
Genomic Evaluation of the Genus Coltivirus Indicates Genetic Diversity among Colorado Tick Fever Virus Strains and Demarcation of a New Species


 , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses and RNA
2.2. Genome Sequencing and Assembly
2.3. Molecular and Phylogenetic Analysis
2.4. Serological Evaluation
3. Results
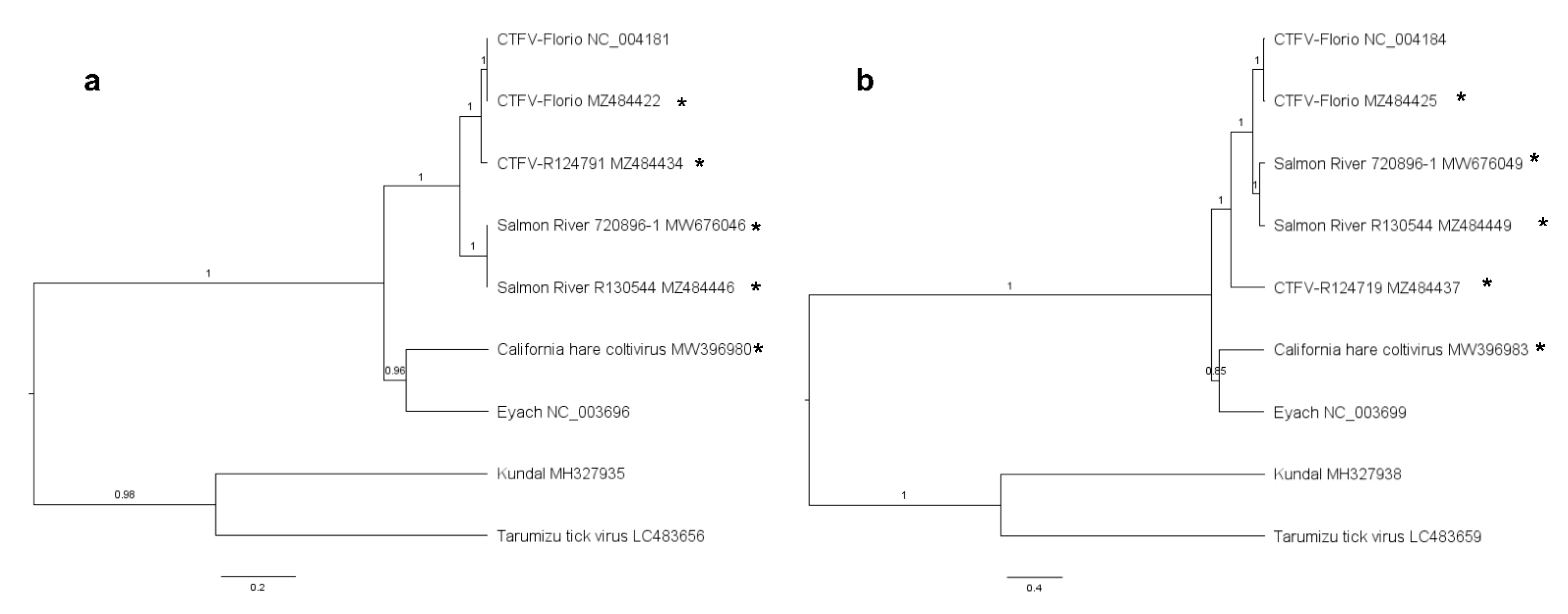
3.1. Phylogenetic Construction of the Genus Coltivirus
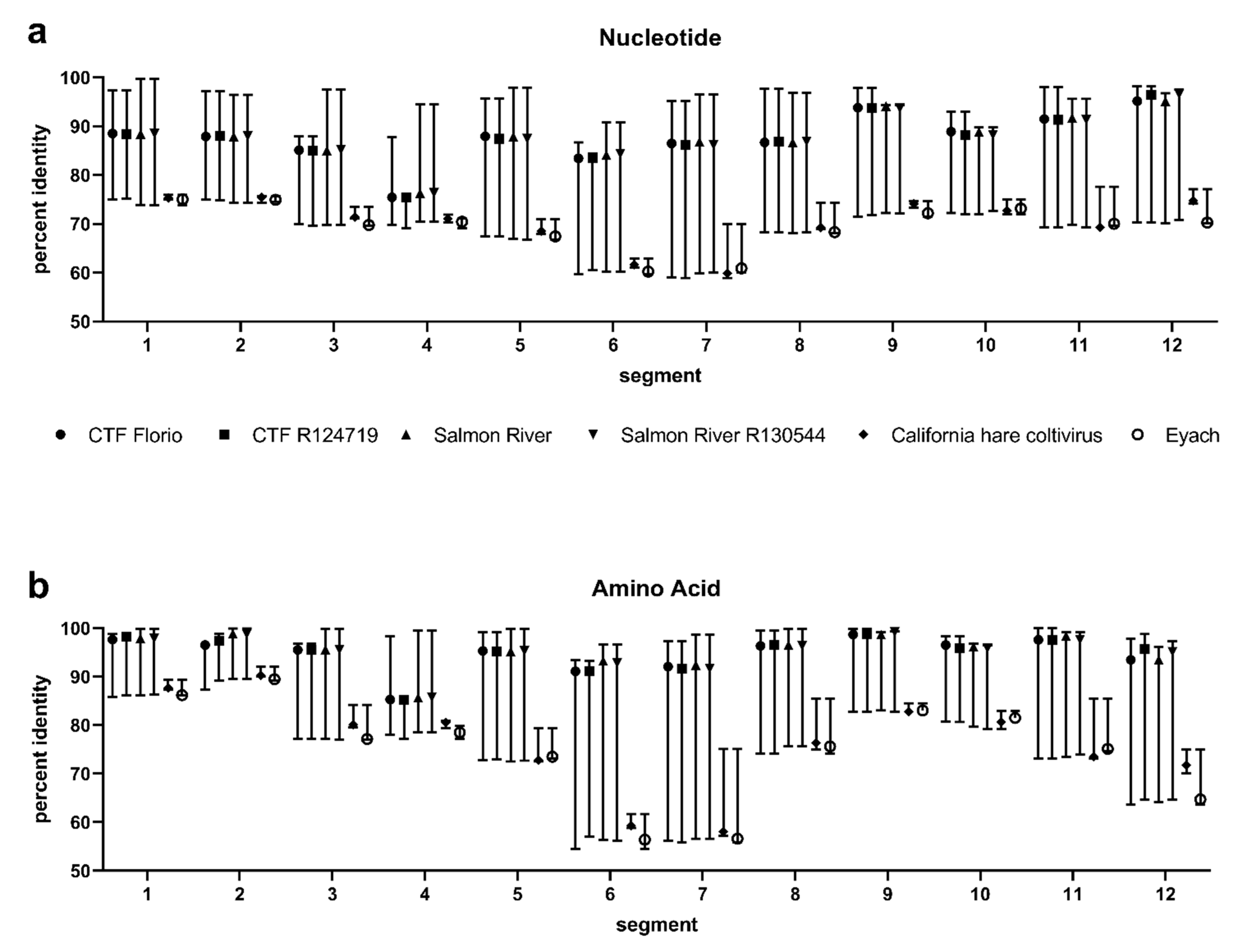
3.2. Segment Analysis
3.3. Evolutionary Distances
3.4. Serological Evaluation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: London, UK, 2011; Volume 9. [Google Scholar]
- Attoui, H.; Billoir, F.; Biagini, P.; de Micco, P.; de Lamballerie, X. Complete sequence determination and genetic analysis of Banna virus and Kadipiro virus: Proposal for assignment to a new genus (Seadornavirus) within the family Reoviridae. J. Gen. Virol. 2000, 81 Pt 6, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Attoui, H.; Billoir, F.; Biagini, P.; Cantaloube, J.F.; de Chesse, R.; de Micco, P.; de Lamballerie, X. Sequence determination and analysis of the full-length genome of colorado tick fever virus, the type species of genus Coltivirus (Family Reoviridae). Biochem. Biophys. Res. Commun. 2000, 273, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, F.M.; Attoui, H.; de Micco, P.; de Lamballerie, X. Termination and read-through proteins encoded by genome segment 9 of Colorado tick fever virus. J. Gen. Virol. 2004, 85 Pt 8, 2237–2244. [Google Scholar] [CrossRef]
- Florio, L.; Miller, M.S.; Mugrage, E.R. Colorado tick fever; isolation of the virus from Dermacentar andersoni in Nature and a laboratory study of the transmission of the virus in the tick. J. Immunol. 1950, 64, 257–263. [Google Scholar] [PubMed]
- Florio, L.; Stewart, M.O.; Mugrage, E.R. The etiology of Colorado tick fever. J. Exp. Med. 1946, 83, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Karabatsos, N.; Lanciotti, R.S. Detection of Colorado tick fever virus by using reverse transcriptase PCR and application of the technique in laboratory diagnosis. J. Clin. Microbiol. 1997, 35, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Karabatsos, N.; Poland, J.D.; Emmons, R.W.; Mathews, J.H.; Calisher, C.H.; Wolff, K.L. Antigenic variants of Colorado tick fever virus. J. Gen. Virol. 1987, 68 Pt 5, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Rehse-Kupper, B.; Casals, J.; Rehse, E.; Ackermann, R. Eyach--an arthropod-borne virus related to Colorado tick fever virus in the Federal Republic of Germany. Acta Virol. 1976, 20, 339–342. [Google Scholar] [PubMed]
