Pulmonary Arterial Hypertension: Pathophysiology and Treatment


Abstract
1. Introduction
2. Risk and Prognostic Factors of PAH
3. Genetics
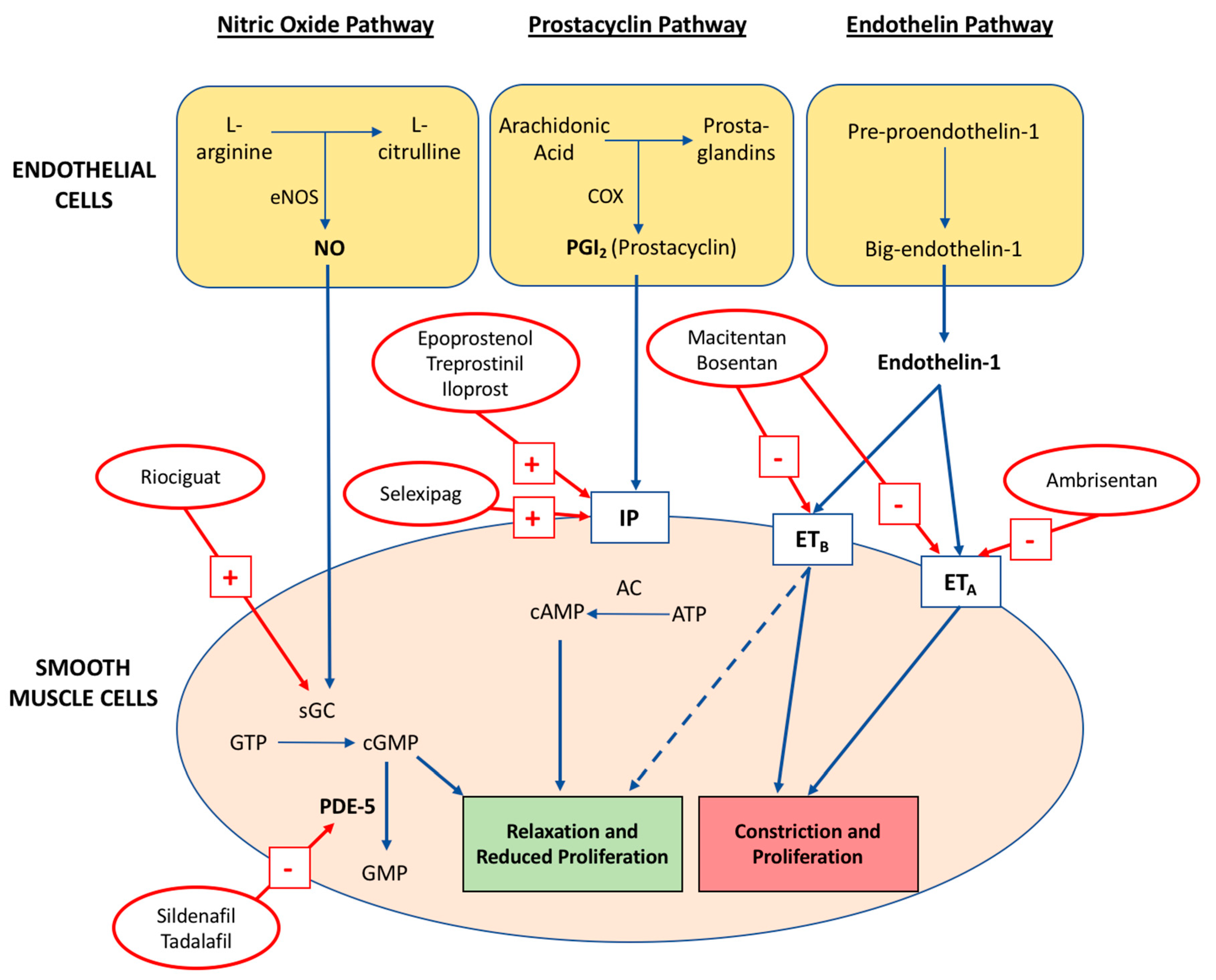
4. Pathophysiology
4.1. Nitric Oxide Pathway
4.2. Prostacyclin-Thromboxane A2 Pathway
4.3. Endothelin-1 Pathway
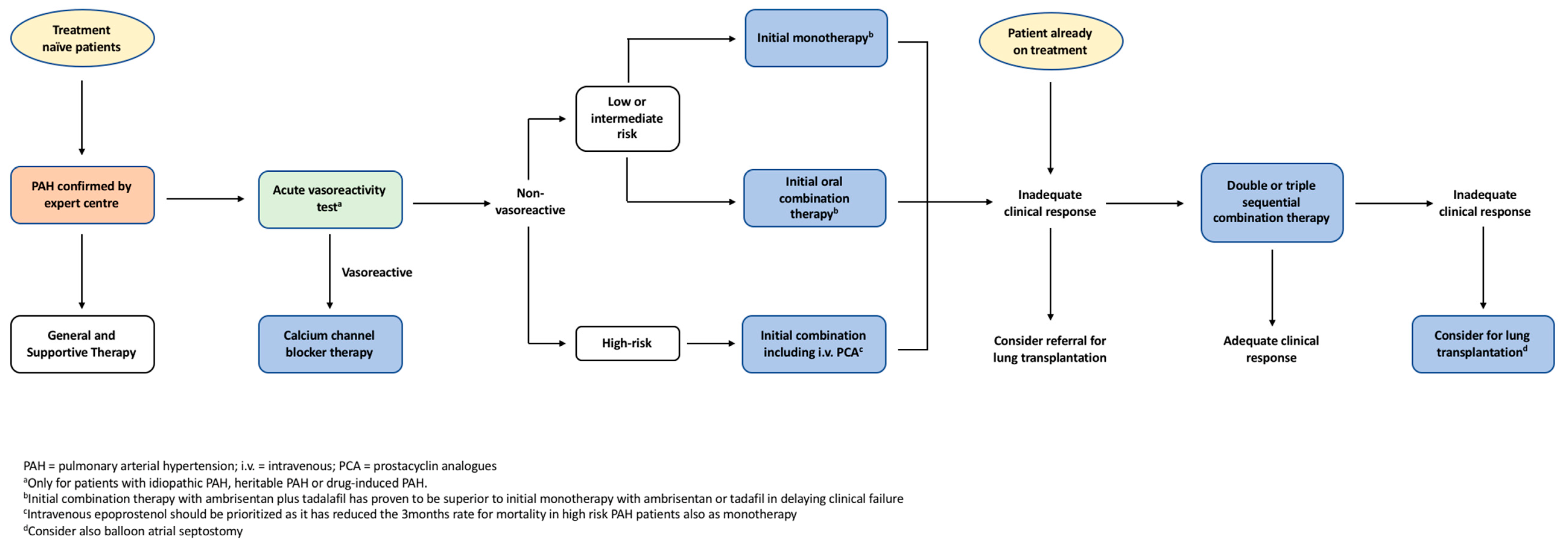
5. Management of PAH
5.1. General and Supportive Therapies
5.2. Calcium Channel Blockers
5.3. Targeted Therapies
5.4. Phosphodiesterase-5 Inhibitors
5.5. Guanylate Cyclase Stimulators
5.6. Prostacyclin Analogues
5.7. Prostacyclin Receptor Agonists
5.8. Endothelin Receptor Antagonists (ERA)
5.9. Combination Therapies
5.10. Clinical Approach to PAH Treatment
5.11. Future Research
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Moreira, E.M.; Gall, H.; Leening, M.J.; Lahousse, L.; Loth, D.W.; Krijthe, B.P.; Kiefte-de Jong, J.C.; Brusselle, G.G.; Hofman, A.; Stricker, B.H. Prevalence of pulmonary hypertension in the general population: The rotterdam study. PLoS ONE 2015, 10, e0130072. [Google Scholar] [CrossRef] [PubMed]
- Strange, G.; Playford, D.; Stewart, S.; Deague, J.A.; Nelson, H.; Kent, A.; Gabbay, E. Pulmonary hypertension: Prevalence and mortality in the armadale echocardiography cohort. Heart 2012, 98, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M. Pulmonary arterial hypertension: Baseline characteristics from the reveal registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Archer, S.L.; Dorfmüller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Ryan, J.J.; Archer, S.L. Evolving epidemiology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Health and Social Care Information Centre. Fifth Annual Report: Key Findings from the National Audit of Pulmonary Hypertension for the United Kingdom, Channel Islands, Gibraltar and Isle of Man; Health and Social Care Information Centre: Leeds, UK, 2015. [Google Scholar]
- Shapiro, S.; Traiger, G.L.; Turner, M.; McGoon, M.D.; Wason, P.; Barst, R.J. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest 2012, 141, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the reveal registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Sanchez, M.A.G.; Kumar, R.K.; Landzberg, M.; Machado, R.F. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Robbins, I.M.; Beghetti, M.; Channick, R.N.; Delcroix, M.; Denton, C.P.; Elliott, C.G.; Gaine, S.P.; Gladwin, M.T.; Jing, Z.-C. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2009, 54, S43–S54. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. Genetics and mediators in pulmonary arterial hypertension. Clin. Chest Med. 2007, 28, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 esc/ers guidelines for the diagnosis and treatment of pulmonary hypertensionthe joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (ESC) and the european respiratory society (ERS): Endorsed by: Association for european paediatric and congenital cardiology (AEPC), international society for heart and lung transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Houtchens, J.; Martin, D.; Klinger, J.R. Diagnosis and management of pulmonary arterial hypertension. Pulm. Med. