
Potential Combination Drug Therapy to Prevent Redox Stress and Mitophagy Dysregulation in Retinal Müller Cells under High Glucose Conditions: Implications for Diabetic Retinopathy


Abstract
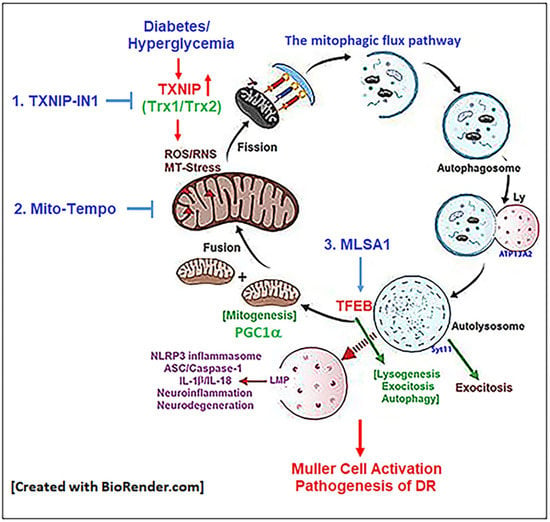
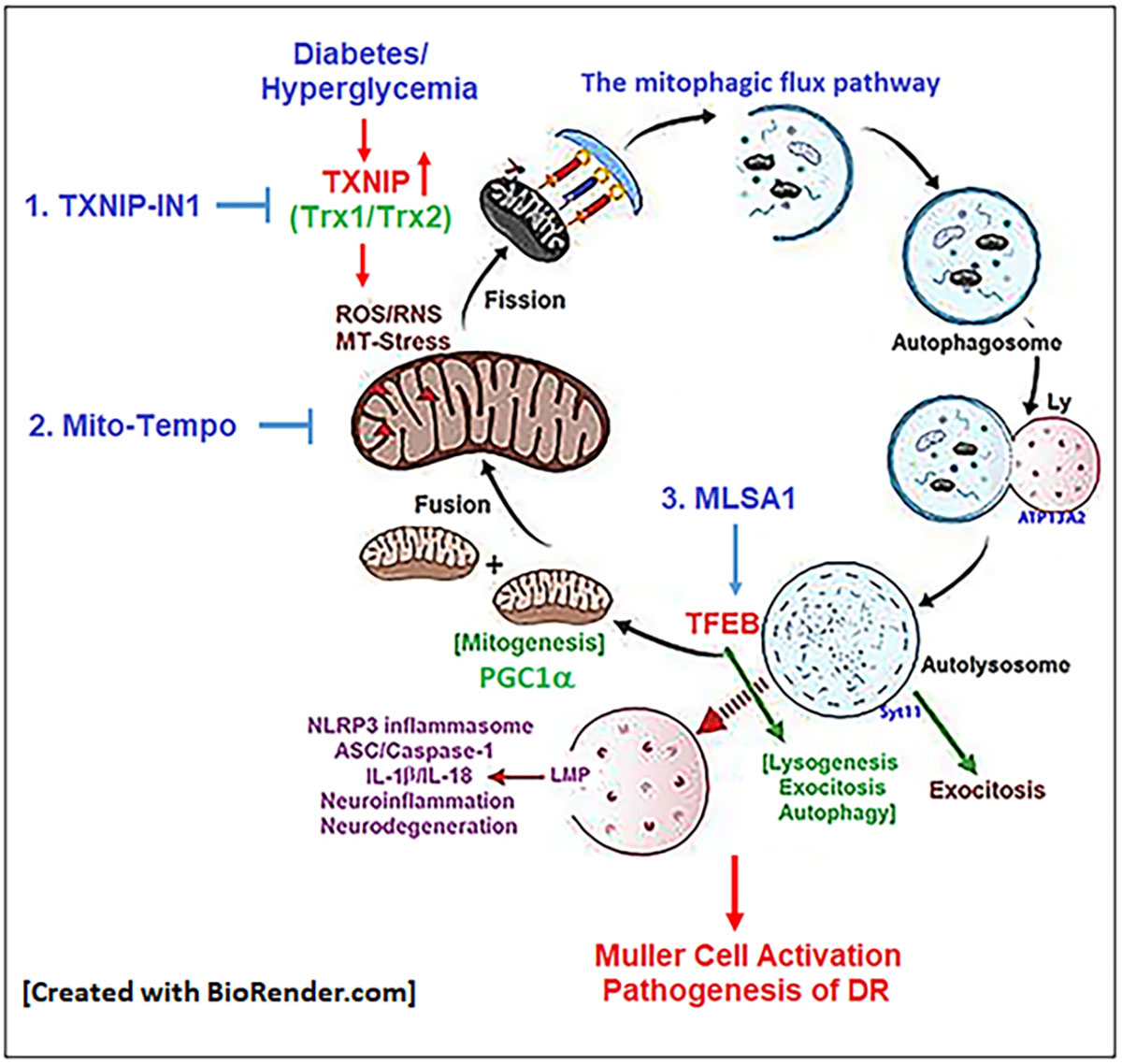
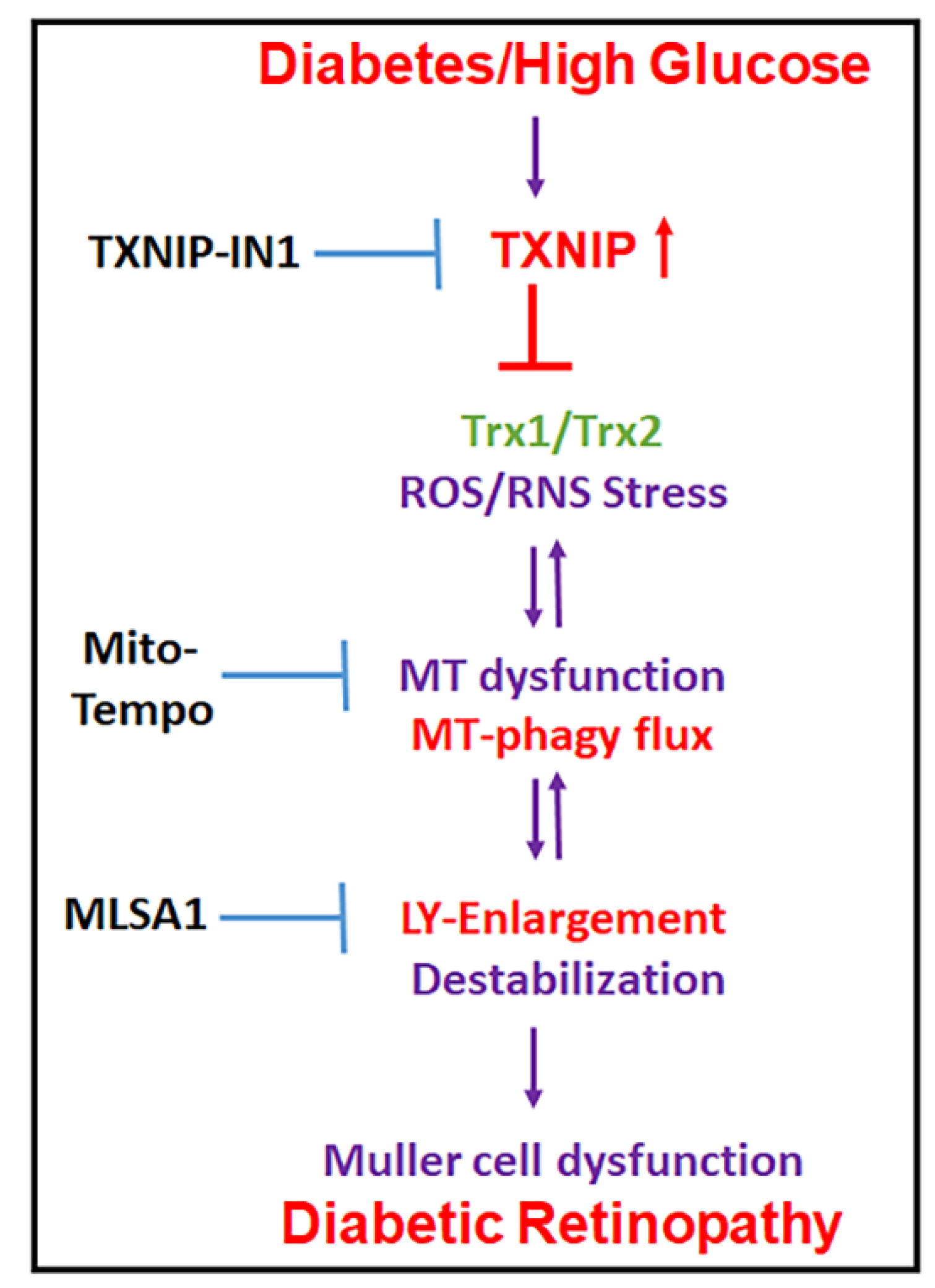
:
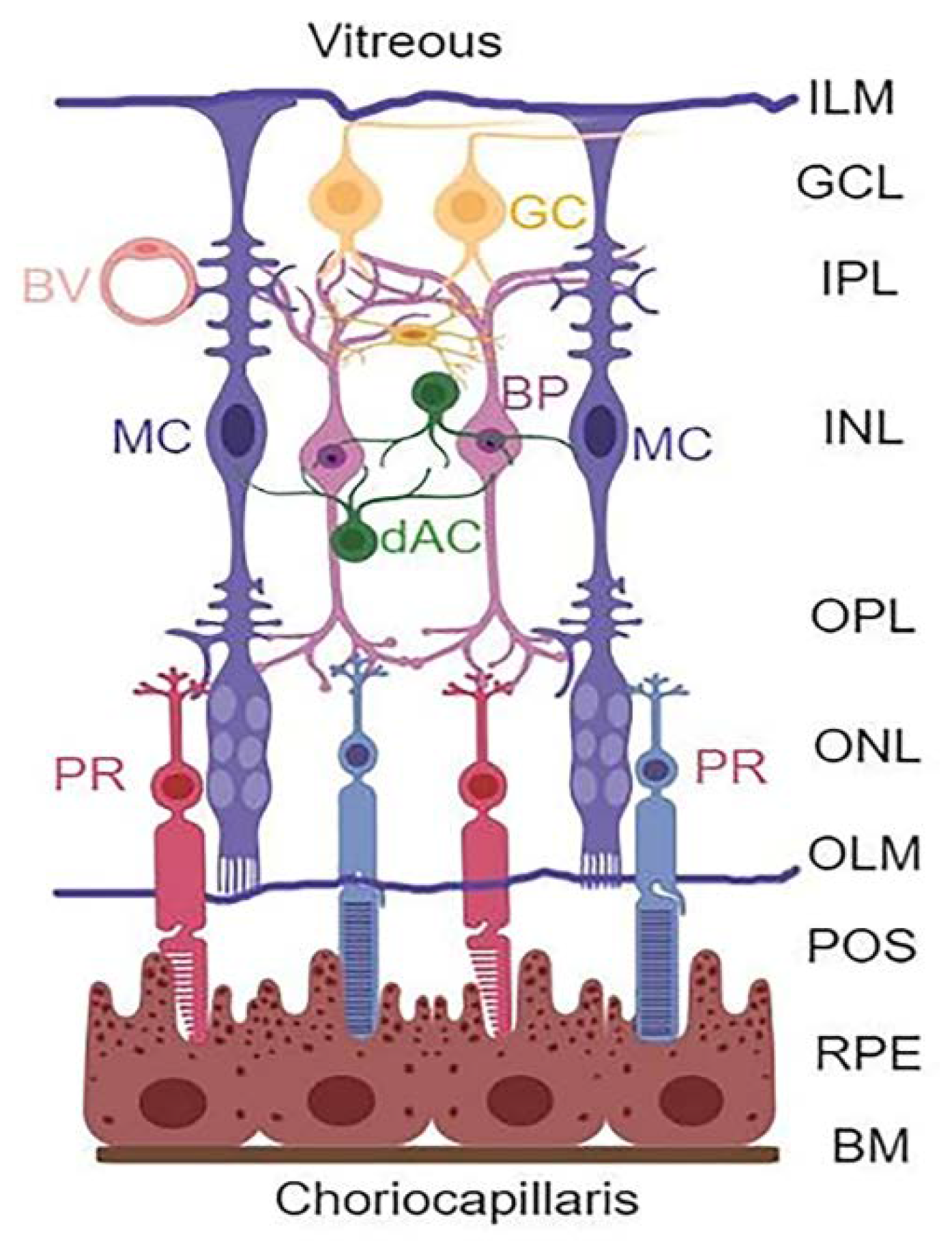
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
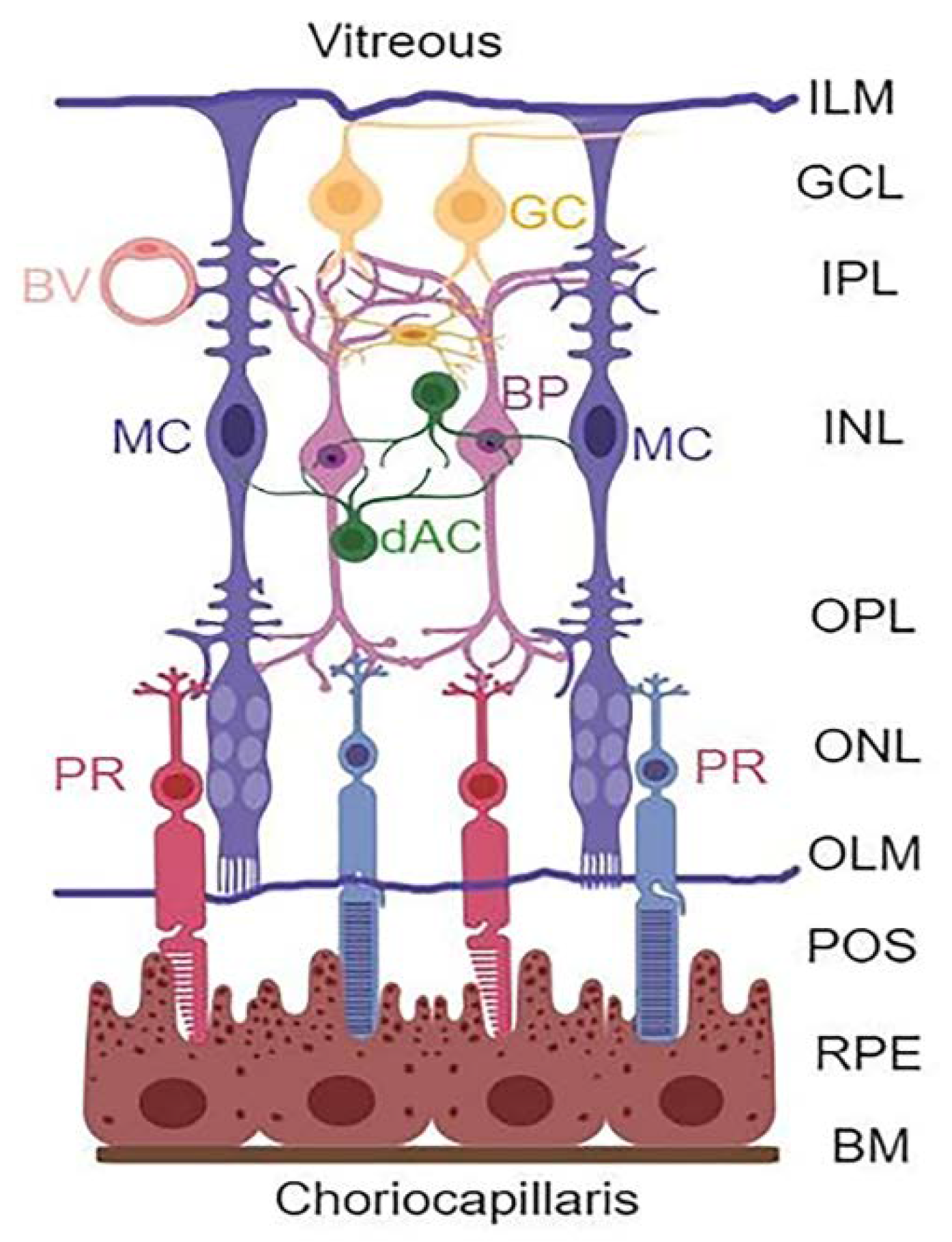
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Live-Cell Imaging
2.3. Fixed-Cell Imaging
2.4. Mitochondrial Isolation
2.5. Western Blotting
2.6. Diabetes Induction in Rats and Immunohistochemistry
2.7. Statistical Analysis
3. Results
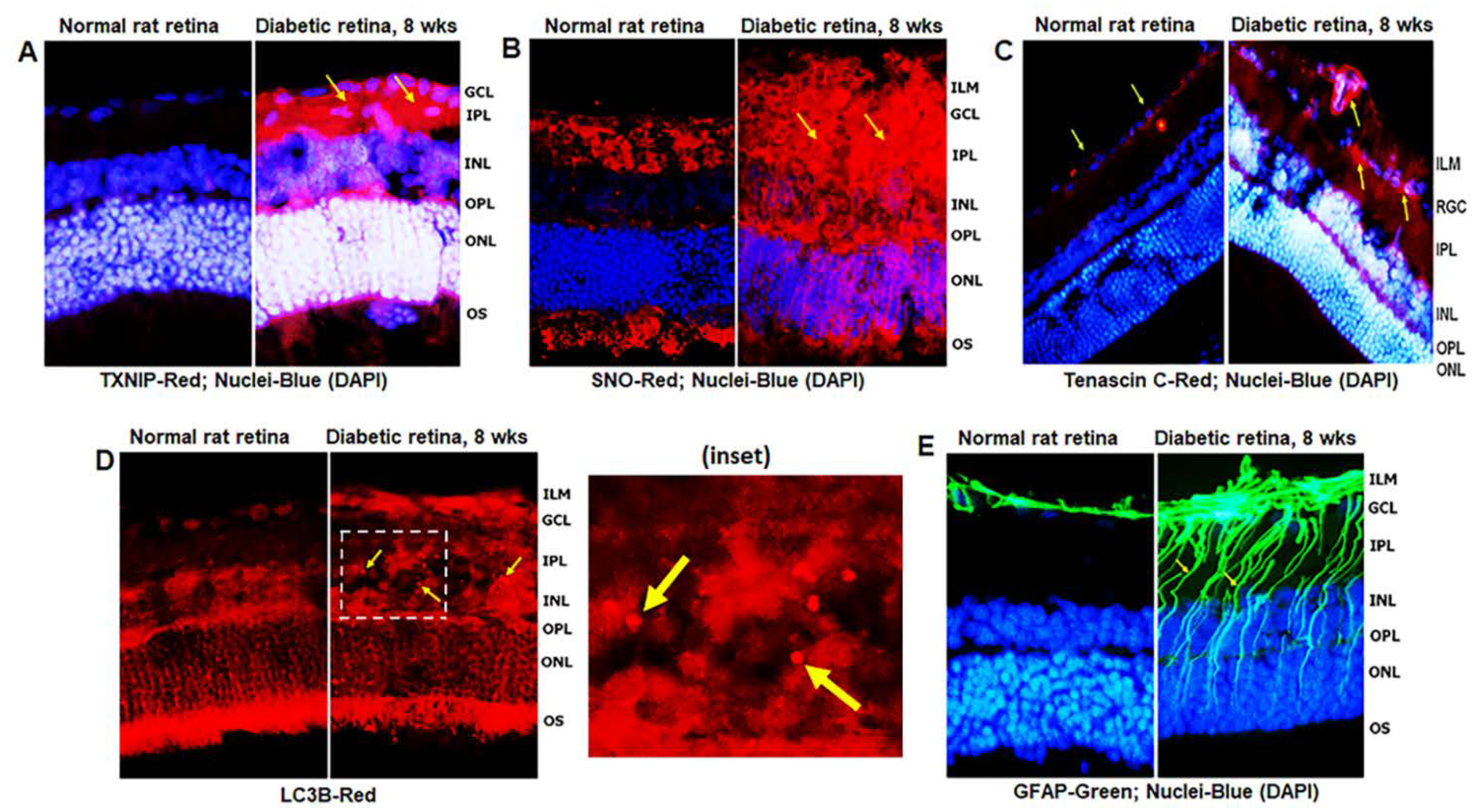
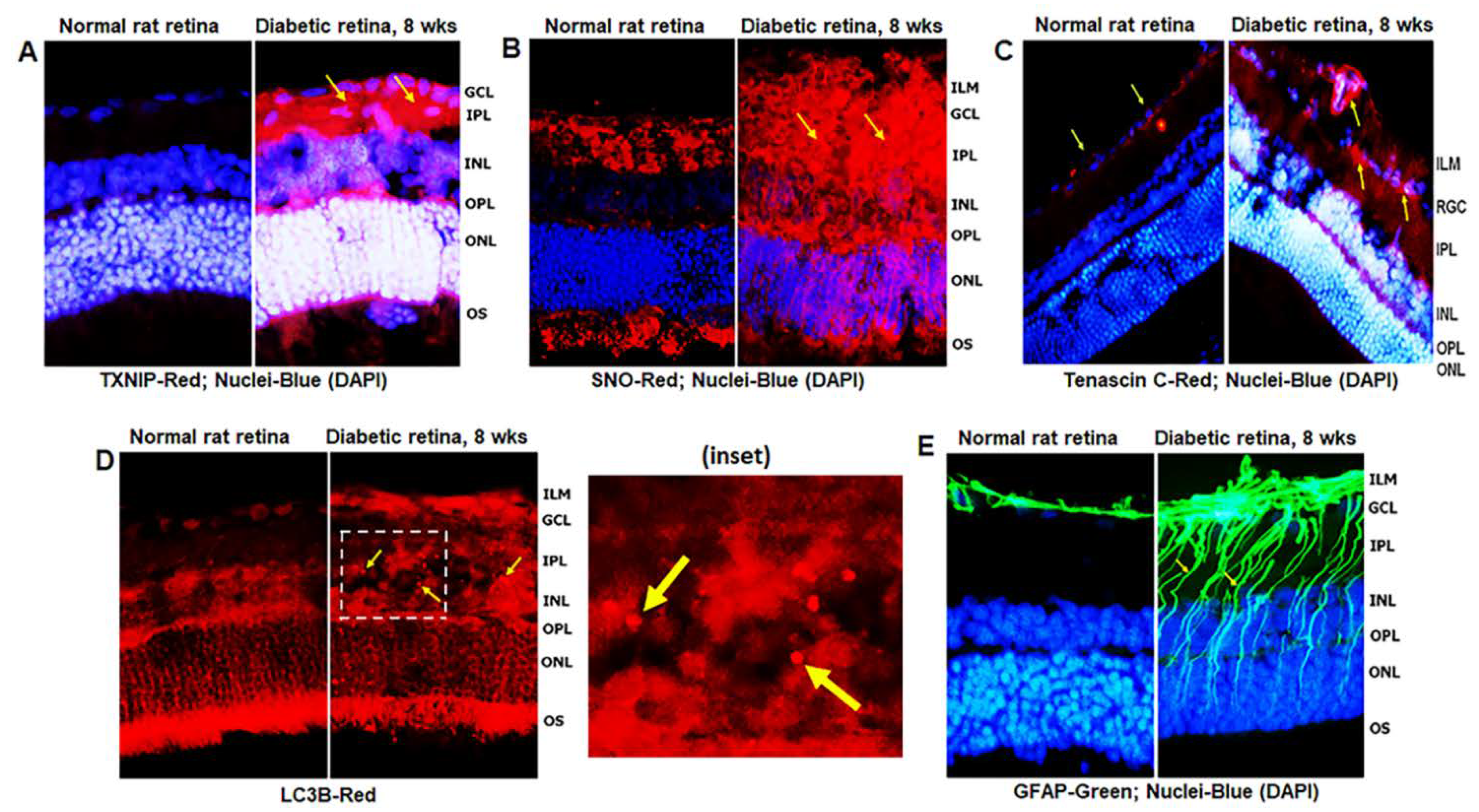
3.1. Diabetes Induces TXNIP Expression, Redox Stress, and Müller Cell Activation in the Diabetic Rat Retina
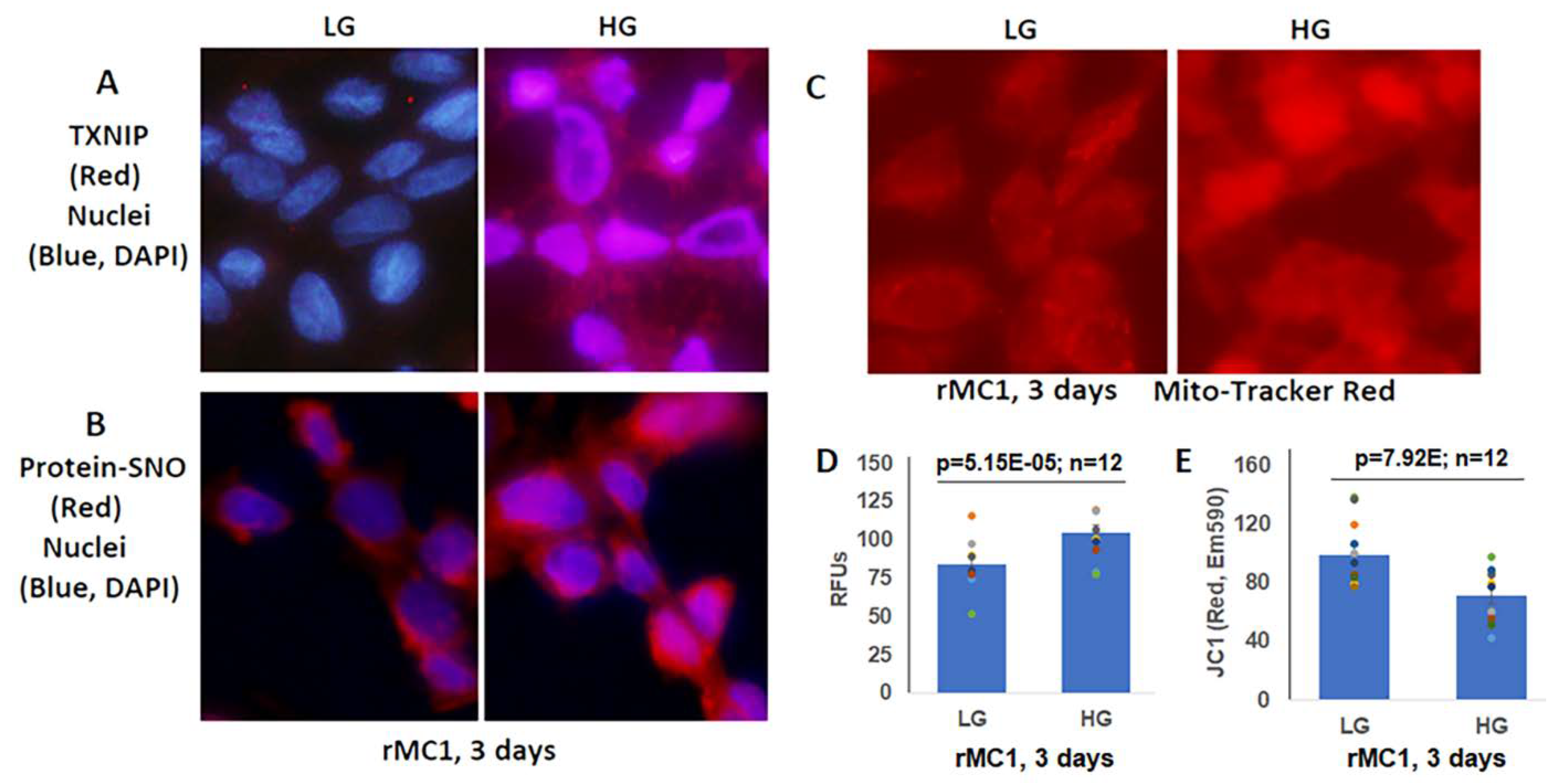
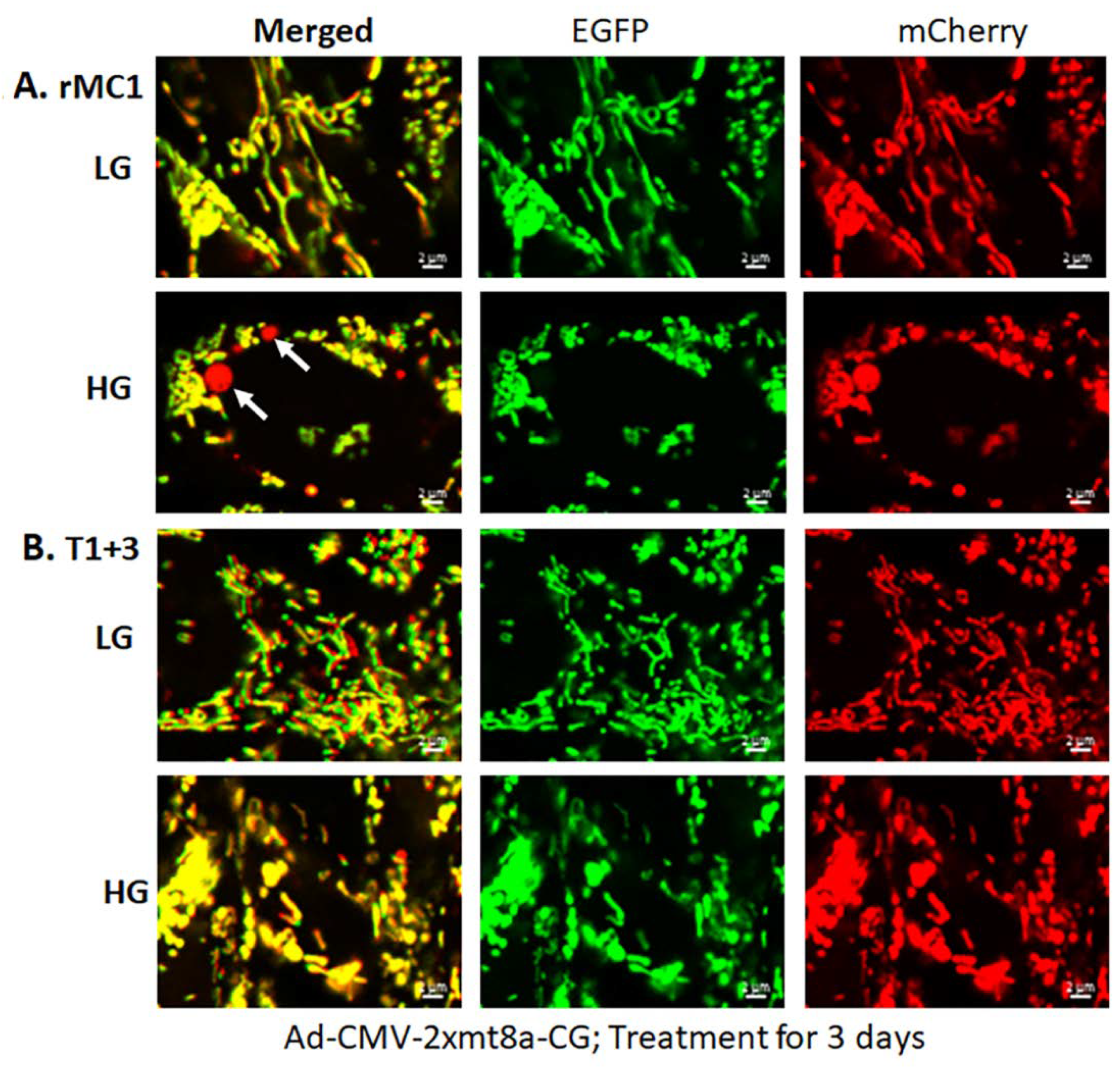
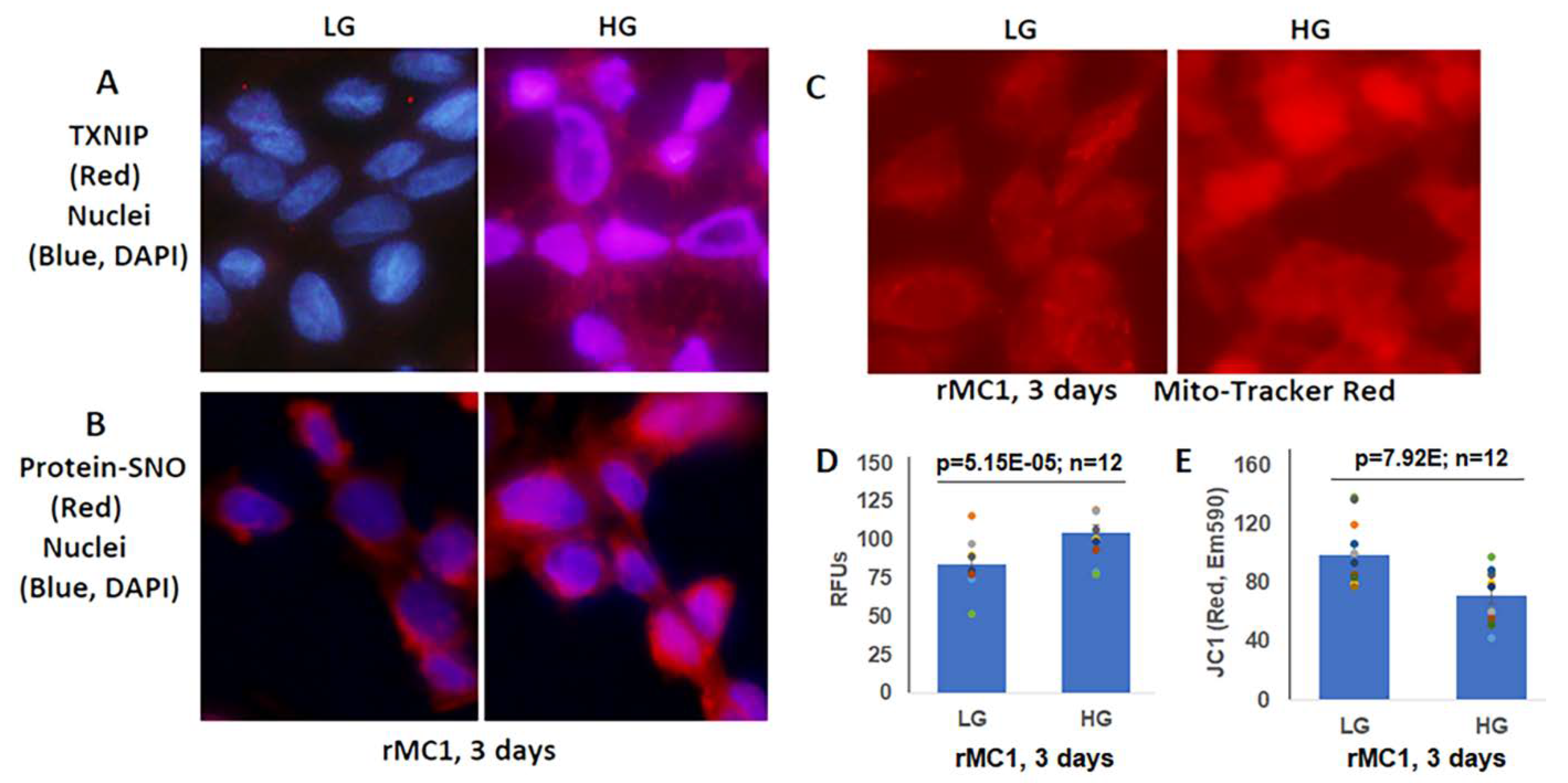
3.2. High Glucose Induces TXNIP Expression, Mitochondrial Stress, and Mitophagic Flux in rMC1 in Culture
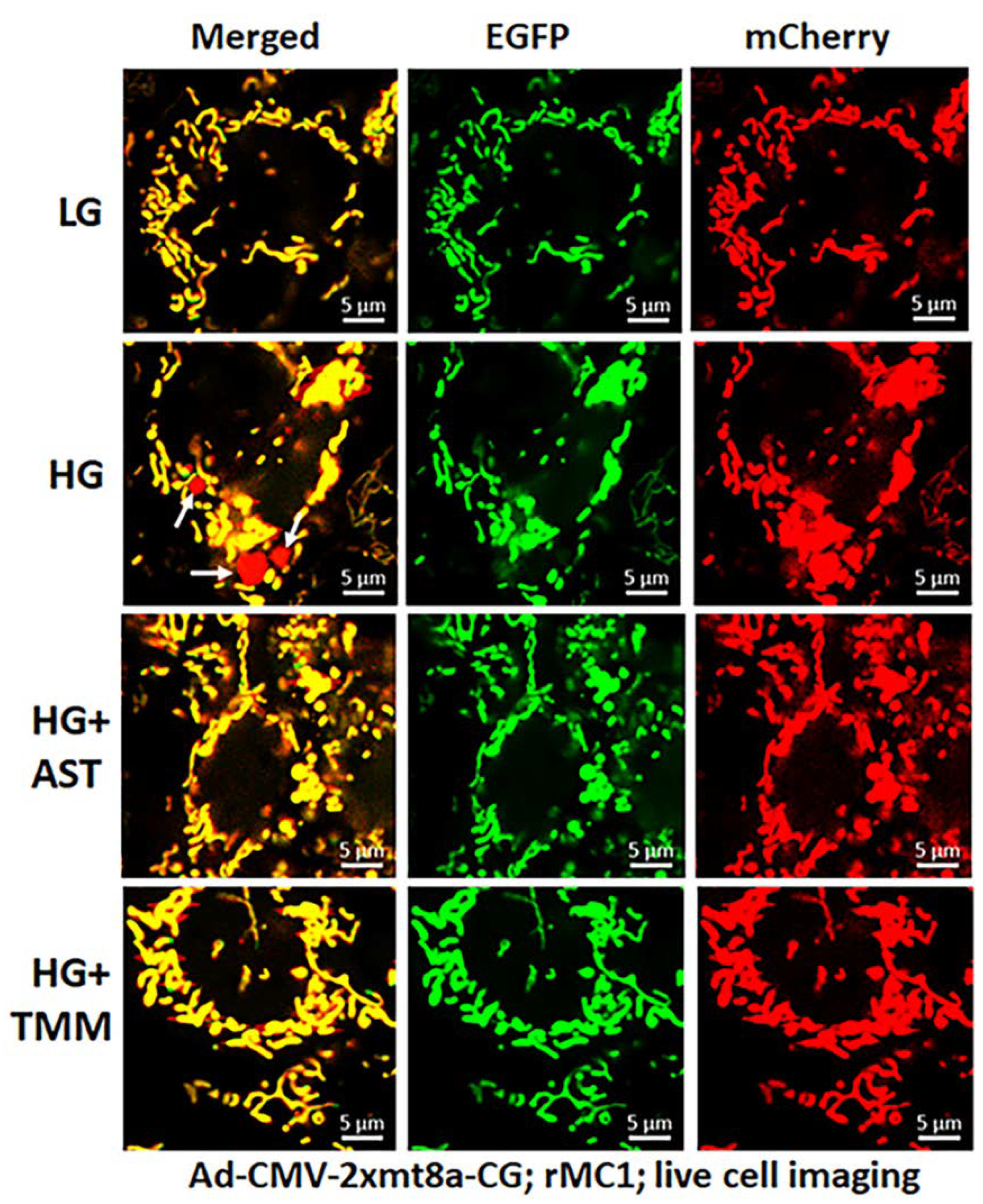
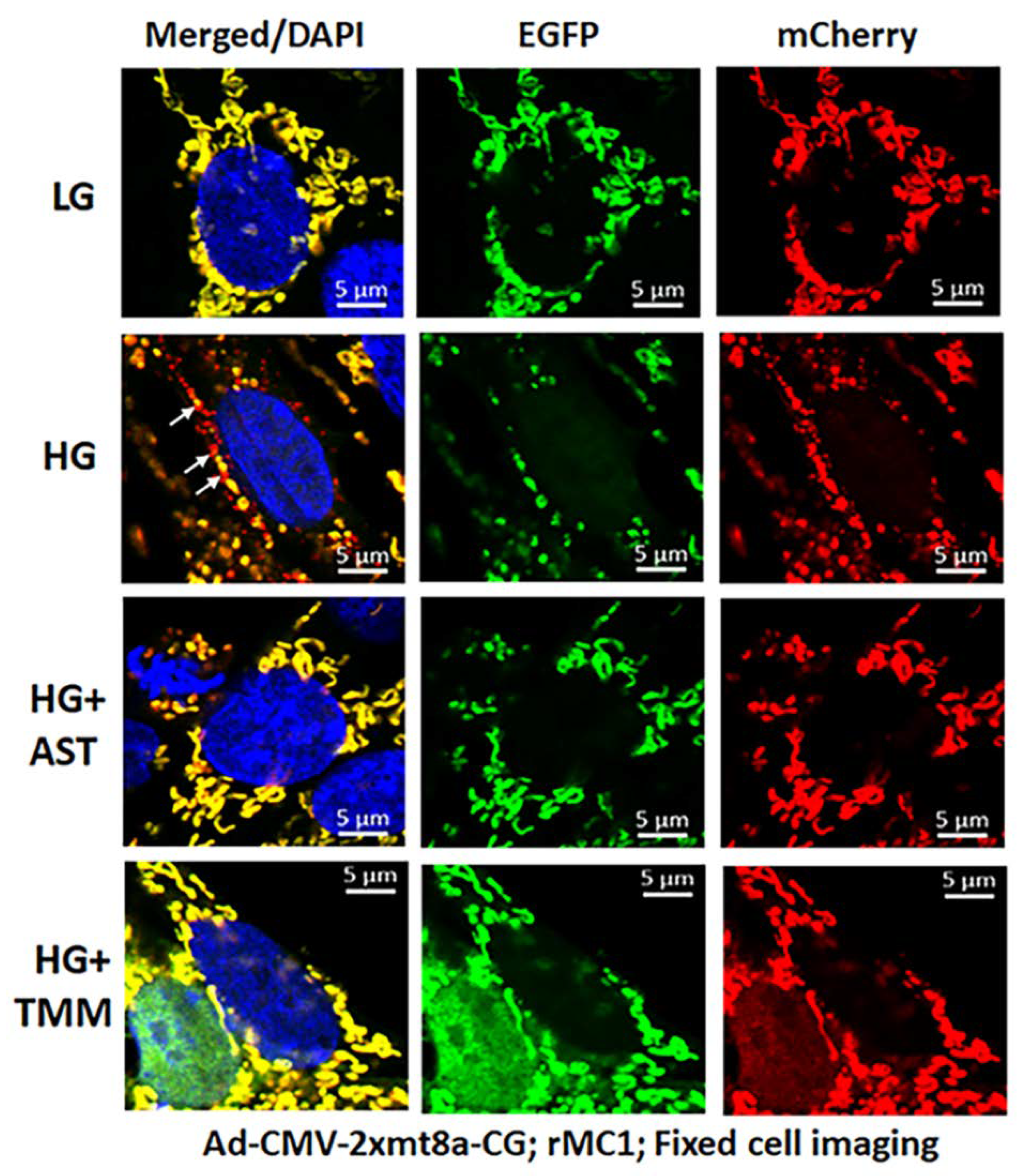
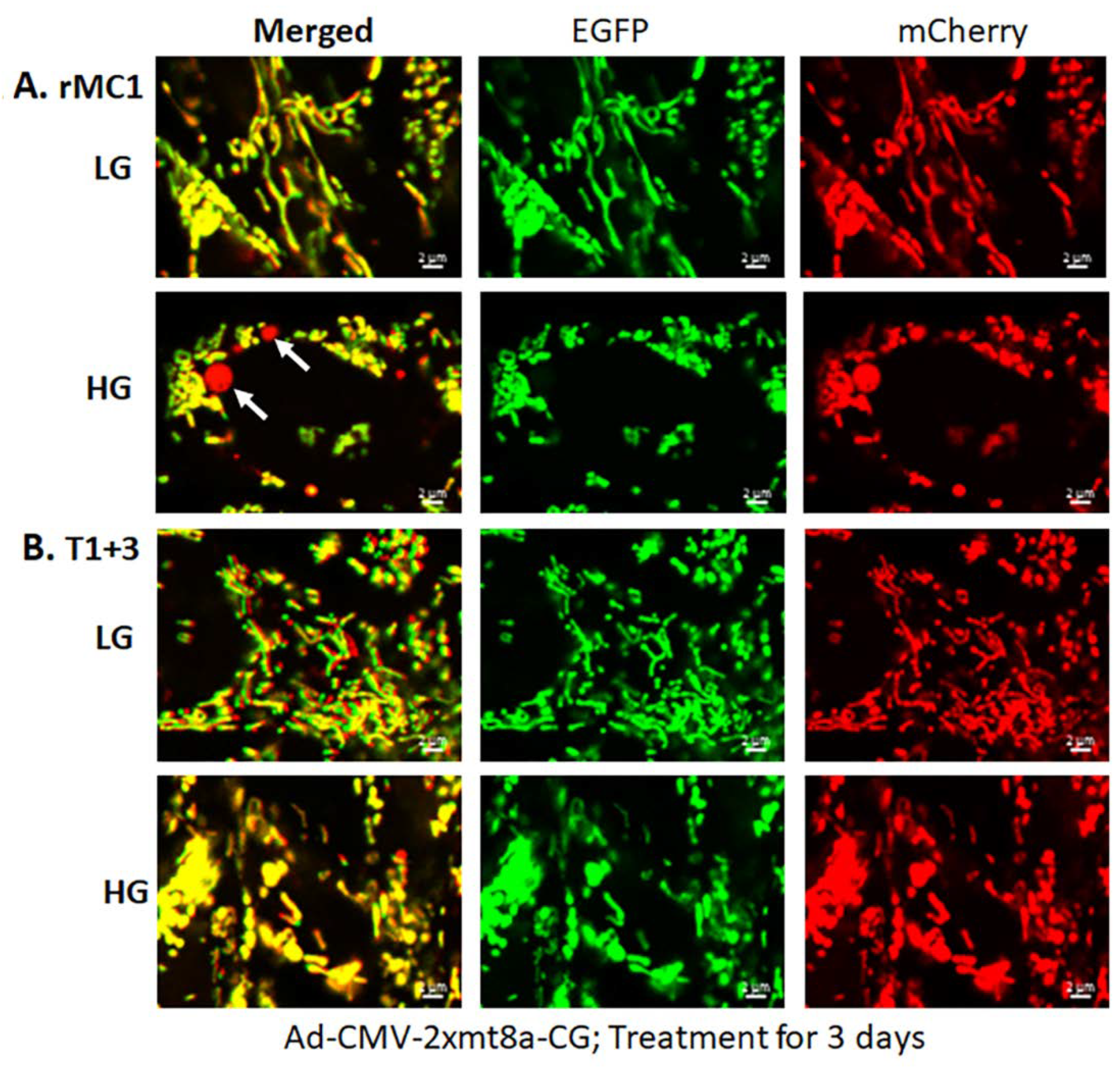
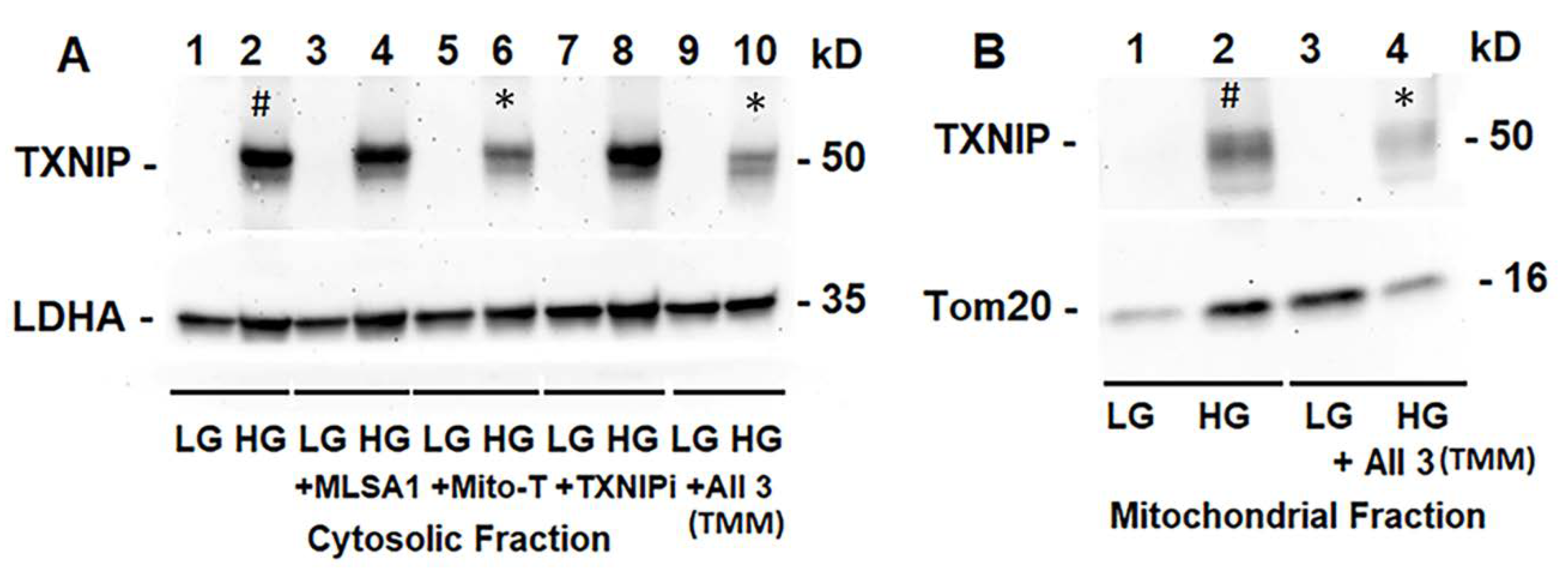
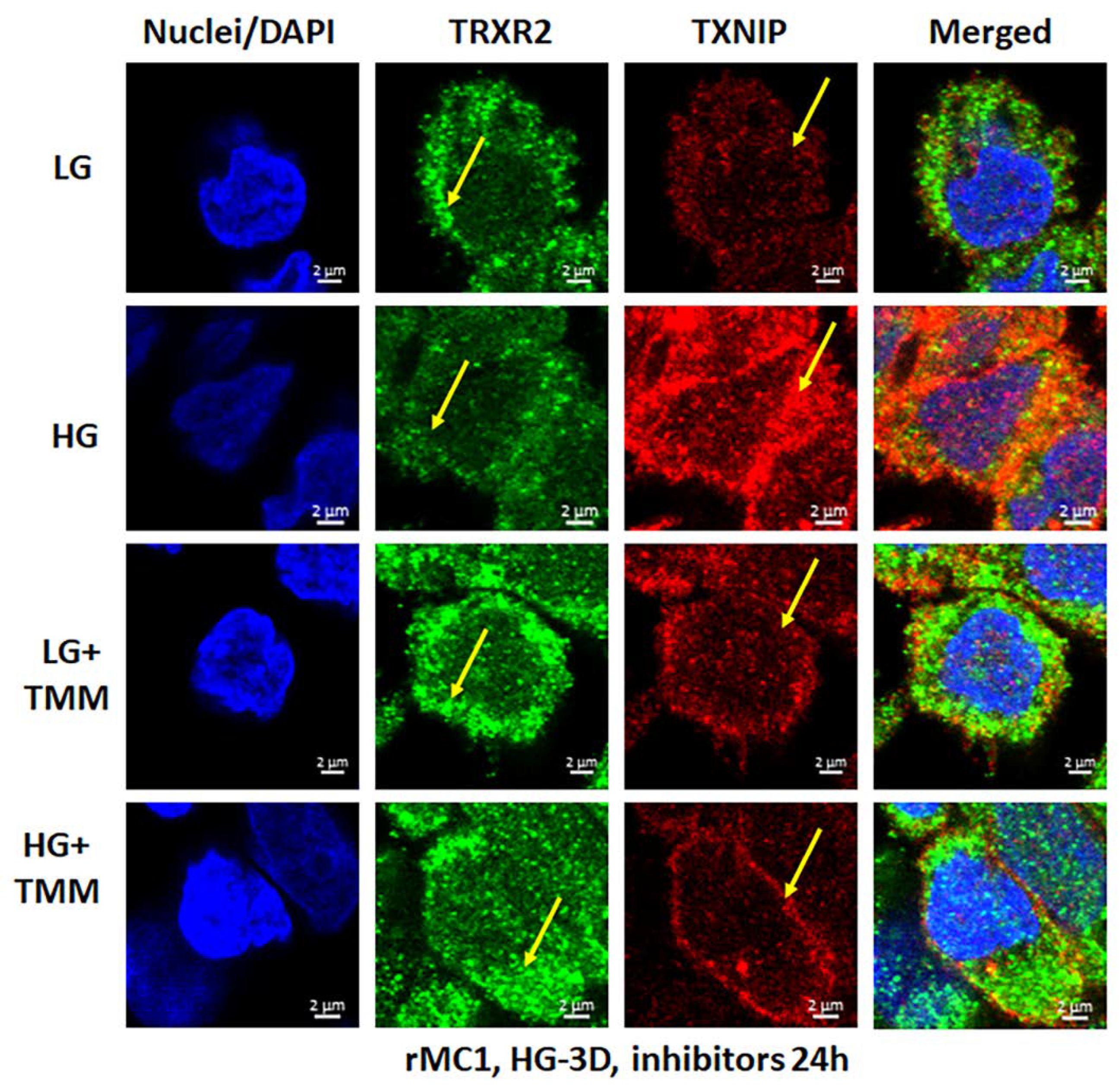
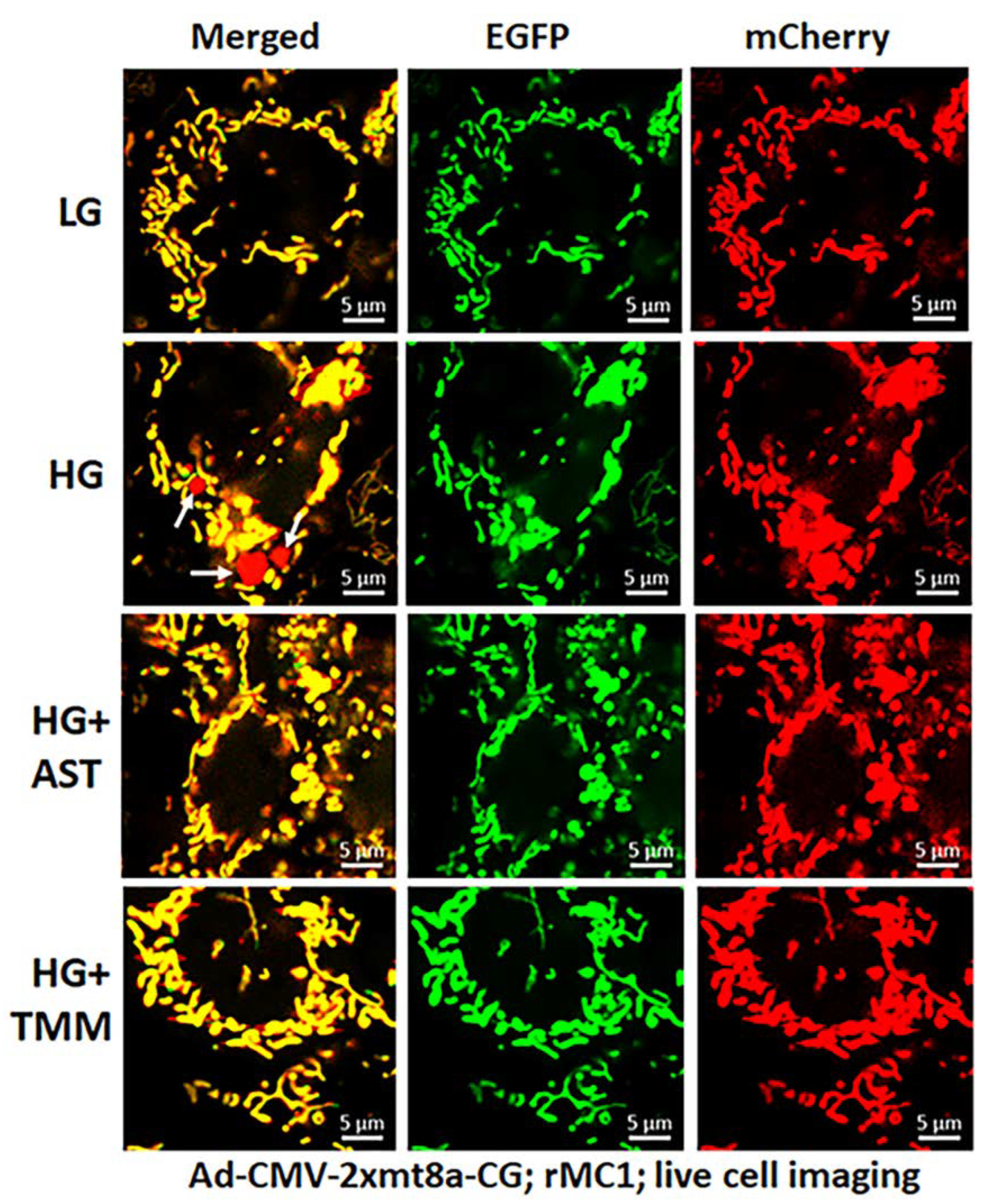
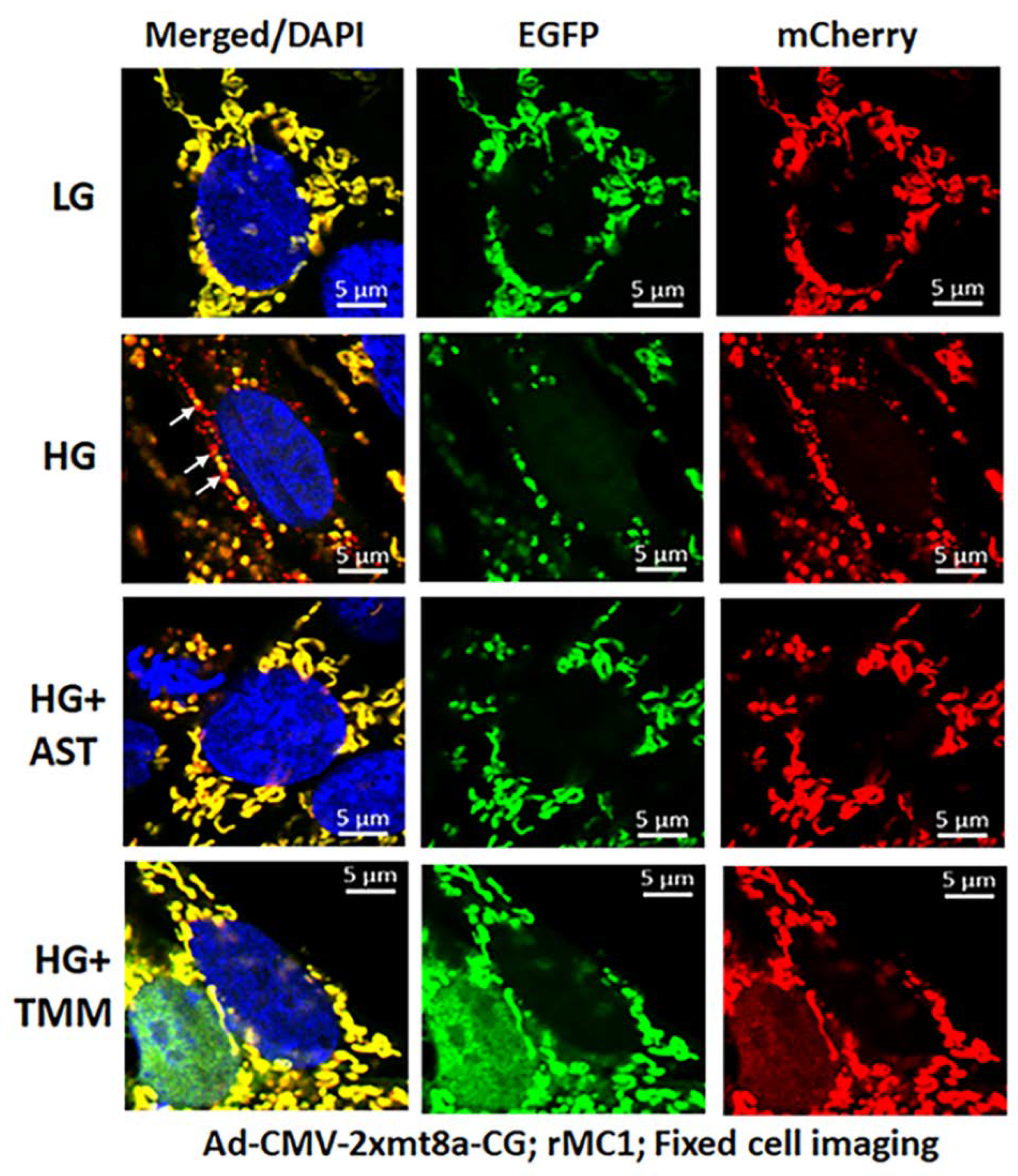
3.3. Combination Drug Treatment Reduces TXNIP Level and Mitophagic Flux in rMC1 under High Glucose Conditions
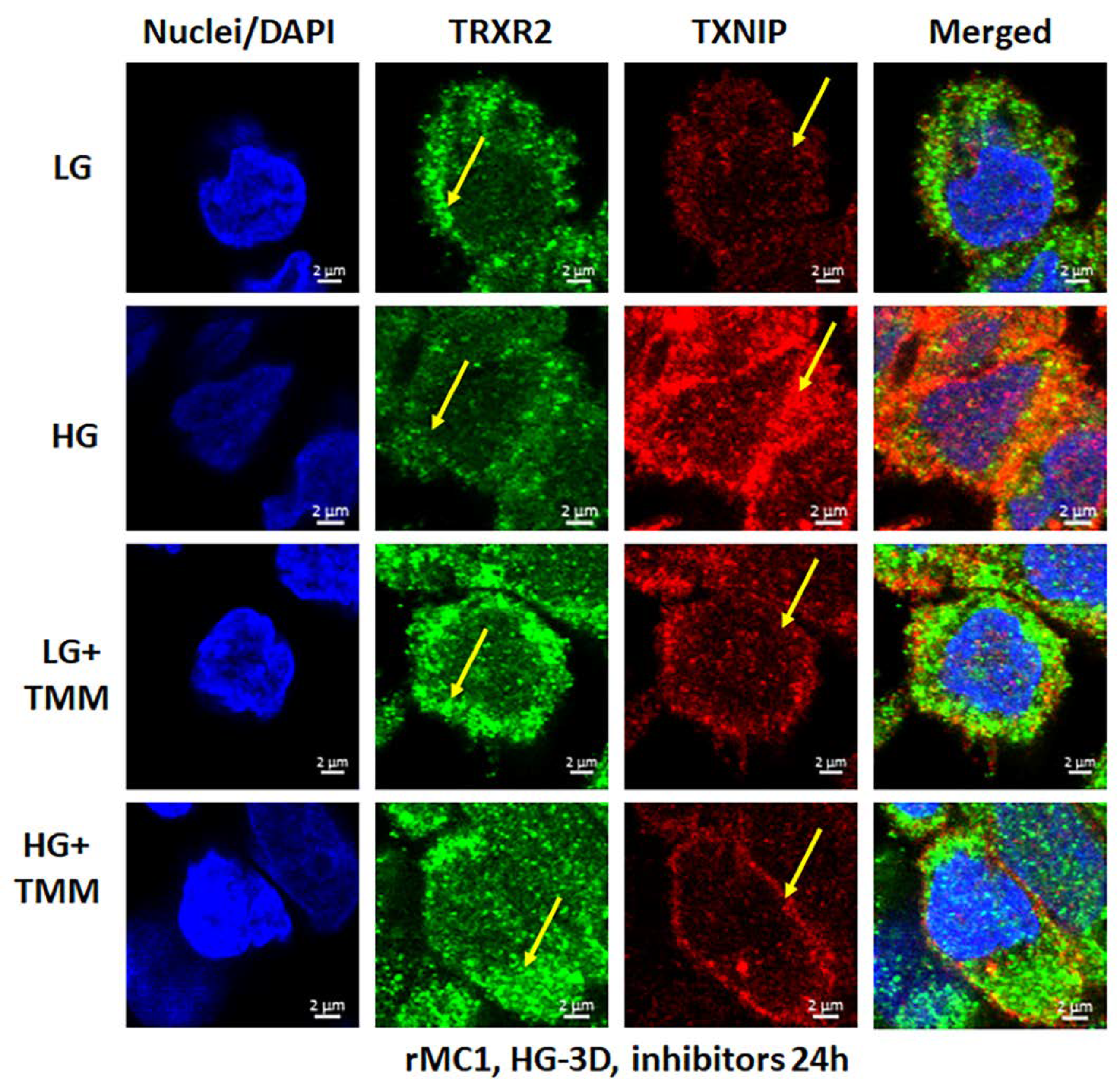