- Chastel, C.; Main, A.J.; Couatarmanac’h, A.; Le Lay, G.; Knudson, D.L.; Quillien, M.C.; Beaucournu, J.C. Isolation of Eyach virus (Reoviridae, Colorado tick fever group) from Ixodes ricinus and I. ventalloi ticks in France. Arch. Virol. 1984, 82, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.D.; Whitmer, S.L.M.; Sarkale, P.; Fei Fan Ng, T.; Goldsmith, C.S.; Nyayanit, D.A.; Esona, M.D.; Shrivastava-Ranjan, P.; Lakra, R.; Pardeshi, P.; et al. Characterization of Novel Reoviruses Wad Medani Virus (Orbivirus) and Kundal Virus (Coltivirus) Collected from Hyalomma anatolicum Ticks in India during Surveillance for Crimean Congo Hemorrhagic Fever. J. Virol. 2019, 93, e00106-19. [Google Scholar] [CrossRef]
- Fujita, R.; Ejiri, H.; Lim, C.K.; Noda, S.; Yamauchi, T.; Watanabe, M.; Kobayashi, D.; Takayama-Ito, M.; Murota, K.; Posadas-Herrera, G.; et al. Isolation and characterization of Tarumizu tick virus: A new coltivirus from Haemaphysalis flava ticks in Japan. Virus Res. 2017, 242, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Dabrowski, P.W.; Kurth, A.; Leendertz, S.A.J.; Leendertz, F.H. A novel Coltivirus-related virus isolated from free-tailed bats from Cote d’Ivoire is able to infect human cells in vitro. Virol. J. 2017, 14, 181. [Google Scholar] [CrossRef]
- Yendell, S.J.; Fischer, M.; Staples, J.E. Colorado tick fever in the United States, 2002–2012. Vector-Borne Zoonotic Dis. 2015, 15, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Stapels, J.E.; Fischer, M. Coltiviruses (Colorado Tick Fever). In Principles and Practice of Pediatric Infectious Diseases, 5th ed.; Long, S.S., Prober, G.C., Fischer, M., Eds.; Elsevier: Philadelphia, PA, USA, 2018; pp. 1119–1121. [Google Scholar]