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Eickelberg, O.; Elliott, C.G.; Geraci, M.W.; Hanaoka, M.; Loyd, J.E.; Newman, J.H.; Phillips, J.A.; Soubrier, F.; Trembath, R.C. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S32–S42. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in bmpr2, encoding a tgf-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.E.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.; Rowell, J.; Machado, R.D.; Elliott, C.; Robbins, I.; Olschewski, H.; McLaughlin, V. Molecular and functional analysis identifies alk-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Chaouat, A.; Coulet, F.; Favre, C.; Simonneau, G.; Weitzenblum, E.; Soubrier, F.; Humbert, M. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004, 59, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.A.; Vijayakrishnan, J.; James, V.; Soubrier, F.; Gomez-Sanchez, M.A.; Martensson, G.; Galie, N.; Manes, A.; Corris, P.; Simonneau, G. Bmpr2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 212–213. [Google Scholar] [CrossRef] [PubMed]
- Cogan, J.D.; Pauciulo, M.W.; Batchman, A.P.; Prince, M.A.; Robbins, I.M.; Hedges, L.K.; Stanton, K.C.; Wheeler, L.A.; Phillips, J.A., III; Loyd, J.E. High frequency of bmpr2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A., III; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Phillips, J.A.; Cogan, J.D.; Hamid, R.; Yu, C.; Stanton, K.C.; Phillips, C.A.; Wheeler, L.A.; Robbins, I.M.; Newman, J.H. Truncating and missense bmpr2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir. Res. 2009, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Jones, P.L.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Fagan, K.; Steudel, W.; Fouty, B.; Lane, K.; Harral, J.; Hoedt-Miller, M.; Tada, Y.; Ozimek, J.; Tuder, R. Pulmonary hypertension in transgenic mice expressing a dominant-negative bmprii gene in smooth muscle. Circ. Res. 2004, 94, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.; Bonnet, S.; Chan, S.Y. Translational advances in the field of pulmonary hypertension. Translating microrna biology in pulmonary hypertension. It will take more than “miR” words. Am. J. Respir. Crit. Care Med. 2017, 195, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial dysfunction in pulmonary hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Prior, D.L.; Adams, H.; Williams, T.J. Update on pharmacotherapy for pulmonary hypertension. Med. J. Aust. 2016, 205, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-Y.; Zhao, Y.D.; Mirza, M.K.; Huang, J.H.; Potula, H.-H.S.; Vogel, S.M.; Brovkovych, V.; Yuan, J.X.-J.; Wharton, J.; Malik, A.B. Persistent enos activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through pkg nitration. J. Clin. Investig. 2009, 119, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N. Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Hoshikawa, Y.; Voelkel, N.F.; Gesell, T.L.; Moore, M.D.; Morris, K.G.; Alger, L.A.; Narumiya, S.; Geraci, M.W. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am. J. Respir. Crit. Care Med. 2001, 164, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med. 2012, 52, 1970–1986. