothyronine and thyronine on ascorbic acid oxidation. Am. J. Physiol. 1951, 167, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Stasilli, N.R.; Kroc, R.L.; Meltzer, R.I. Antigoitrogenic and calorigenic activities of thyroxine analogues in rats. Endocrinology 1959, 64, 62–82. [Google Scholar] [CrossRef] [PubMed]
- Horst, C.; Rokos, H.; Seitz, H.J. Rapid stimulation of hepatic oxygen consumption by 3,5-di-iodo-l-thyronine. Biochem. J. 1989, 261, 945–950. [Google Scholar] [CrossRef]
- Müller, A.; Sies, H. Role of alcohol dehydrogenase activity and the acetaldehyde in ethanol-induced ethane and pentane production by isolated perfused rat liver. Biochem. J. 1982, 206, 153–156. [Google Scholar] [CrossRef]
- Köhrle, J.; Müller, M.J.; Ködding, R.; Seitz, H.J.; Hesch, R. pH-dependency of iodothyronine metabolism in isolated perfused rat liver. Biochem. J. 1982, 202, 667–675. [Google Scholar] [CrossRef]
- Burger, A.G.; O’Connell, M.; Scheidegger, K.; Woo, R.D.; Danforth, E., Jr. Monodeiodination of Triiodothyronine and Reverse Triiodothyronine During Low and High Calorie Diets. J. Clin. Endocrinol. Metab. 1987, 65, 829–835. [Google Scholar] [CrossRef]
- Mendoza, A.; Navarrete-Ramírez, P.; Hernández-Puga, G.; Villalobos, P.; Holzer, G.; Renaud, J.P.; Laudet, V.; Orozco, A. 3,5-T2 Is an Alternative Ligand for the Thyroid Hormone Receptor β1. Endocrinology 2013, 154, 2948–2958. [Google Scholar] [CrossRef]
- Olvera, A.; Martyniuk, C.J.; Buisine, N.; Jiménez-Jacinto, V.; Sanchez-Flores, A.; Sachs, L.M.; Orozco, A. Differential transcriptome regulation by 3,5-T2 and 3′,3,5-T3 in brain and liver uncovers novel roles for thyroid hormones in tilapia. Sci. Rep. 2017, 7, 15043. [Google Scholar] [CrossRef]
- Hernandez-Linares, Y.; Olvera, A.; Villalobos, P.; Lozano-Flores, C.; Varela-Echavarria, A.; Luna, M.; Orozco, A. 3,5-T2 and 3,3′,5-T3 Regulate Cerebellar Thyroid Hormone Signalling and Myelin Molecular Dynamics in Tilapia. Sci. Rep. 2019, 9, 7359. [Google Scholar] [CrossRef] [PubMed]
- Lardy, H.A.; Feldott, G. Metabolic effects of thyroxine in vitro. Ann. N. Y. Acad. Sci. 1951, 54, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Klemperer, H.G. The uncoupling of oxidative phosphorylation in rat-liver mitochondria by thyroxine, triiodothyronine and related substances. Biochem. J. 1955, 60, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zuna, K.; Jovanovic, O.; Khailova, L.S.; Skulj, S.; Brkljaca, Z.; Kreiter, J.; Kotova, E.A.; Vazdar, M.; Antonenko, Y.N.; Pohl, E.E. Mitochondrial Uncoupling Proteins (UCP1-UCP3) and Adenine Nucleotide Translocase (ANT1) Enhance the Protonophoric Action of 2,4-Dinitrophenol in Mitochondria and Planar Bilayer Membranes. Biomolecules 2021, 11, 1178. [Google Scholar] [CrossRef] [PubMed]
- Martius, C.; Hess, B. The Mode of Action of Thyroxin. Arch. Biochem. Biophys. 1951, 33, 486–487. [Google Scholar] [CrossRef]
- Roodyn, D.B.; Freeman, K.B.; Tata, J.R. The Stimulation by Treatment in Vivo with Tri-Iodothyronine of Amino Acid Incorporation into Protein by Isolated Rat-Liver Mitochondria. Biochem. J. 1965, 94, 628–641. [Google Scholar] [CrossRef]
- Lin, C.S.; Klingenberg, M. Isolation of the uncoupling protein from brown adipose tissue mitochondria. FEBS Lett. 1980, 113, 299–303. [Google Scholar] [CrossRef]
- Bianco, A.C.; Silva, J.E. Intracellular conversion of thyroxine to triiodothyronine is required for the optimal thermogenic funciton of brown adipose tissue. J. Clin. Investig. 1987, 79, 295–300. [Google Scholar] [CrossRef]