- McDonald, E.; George, D.; Rekant, S.; Curren, E.; DeBess, E.; Hedberg, K.; Lutz, J.; Faith, J.; Kaisner, H.; Fawcett, R.; et al. Notes from the Field: Investigation of Colorado Tick Fever Virus Disease Cases—Oregon, 2018. Morb. Mortal. Wkly. Rep. 2019, 68, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, A.H.; Delwart, E.; Diaz-Munoz, S.L. Next-generation sequencing of dsRNA is greatly improved by treatment with the inexpensive denaturing reagent DMSO. Microb. Genom. 2019, 5, e000315. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Baele, G.; Lemey, P.; Suchard, M.A. Genealogical Working Distributions for Bayesian Model Testing with Phylogenetic Uncertainty. Syst. Biol. 2016, 65, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Maljkovic Berry, I.; Melendrez, M.C.; Li, T.; Hawksworth, A.W.; Brice, G.T.; Blair, P.J.; Halsey, E.S.; Williams, M.; Fernandez, S.; Yoon, I.K.; et al. Frequency of influenza H3N2 intra-subtype reassortment: Attributes and implications of reassortant spread. BMC Biol. 2016, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Attoui, H.; Mohd Jaafar, F.; Biagini, P.; Cantaloube, J.F.; de Micco, P.; Murphy, F.A.; de Lamballerie, X. Genus Coltivirus (family Reoviridae): Genomic and morphologic characterization of Old World and New World viruses. Arch. Virol. 2002, 147, 533–561. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.S.; Emmons, R.W.; Devlin, V.; Dondero, D.V.; Nelson, B.C. Survey for evidence of Colorado tick fever virus outside of the known endemic area in California. Am. J. Trop. Med. Hyg. 1982, 31, 837–843. [Google Scholar] [CrossRef]
- Chastel, C. Erve and Eyach: Two viruses isolated in France, neuropathogenic for man and widely distributed in Western Europe. Bull. Acad. Natl. Med. 1998, 182, 801–809. [Google Scholar] [PubMed]
- Brown, S.E.; Miller, B.R.; McLean, R.G.; Knudson, D.L. Co-circulation of multiple Colorado tick fever virus genotypes. Am. J. Trop. Med. Hyg. 1989, 40, 94–101. [Google Scholar] [CrossRef]
- Williamson, B.N.; Fischer, R.J.; Lopez, J.E.; Ebihara, H.; Schwan, T.G. Prevalence and Strains of Colorado Tick Fever Virus in Rocky Mountain Wood Ticks in the Bitterroot Valley, Montana. Vector-Borne Zoonotic Dis. 2019, 19, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Bodkin, D.K.; Knudson, D.L. Genetic relatedness of Colorado tick fever virus isolates by RNA-RNA blot hybridization. J. Gen. Virol. 1987, 68 Pt 4, 1199–1204. [Google Scholar] [CrossRef]