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann. Intern. Med. 1991, 114, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.; Markevych, I.; Spiekerkoetter, E.; Welte, T.; Niedermeyer, J. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur. Respir. J. 2005, 26, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, A.C.; Lauzière, G.; Lega, J.-C.; Lacasse, Y.; Martin, S.; Simard, S.; Bonnet, S.; Provencher, S. Combination therapy versus monotherapy for pulmonary arterial hypertension: A meta-analysis. Lancet Respir. Med. 2016, 4, 291–305. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Gomberg-Maitland, M.; Frantz, R.P.; Foreman, A.J.; Coffey, C.S.; Frost, A.; Barst, R.J.; Badesch, D.B.; Elliott, C.G. Predicting survival in pulmonary arterial hypertension. Circulation 2010, 122, 164–172. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Gaine, S.P.; Howard, L.S.; Leuchte, H.H.; Mathier, M.A.; Mehta, S.; Palazzini, M.; Park, M.H.; Tapson, V.F.; Sitbon, O. Treatment goals of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D73–D81. [Google Scholar] [CrossRef] [PubMed]
- Mereles, D.; Ehlken, N.; Kreuscher, S.; Ghofrani, S.; Hoeper, M.M.; Halank, M.; Meyer, F.J.; Karger, G.; Buss, J.; Juenger, J. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation 2006, 114, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.; Chin, L.M.; Kennedy, M.; Woolstenhulme, J.G.; Nathan, S.D.; Weinstein, A.A.; Connors, G.; Weir, N.A.; Drinkard, B.; Lamberti, J. Benefits of intensive treadmill exercise training on cardiorespiratory function and quality of life in patients with pulmonary hypertension. Chest 2013, 143, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.A.; Chin, L.M.; Keyser, R.E.; Kennedy, M.; Nathan, S.D.; Woolstenhulme, J.G.; Connors, G.; Chan, L. Effect of aerobic exercise training on fatigue and physical activity in patients with pulmonary arterial hypertension. Respir. Med. 2013, 107, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.G.; Thomas, S.; Safdar, Z.; Torres, F.; Pacheco, L.D.; Feldman, J. Management of pulmonary arterial hypertension during pregnancy: A retrospective, multicenter experience. Chest 2013, 143, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Ciracì, R.; Tirone, G.; Scaglione, F. The impact of drug–drug interactions on pulmonary arterial hypertension therapy. Pulm. Pharmacol. Ther. 2014, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Theile, D.; Rüppell, M.A.; Speck, T.; Spalwisz, A.; Haefeli, W.E. Interaction profile of macitentan, a new non-selective endothelin-1 receptor antagonist, in vitro. Eur. J. Pharmacol. 2013, 701, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Jaïs, X.; Olsson, K.M.; Barbera, J.A.; Blanco, I.; Torbicki, A.; Peacock, A.; Vizza, C.D.; Macdonald, P.; Humbert, M.; Hoeper, M.M. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur. Respir. J. 2012, 40, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Kiely, D.G.; Cockrill, B.A.; Safdar, Z.; Wilson, V.J.; Al Hazmi, M.; Preston, I.R.; MacLean, M.R.; Lahm, T. Statement on pregnancy in pulmonary hypertension from the pulmonary vascular research institute. Pulm. Circ. 2015, 5, 435–465. [Google Scholar] [CrossRef] [PubMed]