- Silva, J.E.; Rabelo, R. Regulation of the uncoupling protein gene expression. Eur. J. Endocrinol. 1997, 136, 251–264. [Google Scholar] [CrossRef]
- Moreno, M.; Lanni, A.; Lombardi, A.; Goglia, F. How the thyroid controls metabolism in the rat: Different roles for triiodothyronine and diiodothyronines. J. Physiol. 1997, 505, 529–538. [Google Scholar] [CrossRef]
- Lombardi, A.; Lanni, A.; Moreno, M.; Brand, D.M.; Goglia, F. Effect of 3,5-di-iodo-L-thyronine on the mitochondrial energy-transduction apparatus. Biochem. J. 1998, 330, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Lionetti, L.; Moreno, M.; Lombardi, A.; De Lange, P.; Antonelli, A.; Lanni, A.; Cavaliere, G.; Barletta, A.; Goglia, F. 3,5-diiodo-l-thyronine, by modulating mitochondrial functions, reverses hepatic fat accumulation in rats fed a high-fat diet. J. Hepatol. 2009, 51, 363–370. [Google Scholar] [CrossRef] [PubMed]
- de Lange, P.; Cioffi, F.; Senese, R.; Moreno, M.; Lombardi, A.; Silvestri, E.; De Matteis, R.; Lionetti, L.; Mollica, M.P.; Goglia, F.; et al. Nonthyrotoxic Prevention of Diet-Induced Insulin Resistance by 3,5-Diiodo-L-Thyronine in Rats. Diabetes 2011, 60, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, E.; Cioffi, F.; Glinni, D.; Ceccarelli, M.; Lombardi, A.; De Lange, P.; Chambery, A.; Severino, V.; Lanni, A.; Goglia, F.; et al. Pathways affected by 3,5-diiodo-l-thyronine in liver of high fat-fed rats: Evidence from two-dimensional electrophoresis, blue-native PAGE, and mass spectrometry. Mol. BioSyst. 2010, 6, 2256–2271. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, E.; Coppola, M.; Cioffi, F.; Goglia, F. Proteomic approaches for the study of tissue specific effects of 3,5,3′-triiodo-L-thyronine and 3,5-diiodo-L-thyronine in conditions of altered energy metabolism. Front. Physiol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Cavallo, A.; Priore, P.; Gnoni, G.V.; Papa, S.; Zanotti, F.; Gnoni, A. 3,5-Diiodo-L-Thyronine Administration To Hypothyroid Rats Rapidly Enhances Fatty Acid Oxidation Rate and Bioenergetic Parameters in Liver Cells. PLoS ONE 2013, 8, e52328. [Google Scholar] [CrossRef]
- Rochira, A.; Damiano, F.; Marsigliante, S.; Gnoni, G.V.; Siculella, L. 3,5-Diiodo-l-thyronine induces SREBP-1 proteolytic cleavage block and apoptosis in human hepatoma (Hepg2) cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 1679–1689. [Google Scholar] [CrossRef]
- Lietzow, J.; Golchert, J.; Homuth, G.; Völker, U.; Jonas, W.; Köhrle, J. 3,5-T2 alters murine genes relevant for xenobiotic, steroid, and thyroid hormone metabolism. J. Mol. Endocrinol. 2016, 56, 311–323. [Google Scholar] [CrossRef]
- Padron, A.S.; Neto, R.A.L.; Pantaleão, T.U.; De Souza Dos Santos, M.C.; Araujo, R.L.; De Andrade, B.M.; Da Silva Leandro, M.; De Castro, J.P.S.W.; Ferreira, A.C.F.; De Carvalho, D.P. Administration of 3,5-diiodothyronine (3,5-T2) causes central hypothyroidism and stimulates thyroid-sensitive tissues. J. Endocrinol. 2014, 221, 415–427. [Google Scholar] [CrossRef]
- Ball, S.G.; Sokolov, J.; Chin, W.W. 3,5-Diiodo-L-thyronine (T2) has selective thyromimetic effects in vivo and in vitro. J. Mol. Endocrinol. 1997, 19, 137–147. [Google Scholar] [CrossRef]
- da Silva Teixeira, S.; Filgueira, C.; Sieglaff, D.H.; Benod, C.; Villagomez, R.; Minze, L.J.; Zhang, A.; Webb, P.; Nunes, M.T. 3,5-diiodothyronine (3,5-T2) reduces blood glucose independently of insulin sensitization in obese mice. Acta Physiol. 2017, 220, 238–250. [Google Scholar] [CrossRef] [PubMed]