- Suzuki, N.; Supyani, S.; Maruyama, K.; Hillman, B.I. Complete genome sequence of Mycoreovirus-1/Cp9B21, a member of a novel genus within the family Reoviridae, isolated from the chestnut blight fungus Cryphonectria parasitica. J. Gen. Virol. 2004, 85 Pt 11, 3437–3448. [Google Scholar] [CrossRef]
- Attoui, H.; Charrel, R.N.; Billoir, F.; Cantaloube, J.F.; de Micco, P.; de Lamballerie, X. Comparative sequence analysis of American, European and Asian isolates of viruses in the genus Coltivirus. J. Gen. Virol. 1998, 79 Pt 10, 2481–2489. [Google Scholar] [CrossRef][Green Version]
- Forshey, B.M.; Castillo, R.M.; Hang, J. Group C orthobunyavirus genomic sequences require validation. J. Virol. 2014, 88, 3052–3053. [Google Scholar] [CrossRef]
- Nunes, M.R.; Travassos da Rosa, A.P.; Weaver, S.C.; Tesh, R.B.; Vasconcelos, P.F. Reply to “Group C orthobunyavirus genomic sequences require validation”. J. Virol. 2014, 88, 3054. [Google Scholar] [CrossRef] [PubMed][Green Version]



{kind=link}
{kind=link}
| Virus | Isolate Designation | Date | Location | Passage | GenBank No. |
|---|---|---|---|---|---|
| Colorado tick fever | Florio | 1943 | Colorado | SM14 | MZ484422-33 |
| Colorado tick fever | R124719 | 2017 | Wyoming | Vero 1 | MZ484434-45 |
| Salmon River | 720896-1 | 1990 | Idaho | SM3 | MW67046-57 |
| Salmon River | R130544 | 2019 | Oregon | Vero 1 | MZ484446-57 |
| California hare coltivirus | S6-14-03 | 1976 | California | SM5 | MW396980-91 |
| Virus Nucleotide Open Reading Frame Size (Segment Size) | |||||||
|---|---|---|---|---|---|---|---|
| Segment | CTFV-Florio-RefSeq 1 | CTFV-Florio-ARC 2 | CTFV R124719 | Salmon River | Salmon River R130544 | California Hare Coltivirus | Eyach |
| 1 | 4308 (4350) | 4308 (4350) | 4308 (4350) | 4308 (4349) | 4308 (4349) | 4308 (4349) | 4308 (4349) |
| 2 | 3630 4 (3909) | 3804 (3909) | 3804 (3909) | 3804 (3908) | 3804 (3908) | 3858 (3964) | 3828 (3934) |
| 3 | 3549 (3586) | 3549 (3586) | 3549 (3586) | 3549 (3585) | 3549 (3585) | 3549 (3585) | 3549 (3585) |
| 4 | 3084 (3157) | 3084 (3156) | 3087 (3159) | 3084 (3156) | 3084 (3156) | 3084 (3156) | 3084 (3156) |
| 5 | 2256 (2432) | 2256 (2432) | 2256 (2432) | 2256 (2431) | 2256 (2431) | 2256 (2419) | 2256 (2398) |
| 6 3 | 2094 (2141) | 2094 (2141) | 2094 (2141) | 2094 (2140) | 2094 (2140) | 2097 (2175) | 2100 (2178) |