- Bonnin, M.; Mercier, F.J.; Sitbon, O.; Roger-Christoph, S.; Jaïs, X.; Humbert, M.; Audibert, F.; Frydman, R.; Simonneau, G.; Benhamou, D. Severe pulmonary hypertension during pregnancymode of delivery and anesthetic management of 15 consecutive cases. Anesthesiology 2005, 102, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.H.; Lepore, J.J.; Maroo, A.; Semigran, M.J.; Ginns, L.C. Oxygen therapy improves cardiac index and pulmonary vascular resistance in patients with pulmonary hypertension. Chest 2001, 120, 1547–1555. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: A report of the american college of cardiology foundation task force on expert consensus documents and the american heart association developed in collaboration with the american college of chest physicians; american thoracic society, inc.; and the pulmonary hypertension association. J. Am. Coll. Cardiol. 2009, 53, 1573–1619. [Google Scholar] [CrossRef] [PubMed]
- Rich, S.; Seidlitz, M.; Dodin, E.; Osimani, D.; Judd, D.; Genthner, D.; McLaughlin, V.; Francis, G. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest 1998, 114, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Delcroix, M.; Ghofrani, H.-A.; Jansa, P.; Minai, O.; Perchenet, L.; Rubin, L.; Sastry, B.; Torbicki, A.; Simonneau, G. Anticoagulant therapy does not influence long-term outcomes in patients with pulmonary arterial hypertension (PAH): Insights from the randomised controlled SERAPHIN trial of macitentan. Eur. Heart J. 2014, 35, 10. [Google Scholar]
- Herve, P.; Humbert, M.; Sitbon, O.; Parent, F.; Nunes, H.; Legal, C.; Garcia, G.; Simonneau, G. Pathobiology of pulmonary hypertension: The role of platelets and thrombosis. Clin. Chest. Med. 2001, 22, 451–458. [Google Scholar] [CrossRef]
- Huber, K.; Beckmann, R.; Frank, H.; Kneussl, M.; Mlczoch, J.; Binder, B.R. Fibrinogen, t-pa, and pai-1 plasma levels in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1994, 150, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grünig, E.; Staehler, G.; Rosenkranz, S.; Halank, M. Anticoagulation and survival in pulmonary arterial hypertension: Results from the comperative, prospective registry of newly initiated therapies for pulmonary hypertension (COMPERA). Circulation 2013. [Google Scholar] [CrossRef]
- Preston, I.R.; Roberts, K.E.; Miller, D.P.; Sen, G.P.; Selej, M.; Benton, W.W.; Hill, N.S.; Farber, H.W. Effect of warfarin treatment on survival of patients with pulmonary arterial hypertension (PAH) in the registry to evaluate early and long-term PAH disease management (REVEAL). Circulation 2015. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Armstrong, I.; Aldrighetti, R.; Howard, L.S.; Ryftenius, H.; Fischer, A.; Lombardi, S.; Studer, S.; Ferrari, P. Understanding the impact of pulmonary arterial hypertension on patients’ and carers’ lives. Eur. Respir. Rev. 2013, 22, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Löwe, B.; Gräfe, K.; Ufer, C.; Kroenke, K.; Grünig, E.; Herzog, W.; Borst, M.M. Anxiety and depression in patients with pulmonary hypertension. Psychosom. Med. 2004, 66, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Humbert, M.; Jaïs, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Savale, L.; Natali, D.; Jaïs, X.; Herve, P.; Garcia, G.; Humbert, M.; Simonneau, G.; Sitbon, O. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur. Heart J. 2010, 31, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Manes, A.; Negro, L.; Palazzini, M.; Bacchi-Reggiani, M.L.; Branzi, A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur. Heart J. 2009, 30, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, H.; Simonneau, G.; Galiè, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; Nikkho, S.; Speich, R.; Hoeper, M.M.; Behr, J. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.-A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B. Ambrisentan for the treatment of pulmonary arterial hypertension. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Badesch, D.B.; Tapson, V.F.; McGoon, M.D.; Brundage, B.H.; Rubin, L.J.; Wigley, F.M.; Rich, S.; Barst, R.J.; Barrett, P.S.; Kral, K.M. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of diseasea randomized, controlled trial. Ann. Intern. Med. 2000, 132, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Rubin, L.J.; Long, W.A.; McGoon, M.D.; Rich, S.; Badesch, D.B.; Groves, B.M.; Tapson, V.F.; Bourge, R.C.; Brundage, B.H. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N. Engl. J. Med. 1996, 334, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Barst, R.; Robbins, I.; Channick, R.; Galie, N.; Boonstra, A.; Rubin, L.; Horn, E.; Manes, A.; Simonneau, G. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur. Respir. J. 2004, 24, 353–359. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.; Channick, R.N.; Ghofrani, H.-A.; Lemarié, J.-C.; Naeije, R.; Packer, M.; Souza, R.; Tapson, V.F.; Tolson, J.; Al Hiti, H. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur. Respir. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Rubin, L.; Hoeper, M.; Jansa, P.; Al-Hiti, H.; Meyer, G.; Chiossi, E.; Kusic-Pajic, A.; Simonneau, G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): A double-blind, randomised controlled trial. Lancet 2008, 371, 2093–2100. [Google Scholar] [CrossRef]
- Tapson, V.F.; Torres, F.; Kermeen, F.; Keogh, A.M.; Allen, R.P.; Frantz, R.P.; Badesch, D.B.; Frost, A.E.; Shapiro, S.M.; Laliberte, K. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): A randomized controlled trial. Chest 2012, 142, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Tapson, V.F.; Jing, Z.-C.; Xu, K.-F.; Pan, L.; Feldman, J.; Kiely, D.G.; Kotlyar, E.; McSwain, C.S.; Laliberte, K.; Arneson, C. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): A randomized controlled trial. Chest 2013, 144, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; Preiss, R. Selexipag for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Rubin, L.J.; Galie, N.; Barst, R.J.; Fleming, T.R.; Frost, A.E.; Engel, P.J.; Kramer, M.R.; Burgess, G.; Collings, L. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: A randomized trial. Ann. Intern. Med. 2008, 149, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.