- del Viscovo, A.; Secondo, A.; Esposito, A.; Goglia, F.; Moreno, M.; Canzoniero, L.M.T. Intracellular and plasma membrane-initiated pathways involved in the [Ca2+] i elevations induced by iodothyronines (T3 and T2) in pituitary GH 3 cells. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1419–E1430. [Google Scholar] [CrossRef] [PubMed]
- Horst, C.; Harneit, A.; Seitz, H.J.; Rokos, H. 3,5-Di-iodo-L-thyronine suppresses TSH in rats in vivo and in rat pituitary fragments in vitro. J. Endocrinol. 1995, 145, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Baur, A.; Bauer, K.; Jarry, H.; Köhrle, J. 3,5-Diiodo-L-thyronine stimulates type 1 5′deiodinase activity in rat anterior pituitaries in vivo and in reaggregate cultures and GH3 cells in vitro. Endocrinology 1997, 138, 3242–3248. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Huang, L.S.; Huggins, L.A.; Yu, S.; Nagareddy, P.R.; Scanlan, T.S.; Ehrenkranz, J.R. Thyroid hormone reduces cholesterol via a non-LDL receptor-mediated pathway. Endocrinology 2012, 153, 5143–5149. [Google Scholar] [CrossRef]
- Lietzow, J.; Golchert, J.; Pietzner, M.; Völker, U.; Poutanen, M.; Ohlsson, C.; Homuth, G.; Köhrle, J. Comparative Analysis of the Effects of Long-Term 3,5-diiodothyronine Treatment on the Murine Hepatic Proteome and Transcriptome Under Conditions of Normal Diet and High-Fat Diet. Thyroid 2021, 31, 1135–1146. [Google Scholar] [CrossRef]
- Piehl, S.; Heberer, T.; Balizs, G.; Scanlan, T.S.; Köhrle, J. Development of a validated liquid chromatography/tandem mass spectrometry method for the distinction of thyronine and thyronamine constitutional isomers and for the identification of new deiodinase substrates. Rapid Commun. Mass Spectrom. 2008, 22, 3286–3296. [Google Scholar] [CrossRef]
- Renko, K.; Schäche, S.; Hoefig, C.S.; Welsink, T.; Schwiebert, C.; Braun, D.; Becker, N.P.; Köhrle, J.; Schomburg, L. An Improved Nonradioactive Screening Method Identifies Genistein and Xanthohumol as Potent Inhibitors of Iodothyronine Deiodinases. Thyroid 2015, 25, 962–968. [Google Scholar] [CrossRef]
- Köhrle, J. The Colorful Diversity of Thyroid Hormone Metabolites. Eur. Thyroid J. 2019, 8, 115–129. [Google Scholar] [CrossRef]
- Xu, C.; Li, C.Y.-T.; Kong, A.-N.T. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharmacal Res. 2005, 28, 249–268. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. 2010, 154, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M.; Feng, X.; Flamant, F.; Chen, Y.; Walker, R.L.; Weiss, R.E.; Chassande, O.; Samarut, J.; Refetoff, S.; Meltzer, P.S. Effects of ligand and thyroid hormone receptor isoforms on hepatic gene expression profiles of thyroid hormone receptor knockout mice. EMBO Rep. 2003, 4, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kobayashi, M.; Kitagishi, Y. Roles for PI3K/AKT/PTEN Pathway in Cell Signaling of Nonalcoholic Fatty Liver Disease. ISRN Endocrinol. 2013, 2013, 472432. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Kambe, F.; Moeller, L.C.; Refetoff, S.; Seo, H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol. Endocrinol. 2005, 19, 102–112. [Google Scholar] [CrossRef]
- Giammanco, M.; Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Genomic and non-genomic mechanisms of action of thyroid hormones and their catabolite 3,5-diiodo-l-thyronine in Mammals. Int. J. Mol. Sci. 2020, 21, 4140. [Google Scholar] [CrossRef]