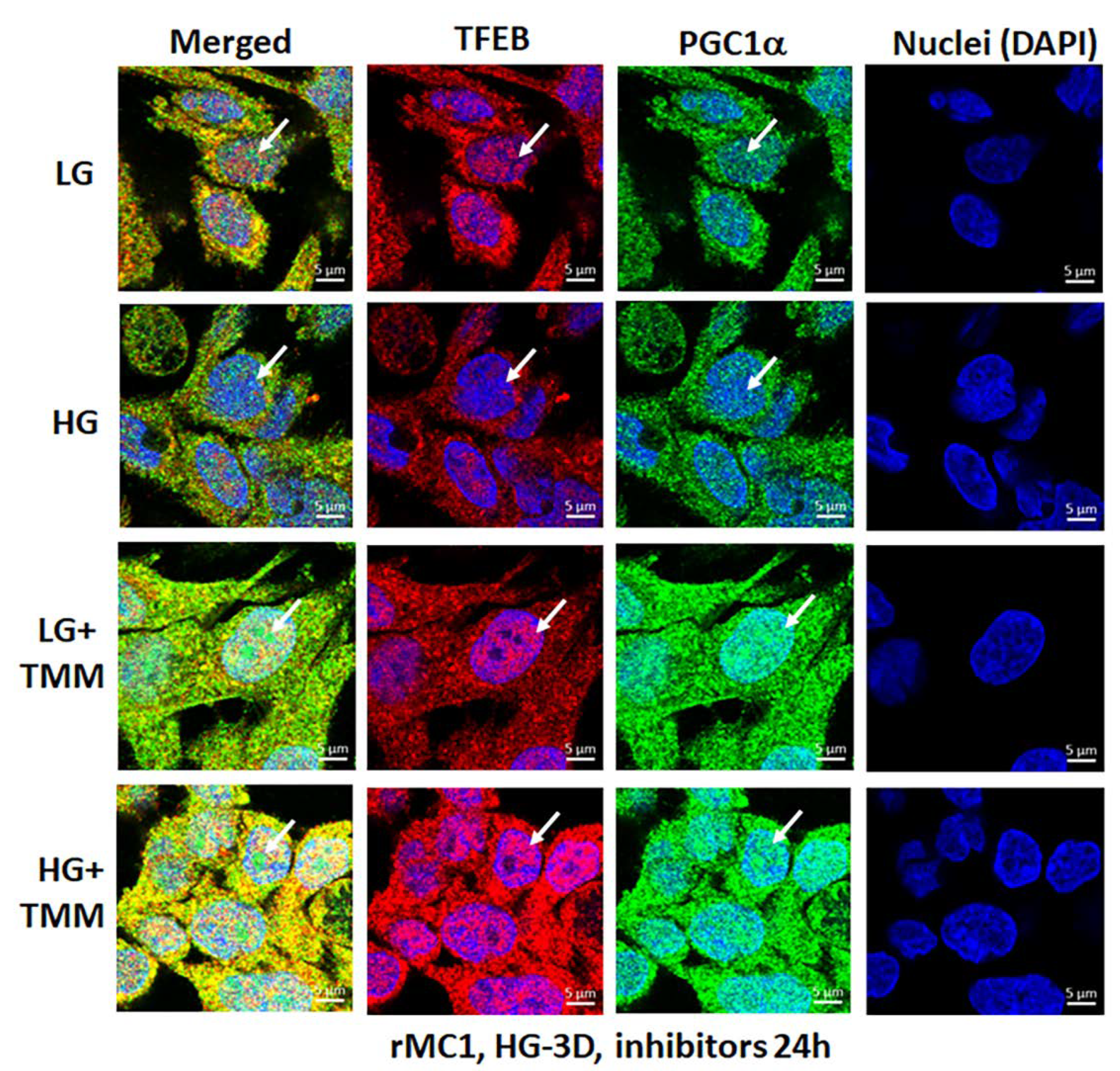
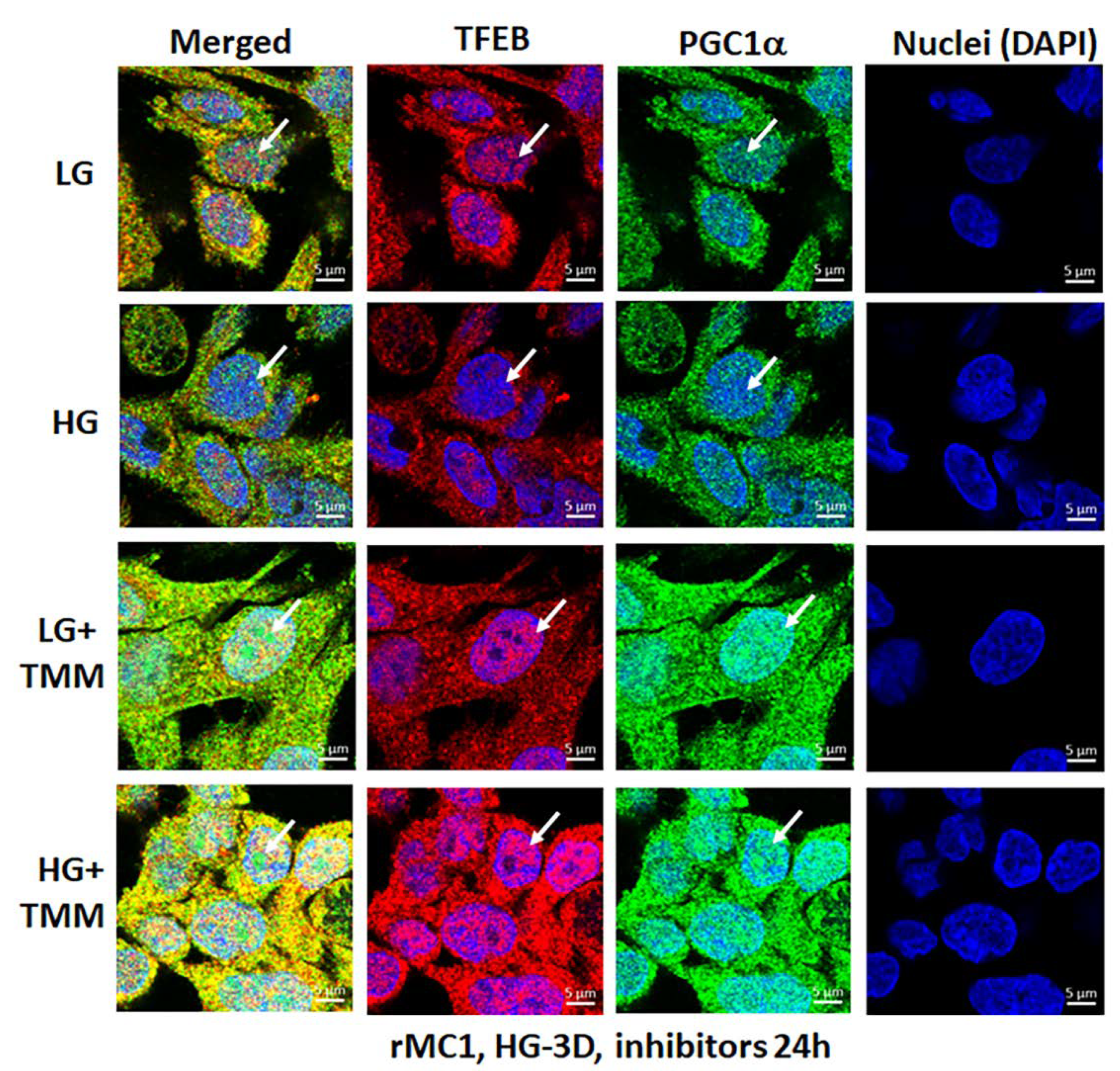
3.4. Combination TXNIP-IN1, Mito-Tempo, and ML-SA1 (TMM) Treatment Induces Transcription Factor TFEB and PGC1α Nuclear Localization in rMC1 Cells under High Glucose Conditions
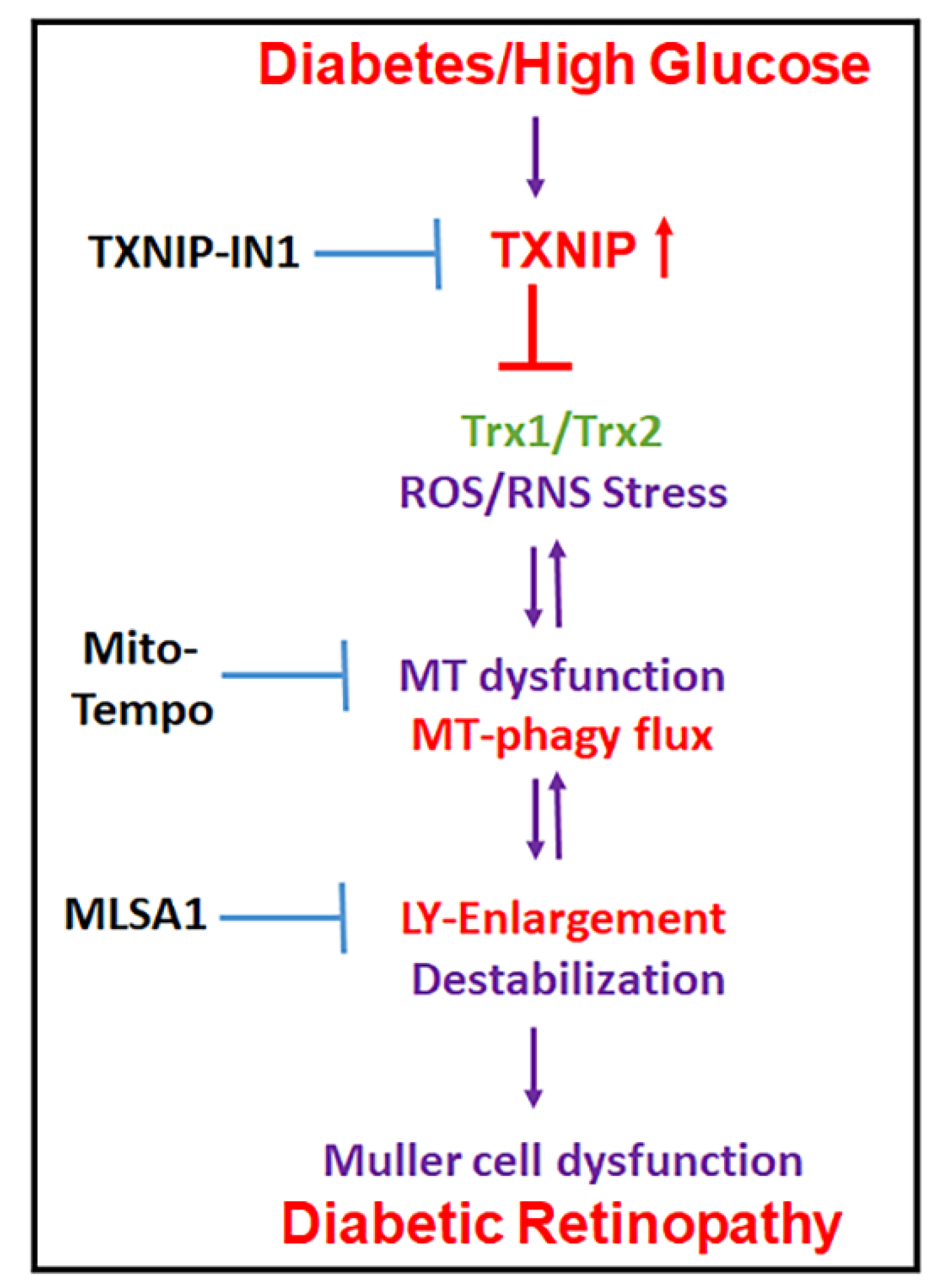
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoon, M.; Okawa, H.; Santina, L.D.; Wong, R.L. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheskey, S.R.; Antonetti, D.A.; Rentería, R.C.; Lin, C.M. Correlation of retinal rtructure and visual function assessments in mouse diabetes models. Investig. Ophthalmol. Vis. Sci. 2021, 62, 20. [Google Scholar] [CrossRef] [PubMed]
- Tonade, D.; Kern, T.S. Photoreceptor cells and RPE contribute to the development of diabetic retinopathy. Prog. Retin. Eye Res. 2021, 83, 100919. [Google Scholar] [CrossRef]
- Hazim, R.A.; Williams, D.S. Cell Culture Analysis of the Phagocytosis of Photoreceptor Outer Segments by Primary Mouse RPE Cells. Methods Mol. Biol. 2018, 1753, 63–71. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Muller Cell Metabolic Signatures: Evolutionary Conservation and Disruption in Disease. Trends Endocrinol. Metab. 2020, 31, 320–329. [Google Scholar] [CrossRef]
- Dyer, M.A.; Cepko, C.L. Control of Muller glial cell proliferation and activation following retinal injury. Nat. Neurosci. 2000, 3, 873–880. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.L.; Terasaki, T.; Singh, L.P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010, 1, e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deliyanti, D.; Zhang, Y.; Khong, F.; Berka, D.R.; Stapleton, D.I.; Kelly, D.J.; Wilkinson-Berka, J.L. FT011, a Novel Cardiorenal Protective Drug, Reduces Inflammation, Gliosis and Vascular Injury in Rats with Diabetic Retinopathy. PLoS ONE 2015, 10, e0134392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.W.; Jiang, Y.; Shalev, A.; Kowluru, R.; Crook, E.D.; Singh, L.P. An analysis of high glucose and glucosamine-induced gene expression and oxidative stress in renal mesangial cells. Arch. Physiol. Biochem. 2006, 112, 189–218. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.P. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Perrone, L.; Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef]
- Tanito, M.; Agbaga, M.P.; Anderson, R.E. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic. Biol. Med. 2007, 42, 1838–1850. [Google Scholar] [CrossRef]
- Yumnamcha, T.; Devi, T.S.; Singh, L.P. Auranofin Mediates Mitochondrial Dysregulation and Inflammatory Cell Death in Human Retinal Pigment Epithelial Cells: Implications of Retinal Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1065. [Google Scholar] [CrossRef]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular matrix remodeling regulates glucose metabolism through TXNIP destabilization. Cell 2018, 175, 117–132.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Min Kim, S.; Dotimas, J.; Li, L.; Feener, E.P.; Baldus, S.; Myers, R.B.; Chutkow, W.A.; Patwari, P.; Yoshioka, J.; et al. Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Mol. Med. 2014, 6, 732–743. [Google Scholar] [CrossRef]
- Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Critical role of TXNIP in oxidative stress, DNA damage and retinal pericyte apoptosis under high glucose: Implications for diabetic retinopathy. Exp. Cell Res. 2013, 319, 1001–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, T.S.; Lee, I.; Hüttemann, M.; Kumar, A.; Nantwi, K.D.; Singh, L.P. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Müller glia under chronic hyperglycemia: Implications for diabetic retinopathy. Exp. Diabetes Res. 2012, 2012, 438238. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef] [Green Version]
- Singh, L.P.; Devi, T.S.; Yumnamcha, T. The Role of Txnip in Mitophagy Dysregulation and Inflammasome Activation in Diabetic Retinopathy: A New Perspective. JOJ Ophthalmol. 2017, 4, 555643. [Google Scholar] [CrossRef]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef] [PubMed]
- Lysyk, L.; Brassard, R.; Touret, N.; Lemieux, M.J. PARL Protease: A Glimpse at Intramembrane Proteolysis in the Inner Mitochondrial Membrane. J. Mol. Biol. 2020, 432, 5052–5062. [Google Scholar] [CrossRef]
- Goudarzi, S.; Hosseini, A.; Abdollahi, M.; Haghi-Aminjan, H. Insights into Parkin-Mediated Mitophagy in Alzheimer’s Disease: A Systematic Review. Front. Aging Neurosci. 2021, 13, 674071. [Google Scholar] [CrossRef]