-A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Mendoza, J.; Hood, M.; McGoon, M.; Barst, R.; Williams, W.B.; Diehl, J.H.; Crow, J.; Long, W. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Ann. Intern. Med. 1990, 112, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.-A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Benza, R.L.; Rubin, L.J.; Channick, R.N.; Voswinckel, R.; Tapson, V.F.; Robbins, I.M.; Olschewski, H.; Rubenfire, M.; Seeger, W. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J. Am. Coll. Cardiol. 2010, 55, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Fleming, T.R.; Galiè, N.; Simonneau, G.; Ghofrani, H.A.; Oakes, M.; Layton, G.; Serdarevic-Pehar, M.; McLaughlin, V.V. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: The super-2 study. Chest 2011, 140, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Oudiz, R.J.; Brundage, B.H.; Galiè, N.; Ghofrani, H.A.; Simonneau, G.; Botros, F.T.; Chan, M.; Beardsworth, A.; Barst, R.J.; Group, P.S. Tadalafil for the treatment of pulmonary arterial hypertension: A double-blind 52-week uncontrolled extension study. J. Am. Coll. Cardiol. 2012, 60, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.; Langleben, D.; Fritsch, A.; Menezes, F. Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur. Respir. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Müller, K.; Scalise, A.-V.; Grünig, E. Patent plus: A blinded, randomised and extension study of riociguat plus sildenafil in pulmonary arterial hypertension. Eur. Respir. J. 2015, 45, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, J.; Thanikachalam, S.; Parikh, K.; Shanmugasundaram, S.; Bangera, S.; Shapiro, L.; Pott, G.B.; Vnencak-Jones, C.L.; Arneson, C.; Wade, M. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: A placebo-controlled trial. J. Heart Lung Transplant. 2010, 29, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Manes, A.; Jais, X.; Pallazini, M.; Humbert, M.; Presotto, L.; de Paillette, L.; Zaccardelli, D.; Davis, G.; Jeffs, R. Rapid switch from intravenous epoprostenol to intravenous treprostinil in patients with pulmonary arterial hypertension. J. Cardiovasc. Pharmacol. Ther. 2007, 49, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tapson, V.F.; Gomberg-Maitland, M.; McLaughlin, V.V.; Benza, R.L.; Widlitz, A.C.; Krichman, A.; Barst, R.J. Safety and efficacy of iv treprostinil for pulmonary arterial hypertension: A prospective, multicenter, open-label, 12-week trial. Chest 2006, 129, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.-C.; Parikh, K.; Pulido, T.; Jerjes-Sanchez, C.; White, R.J.; Allen, R.; Torbicki, A.; Xu, K.-F.; Yehle, D.; Laliberte, K. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: A randomized controlled trial. Circulation 2013. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.; Gall, H.; Seyfarth, H.; Halank, M.; Ghofrani, H.; Winkler, J.; Golpon, H.; Olsson, K.; Nickel, N.; Opitz, C. Long-term outcome with intravenous iloprost in pulmonary arterial hypertension. Eur. Respir. J. 2009, 34, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Palazzini, M.; Manes, A. Pulmonary arterial hypertension: From the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur. Heart J. 2010, 31, 2080–2086. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Badesch, D.B.; Delcroix, M.; Fleming, T.R.; Gaine, S.P.; Galiè, N.; Gibbs, J.S.R.; Kim, N.H.; Oudiz, R.J.; Peacock, A. End points and clinical trial design in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S97–S107. [Google Scholar] [CrossRef] [PubMed]