- Hönes, G.S.; Rakov, H.; Logan, J.; Liao, X.H.; Werbenko, E.; Pollard, A.S.; Præstholm, S.M.; Siersbæk, M.S.; Rijntjes, E.; Gassen, J.; et al. Noncanonical thyroid hormone signaling mediates cardiometabolic effects in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E11323–E11332. [Google Scholar] [CrossRef]
- Mason, R.; Hunt, H.; HurxTHAL, L. Blood cholesterol values in hyperthyroidism and hypothyroidism—Their significance. N. Engl. J. Med. 1930, 203, 1273–1278. [Google Scholar] [CrossRef]
- Van Der Valk, F.; Hassing, C.; Visser, M.; Thakkar, P.; Mohanan, A.; Pathak, K.; Dutt, C.; Chauthaiwale, V.; Ackermans, M.; Nederveen, A.; et al. The effect of a diiodothyronine mimetic on insulin sensitivity in male cardiometabolic patients: A double-blind randomized controlled trial. PLoS ONE 2014, 9, e86890. [Google Scholar] [CrossRef]
- Vatner, D.F.; Snikeris, J.; Popov, V.; Perry, R.J.; Rahimi, Y.; Samuel, V.T. 3,5 Diiodo-L-Thyronine (T2) does not prevent hepatic steatosis or insulin resistance in fat-fed sprague dawley rats. PLoS ONE 2015, 10, e0140837. [Google Scholar] [CrossRef]
- Groeneweg, S.; Van Geest, F.S.; Peeters, R.P.; Heuer, H.; Visser, W.E. Thyroid Hormone Transporters. Endocr. Rev. 2020, 41, 146–201. [Google Scholar] [CrossRef]
- Krause, G.; Hinz, K.M. Thyroid hormone transport across L-type amino acid transporters: What can molecular modelling tell us? Mol. Cell. Endocrinol. 2017, 458, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Angelin, B.; Kristensen, J.D.; Eriksson, M.; Carlsson, B.; Klein, I.; Olsson, A.G.; Ridgway, E.R.; Ladenson, P.W. Reductions in serum levels of LDL cholesterol, apolipoprotein B, triglycerides and lipoprotein(a) in hypercholesterolaemic patients treated with the liver-selective thyroid hormone receptor agonist eprotirome. J. Intern. Med. 2015, 277, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Babbitt, L.L.; Liebentritt, D.K. Human liver triiodothyronine sulfotransferase: Copurification with phenol sulfotransferases. Thyroid 1995, 5, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Runge-Morris, M.; Barrett, K.G.; Fang, H.; Cukovic, D.; Dombkowski, A.A.; Kocarek, T.A. Upregulation of UGT2B4 Expression by 3′-Phosphoadenosine-5′-Phosphosulfate Synthase Knockdown: Implications for Coordinated Control of Bile Acid Conjugation. Drug Metab. Dispos. 2015, 43, 1061–1070. [Google Scholar] [CrossRef]
- Visser, T.J. Role of sulfation in thyroid hormone metabolism. Chem. Biol. Interact. 1994, 92, 293–303. [Google Scholar] [CrossRef]
- Wu, S.Y.; Green, W.L.; Huang, W.S.; Hays, M.T.; Chopra, I.J. Alternate pathways of thyroid hormone metabolism. Thyroid 2005, 15, 943–958. [Google Scholar] [CrossRef]
- Silvestri, E.; Senese, R.; Cioffi, F.; De Matteis, R.; Lattanzi, D.; Lombardi, A.; Giacco, A.; Salzano, A.M.; Scaloni, A.; Ceccarelli, M.; et al. 3,5-Diiodo-L-Thyronine Exerts Metabolically Favorable Effects on Visceral Adipose Tissue of Rats Receiving a High-Fat Diet. Nutrients 2019, 11, 278. [Google Scholar] [CrossRef]
- Grasselli, E.; Voci, A.; Demori, I.; Canesi, L.; De Matteis, R.; Goglia, F.; Lanni, A.; Gallo, G.; Vergani, L. 3,5-Diiodo-L-thyronine modulates the expression of genes of lipid metabolism in a rat model of fatty liver. J. Endocrinol. 2012, 212, 149–158. [Google Scholar] [CrossRef]