- Liu, Y.T.; Sliter, D.A.; Shammas, M.K.; Huang, X.; Wang, C.; Calvelli, H.; SMaric, D.; Narendra, P.D. Mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC. Autophagy 2021, 8, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, L.P.; Yumnamcha, T.; Devi, T.S. Mitophagic flux deregulation, lysosomal destabilization and NLRP3 inflammasome activation in diabetic retinopathy: Potentials of gene therapy targeting TXNIP and the edox system. Ophthalmol. Res. Rep. 2018, 3, ORRT-126. [Google Scholar] [PubMed]
- Ko, M.S.; Yun, J.Y.; Baek, I.J.; Jang, J.E.; Hwang, J.J.; Lee, S.E.; Heo, S.H.; Bader, D.A.; Lee, C.H.; Han, J.; et al. Mitophagy deficiency increases NLRP3 to induce brown fat dysfunction in mice. Autophagy 2021, 17, 1205–1221. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring in vivo mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosignol, I.; Villarejo-Zori, B.; Teresak, P.; Sierra-Filardi, E.; Pereiro, X.; Rodríguez-Muela, N.; Vecino, E.; Vieira, H.L.A.; Bell, K.; Boya, P. The mito-QC reporter for quantitative mitophagy assessment in primary retinal ganglion cells and experimental glaucoma models. Int. J. Mol. Sci. 2020, 21, 1882. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Hama, H.; Nagasawa, K.; Kurokawa, H.; Sugiyama, M.; Ando, R.; Funata, M.; Yoshida, N.; Homma, M.; Nishimura, T.; et al. Visualizing and modulating mitophagy for therapeutic studies of neurodegeneration. Cell 2020, 181, 1176–1187.e16. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, S.; Sohn, H.Y.; Koh, Y.H.; Jo, C. TFEB activates Nrf2 by repressing its E3 ubiquitin ligase DCAF11 and promoting phosphorylation of p62. Sci. Rep. 2019, 9, 14354. [Google Scholar] [CrossRef]