| 7 | 2055 (2133) | 2055 (2133) | 2055 (2133) | 2055 (2133) | 2055 (2132) | 2094 (2139) | 2094 (2139) |
| 8 | 1983 (2029) | 1983 (2029) | 1983 (2029) | 1983 (2028) | 1983 (2028) | 1983 (2029) | 1983 (2028) |
| 9 | 1809 (1884) | 1809 (1884) | 1809 (1884) | 1809 (1884) | 1809 (1884) | 1809 (1884) | 1809 (1884) |
| 10 | 1818 (1880) | 1818 (1880) | 1818 (1880) | 1818 (1880) | 1818 (1880) | 1818 (1879) | 1818 (1879) |
| 11 | 750 (998) | 927 (1002) | 927 (1002) | 927 (1002) | 927 (1002) | 927 (1002) | 927 (1002) |
| 12 | 558 (675) | 558 (675) | 558 (676) | 558 (675) | 558 (675) | 558 (678) | 555 (678) |
| Plaque Reduction Neutralization Test 90% | ||||
|---|---|---|---|---|
| Hyperimmune Fluid Antibody | Salmon River Virus 1 | CTFV-Florio | California Hare Coltivirus | Eyach Virus 3 |
| Salmon River | 6402 | 320 | <10 | 10 |
| CTFV Florio | 640 | 5120 | <10 | 10 |
| California hare coltivirus | 20 | <10 | 160 | <10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, H.R.; Velez, J.O.; Fitzpatrick, K.; Davis, E.H.; Russell, B.J.; Lambert, A.J.; Staples, J.E.; Brault, A.C. Genomic Evaluation of the Genus Coltivirus Indicates Genetic Diversity among Colorado Tick Fever Virus Strains and Demarcation of a New Species. Diseases 2021, 9, 92. https://doi.org/10.3390/diseases9040092
Hughes HR, Velez JO, Fitzpatrick K, Davis EH, Russell BJ, Lambert AJ, Staples JE, Brault AC. Genomic Evaluation of the Genus Coltivirus Indicates Genetic Diversity among Colorado Tick Fever Virus Strains and Demarcation of a New Species. Diseases. 2021; 9(4):92. https://doi.org/10.3390/diseases9040092
Chicago/Turabian StyleHughes, Holly R., Jason O. Velez, Kelly Fitzpatrick, Emily H. Davis, Brandy J. Russell, Amy J. Lambert, J. Erin Staples, and Aaron C. Brault. 2021. "Genomic Evaluation of the Genus Coltivirus Indicates Genetic Diversity among Colorado Tick Fever Virus Strains and Demarcation of a New Species" Diseases 9, no. 4: 92. https://doi.org/10.3390/diseases9040092
APA StyleHughes, H. R., Velez, J. O., Fitzpatrick, K., Davis, E. H., Russell, B. J., Lambert, A. J., Staples, J. E., & Brault, A. C. (2021). Genomic Evaluation of the Genus Coltivirus Indicates Genetic Diversity among Colorado Tick Fever Virus Strains and Demarcation of a New Species. Diseases, 9(4), 92. https://doi.org/10.3390/diseases9040092