- Lombardi, A.; De Matteis, R.; Moreno, M.; Napolitano, L.; Busiello, R.A.; Senese, R.; de Lange, P.; Lanni, A.; Goglia, F. Responses of skeletal muscle lipid metabolism in rat gastrocnemius to hypothyroidism and iodothyronine administration: A putative role for FAT/CD36. Am. J. Physiol. Endocrinol. Metab. 2012, 303, 1222–1233. [Google Scholar] [CrossRef]
- Ghose, R.; Omoluabi, O.; Gandhi, A.; Shah, P.; Strohacker, K.; Carpenter, K.C.; McFarlin, B.; Guo, T. Role of high-fat diet in regulation of gene expression of drug metabolizing enzymes and transporters. Life Sci. 2011, 89, 57–64. [Google Scholar] [CrossRef]
- Nemoto, K.; Ikeda, A.; Ito, S.; Miyata, M.; Yoshida, C.; Degawa, M. Comparison of Constitutive Gene Expression Levels of Hepatic Cholesterol Biosynthetic Enzymes between Wistar-Kyoto and Stroke-Prone Spontaneously Hypertensive Rats. Biol. Pharm. Bull. 2013, 36, 1216–1220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goedeke, L.; Perry, R.J.; Shulman, G.I. Emerging Pharmacological Targets for the Treatment of Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Type 2 Diabetes. Annu. Rev. Pharmacol. Toxicol. 2016, 59, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, F.; Sestito, S.; Runfola, M.; Rapposelli, S.; Chiellini, G. Selective thyroid hormone receptor-beta (TRβ) agonists: New perspectives for the treatment of metabolic and neurodegenerative disorders. Front. Med. 2020, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Goglia, F.; Lanni, A.; Horst, C.; Moreno, M.; Thoma, R. In vitro binding of 3,5-di-iodo-L-thyronine to rat liver mitochondria. J. Mol. Endocrinol. 1994, 13, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lanni, A.; Moreno, M.; Lombardi, A.; De Lange, P.; Goglia, F. Control of energy metabolism by iodothyronines. J. Endocrinol. Investig. 2001, 24, 897–913. [Google Scholar] [CrossRef]
- Casas, F.; Rochard, P.; Rodier, A.; Cassar-Malek, I.; Marchal-Victorion, S.; Wiesner, R.J.; Cabello, G.; Wrutniak, C. A variant form of the nuclear triiodothyronine receptor c-ErbAα1 plays a direct role in regulation of mitochondrial RNA synthesis. Mol. Cell. Biol. 1999, 19, 7913–7924. [Google Scholar] [CrossRef]
- Tata, J.R. Basal Metabolic Rate and Thyroid Hormones. Adv. Metab. Disord. 1964, 15, 153–189. [Google Scholar] [CrossRef]
- Sacripanti, G.; Nguyen, N.M.; Lorenzini, L.; Frascarelli, S.; Saba, A.; Zucchi, R.; Ghelardoni, S. 3, 5-Diiodo-l-thyronine increases glucose consumption in cardiomyoblasts without affecting the contractile performance in rat heart. Front. Endocrinol. 2018, 9, 282. [Google Scholar] [CrossRef]
- Lanni, A.; Moreno, M.; Lombardi, A.; Goglia, F. 3,5-diiodo-L-thyronine and 3,5,3′-triiodo-L-thyronine both improve the cold tolerance of hypothyroid rats, but possibly via different mechanisms. Pflug. Arch. Eur. J. Physiol. 1998, 436, 407–414. [Google Scholar] [CrossRef]
- Sheehan, T.E.; Kumar, P.A.; Hood, D.A. Tissue-specific regulation of cytochrome c oxidase subunit expression by thyroid hormone. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E968–E974. [Google Scholar] [CrossRef]
- Louzada, R.A.; Padron, A.S.; Marques-Neto, S.R.; Maciel, L.; Werneck-de-Castro, J.P.; Ferreira, A.C.F.; Nascimento, J.H.M.; Carvalho, D.P. 3,5-Diiodothyronine protects against cardiac ischaemia–reperfusion injury in male rats. Exp. Physiol. 2021, 106, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Geist, D.; Hönes, G.S.; Gassen, J.; Kerp, H.; Kleinbongard, P.; Heusch, G.; Führer, D.; Moeller, L.C. Noncanonical Thyroid Hormone Receptor α Action Mediates Arterial Vasodilation. Endocrinology 2021, 162, bqab099. [Google Scholar] [CrossRef] [PubMed]