- Evans, T.D.; Zhang, X.; Jeong, S.J.; He, A.; Song, E.; Bhattacharya, S.; Holloway, K.B.; Lodhi, I.J.; Razani, B. TFEB drives PGC-1α expression in adipocytes to protect against diet-induced metabolic dysfunction. Sci. Signal. 2019, 12, eaau2281. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.Y.; Alamri, A.H.; Valapala, M. Nutrient deprivation and lysosomal stress induce activation of TFEB in retinal pigment epithelial cells. Cell. Mol. Biol. Lett. 2019, 24, 33. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Barrios, A.; Álvarez, L.; García, M.; Artime, E.; Pereiro, R.; González-Iglesias, H. Antioxidant Defenses in the Human Eye: A focus on metallothioneins. Antioxidants 2021, 10, 89. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.M.; Tewari, S.; Goldberg, A.F.; Kowluru, R.A. Mitochondrial biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011, 51, 1849–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thounaojam, M.C.; Jadeja, R.N.; Warren, M.; Powell, F.L.; Raju, R.; Gutsaeva, D.; Khurana, S.; Martin, P.M.; Bartoli, M. MicroRNA-34a (miR-34a) mediates retinal endothelial cell premature senescence through mitochondrial dysfunction and loss of antioxidant activities. Antioxidants 2019, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. A new human blood-retinal barrier model based on endothelial cells, pericytes, and astrocytes. Int. J. Mol. Sci. 2020, 21, 1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theofani, E.; Xanthou, G. Autophagy: A friend or foe in allergic asthma? Int. J. Mol. Sci. 2021, 22, 6314. [Google Scholar] [CrossRef]