- Kerp, H.; Hönes, G.S.; Tolstik, E.; Hönes-Wendland, J.; Gassen, J.; Moeller, L.C.; Lorenz, K.; Führer, D. Protective Effects of Thyroid Hormone Deprivation on Progression of Maladaptive Cardiac Hypertrophy and Heart Failure. Front. Cardiovasc. Med. 2021, 8, 683522. [Google Scholar] [CrossRef] [PubMed]
- Little, A.G.; Seebacher, F. Thyroid hormone regulates cardiac performance during cold acclimation in zebrafish (Danio rerio). J. Exp. Biol. 2014, 217, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Little, A.G.; Kunisue, T.; Kannan, K.; Seebacher, F. Thyroid hormone actions are temperature-specific and regulate thermal acclimation in zebrafish (Danio rerio). BMC Biol. 2013, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Orozco, A.; Navarrete-Ramírez, P.; Olvera, A.; García-G, C. 3,5-Diiodothyronine (T2) is on a role. A new hormone in search of recognition. Gen. Comp. Endocrinol. 2014, 203, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Lanni, A.; Goglia, F. Thyroid hormones, mitochondrial bioenergetics and lipid handling. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 402–407. [Google Scholar] [CrossRef]
- Moreno, M.; Lombardi, A.; Beneduce, L.; Silvestri, E.; Pinna, G.; Goglia, F.; Lanni, A.; Biologiche, S.; La, V.A.; Universita, S.; et al. Are the Effects of T 3 on Resting Metabolic Rate in Euthyroid Rats Entirely Caused by T 3 Itself? Endocrinology 2002, 143, 504–510. [Google Scholar] [CrossRef]
- Silvestri, E.; Lombardi, A.; Coppola, M.; Gentile, A.; Cioffi, F.; Senese, R.; Goglia, F.; Lanni, A.; Moreno, M.; De Lange, P. Differential effects of 3,5-Diiodo-L-Thyronine and 3,5,3′-Triiodo-L-Thyronine on mitochondrial respiratory pathways in liver from hypothyroid rats. Cell. Physiol. Biochem. 2018, 47, 2471–2483. [Google Scholar] [CrossRef]
- Hoch, F.L. Thyrotoxicosis as a disease of mitochondria. N. Engl. J. Med. 1962, 266, 446–454. [Google Scholar] [CrossRef]
- Rabah, S.A.; Gowan, I.L.; Pagnin, M.; Osman, N.; Richardson, S.J. Thyroid hormone distributor proteins during development in vertebrates. Front. Endocrinol. 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. Thyroid Hormones and Derivatives: Endogenous Thyroid Hormones and Their Targets. Methods Mol. Biol. 2018, 1801, 85–104. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.H.; Monk, R.; Renko, K.; Rathmann, D.; Rijntjes, E.; Köhrle, J. A combined LC-MS/MS and LC-MS3 multi-method for the quantification of iodothyronines in human blood serum. Anal. Bioanal. Chem. 2019, 411, 5605–5616. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, L.; Nguyen, N.M.; Sacripanti, G.; Serni, E.; Borsò, M.; Saponaro, F.; Cecchi, E.; Simoncini, T.; Ghelardoni, S.; Zucchi, R.; et al. Assay of endogenous 3,5-diiodo-L-thyronine (3,5-T2) and 3,3′-diiodo-L-thyronine (3,3′-T2) in human serum: A feasibility study. Front. Endocrinol. 2019, 10, 88. [Google Scholar] [CrossRef]
- Jongejan, R.M.S.; Van Velsen, E.F.S.; Meima, M.E.; Klein, T.; Van Den Berg, S.A.A.; Massolt, E.T.; Visser, W.E.; Peeters, R.P.; De Rijke, Y.B. Change in Thyroid Hormone Metabolite Concentrations Across Different Thyroid States. Thyroid 2022, 32, 119–127. [Google Scholar] [CrossRef]
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Müller, L.; De Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell 2016, 167, 843–857.e14. [Google Scholar] [CrossRef]