- Pickles, S.; Pierre Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef] [PubMed]
- Rigo, A.; Ferrarini, I.; Lorenzetto, E.; Darra, E.; Liparulo, I.; Bergamini, C.; Sissa, C.; Cavalieri, E.; Vinante, F. BID and the α-bisabolol-triggered cell death program: Converging on mitochondria and lysosomes. Cell Death Dis. 2019, 10, 889. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [Green Version]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef] [Green Version]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [Green Version]
- Humphries, F.; Bergin, R.; Jackson, R.; Delagic, N.; Wang, B.; Yang, S.; Dubois, A.V.; Ingram, R.J.; Moynagh, P.N. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 2018, 9, 1560. [Google Scholar] [CrossRef] [PubMed]
- Akther, M.; Haque, M.E.; Park, J.; Kang, T.B.; Lee, K.H. NLRP3 Ubiquitination-A New Approach to Target NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2021, 22, 8780. [Google Scholar] [CrossRef] [PubMed]
- Eastlake, K.; Heywood, W.E.; Banerjee, P.; Bliss, E.; Mills, K.; Khaw, P.T.; Charteris, D.; Limb, G.A. Comparative proteomic analysis of normal and gliotic PVR retina and contribution of Müller glia to this profile. Exp. Eye Res. 2018, 177, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Hippert, C.; Graca, A.B.; Basche, M.; Kalargyrou, A.A.; Georgiadis, A.; Ribeiro, J.; Matsuyama, A.; Aghaizu, N.; Bainbridge, J.W.; Smith, A.J.; et al. RNAi-mediated suppression of vimentin or glial fibrillary acidic protein prevents the establishment of Müller glial cell hypertrophy in progressive retinal degeneration. Glia 2021, 69, 2272–2290. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; De la Cruz-Ojeda, P.; De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; De Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial dynamics in mitochondrial diseases. Diseases 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, L.P.; Devi, T.S. Potential Combination Drug Therapy to Prevent Redox Stress and Mitophagy Dysregulation in Retinal Müller Cells under High Glucose Conditions: Implications for Diabetic Retinopathy. Diseases 2021, 9, 91. https://doi.org/10.3390/diseases9040091
Singh LP, Devi TS. Potential Combination Drug Therapy to Prevent Redox Stress and Mitophagy Dysregulation in Retinal Müller Cells under High Glucose Conditions: Implications for Diabetic Retinopathy. Diseases. 2021; 9(4):91. https://doi.org/10.3390/diseases9040091
Chicago/Turabian StyleSingh, Lalit Pukhrambam, and Takhellambam S. Devi. 2021. "Potential Combination Drug Therapy to Prevent Redox Stress and Mitophagy Dysregulation in Retinal Müller Cells under High Glucose Conditions: Implications for Diabetic Retinopathy" Diseases 9, no. 4: 91. https://doi.org/10.3390/diseases9040091
APA StyleSingh, L. P., & Devi, T. S. (2021). Potential Combination Drug Therapy to Prevent Redox Stress and Mitophagy Dysregulation in Retinal Müller Cells under High Glucose Conditions: Implications for Diabetic Retinopathy. Diseases, 9(4), 91. https://doi.org/10.3390/diseases9040091