- Zhao, M.; Xie, H.; Shan, H.; Zheng, Z.; Li, G.; Li, M.; Hong, L. Development of Thyroid Hormones and Synthetic Thyromimetics in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 1102. [Google Scholar] [CrossRef]
- Hackenmueller, S.A.; Scanlan, T.S. Identification and quantification of 3-iodothyronamine metabolites in mouse serum using liquid chromatography-tandem mass spectrometry. J. Chromatogr. 2012, 1256, 89–97. [Google Scholar] [CrossRef]
- Piehl, S.; Hoefig, C.S.; Scanlan, T.S.; Köhrle, J. Thyronamines—Past, present, and future. Endocr. Rev. 2011, 32, 64–80. [Google Scholar] [CrossRef]
- Cioffi, F.; Zambad, S.P.; Chhipa, L.; Senese, R.; Busiello, R.A.; Tuli, D.; Munshi, S.; Moreno, M.; Lombardi, A.; Gupta, R.C.; et al. TRC150094, a novel functional analog of iodothyronines, reduces adiposity by increasing energy expenditure and fatty acid oxidation in rats receiving a high-fat diet. FASEB J. 2010, 24, 3451–3461. [Google Scholar] [CrossRef]
- Silvestri, E.; Glinni, D.; Cioffi, F.; Moreno, M.; Lombardi, A.; De Lange, P.; Senese, R.; Ceccarelli, M.; Salzano, A.M.; Scaloni, A.; et al. Metabolic effects of the iodothyronine functional analogue TRC150094 on the liver and skeletal muscle of high-fat diet fed overweight rats: An integrated proteomic study. Mol. BioSyst. 2012, 8, 1987–2000. [Google Scholar] [CrossRef] [PubMed]
- Zambad, S.P.; Munshi, S.; Dubey, A.; Gupta, R.; Busiello, R.A.; Lanni, A.; Goglia, F.; Gupta, R.C.; Chauthaiwale, V.; Dutt, C. TRC150094 attenuates progression of nontraditional cardiovascular risk factors associated with obesity and type 2 diabetes in obese ZSF1 rats. Diabetes Metab. Syndr. Obes. Targets Ther. 2011, 4, 5–16. [Google Scholar] [CrossRef][Green Version]
- Joshi, D.; Jamadarkhana, P.; Kumbhare, S.; Singh, A.; Kotecha, J.; Bunger, D.; Shiwalkar, A.; Mohanan, A.; Dutt, C. Safety, Tolerability, and Pharmacokinetics of a Novel Mitochondrial Modulator, TRC150094, in Overweight and Obese Subjects: A Randomized Phase-I Clinical Trial. Front. Pharmacol. 2021, 12, 729424. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | C | 3,5-T2 |
|---|---|---|
| Hepatic CS activity (µmol·min−1·min−1) | 700 ± 30.0 | 1024 ± 35.7 ** |
| Hepatic D1 activity (pmol·mg−1·min−1) | 150 ± 52 | 380 ± 93 *** |
| Serum triglycerides (µg/mL) | 80 ± 7.3 | 97 ± 12.1 |
| Free fatty acids (mM) | 0.65 ± 0.04 | 0.68 ± 0.04 |
| Total cholesterol (mg/dL) | 181 ± 7.1 | 104 ± 8.0 *** |
| Liver triglycerides (µg/g protein) | 1.18 ± 0.39 | 0.73 ± 0.13 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sane, R.; Wirth, E.K.; Köhrle, J. 3,5-T2-an Endogenous Thyroid Hormone Metabolite as Promising Lead Substance in Anti-Steatotic Drug Development? Metabolites 2022, 12, 582. https://doi.org/10.3390/metabo12070582
Sane R, Wirth EK, Köhrle J. 3,5-T2-an Endogenous Thyroid Hormone Metabolite as Promising Lead Substance in Anti-Steatotic Drug Development? Metabolites. 2022; 12(7):582. https://doi.org/10.3390/metabo12070582
Chicago/Turabian StyleSane, Rajas, Eva K. Wirth, and Josef Köhrle. 2022. "3,5-T2-an Endogenous Thyroid Hormone Metabolite as Promising Lead Substance in Anti-Steatotic Drug Development?" Metabolites 12, no. 7: 582. https://doi.org/10.3390/metabo12070582
APA StyleSane, R., Wirth, E. K., & Köhrle, J. (2022). 3,5-T2-an Endogenous Thyroid Hormone Metabolite as Promising Lead Substance in Anti-Steatotic Drug Development? Metabolites, 12(7), 582. https://doi.org/10.3390/metabo12070582