- Kawut, S.M.; Archer-Chicko, C.L.; DeMichele, A.; Fritz, J.S.; Klinger, J.R.; Ky, B.; Palevsky, H.I.; Palmisciano, A.J.; Patel, M.; Pinder, D. Anastrozole in pulmonary arterial hypertension. A randomized, double-blind, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2017, 195, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized impres study. Circulation 2013. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of fk506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602449. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
|
|
|
|
|
| Determinants of Prognosis | Low Risk: <5% | Intermediate Risk: 5–10% | High Risk: >10% |
|---|---|---|---|
| Clinical signs of RHF | Absent | Absent | Present |
| Progression of symptoms | No | Slow | Rapid |
| Episodes of syncope | None | Occasional a | Recurrent b |
| WHO functional class | I, II | III | IV |
| 6-minute walk distance | >440 m | 165–440 m | <165 m |
| Cardiopulmonary exercise testing | Peak VO2: >15 mL/min/kg (>65% pred.) VE/VCO2: slope < 36 | Peak VO2: 11–15 mL/min/kg (35–65% pred.) VE/VCO2: slope 36–44.9 | Peak VO2: <11 mL/min/kg (<35% pred.) VE/VCO2: slope ≥ 45 |
| NT-proBNP plasma levels | BNP: <50 ng/L NT-proBNP: <300 ng/L | BNP: 50–300 ng/L NT-proBNP: 300–1400 ng/L | BNP: >300 ng/L NT-proBNP: >1400 ng/L |
| Imaging (Echocardiogram, CMR imaging) | RA area: <18 cm2 No pericardial effusion | RA area: 18–26 cm2 No or minimal pericardial effusion | RA area: >26 cm2 Pericardial effusion |
| Haemodynamics | RAP: <8 mmHg CI: ≥ 2.5 L/min/m2 SvO2: >65% | RAP: 8–14 mmHg CI: 2.0–2.4 L/min/m2 SvO2: 60–65% | RAP: >14 mmHg CI: <2.0 L/min/m2 SvO2: <60% |
| Background Therapy | Number of Participants | Study Duration (Weeks) | Primary Endpoint | Secondary Endpoint | Main Adverse Events | |
|---|---|---|---|---|---|---|
| AIR [64] (iloprost) | None | 203 | 12 | 6MWD | NYHA functional class Mahler dyspnoea index Quality of life Death (NS) | Flushing Jaw pain |
| AMBITION [65] (ambrisentan vs. tadalafil vs. dual) | None | 500 | 74 | Time to first clinical failure | 6MWD WHO-FC (NS) Borg dyspnoea index | Peripheral oedema Headache Nasal congestion |
| ARIES-1 [66] (ambrisentan) | None | 202 | 12 | 6MWD | TTCW (NS) WHO-FC Quality of life (NS) Borg dyspnoea score BNP | Peripheral oedema Headache Flushing |
| ARIES-2 [66] (ambrisentan) | None | 192 | 12 | 6MWD | TTCW WHO-FC (NS) Quality of life Borg dyspnoea score BNP | Peripheral oedema Headache Nasal congestion |
| Badesch and colleagues [67] (epoprostenol) | None | 111 | 12 | 6MWD | Haemodynamics NYHA functional class | Jaw pain Diarrhoea Nausea and vomiting infection |
| Barst and colleagues [68] (epoprostenol) | none | 81 | 12 | 6MWD | WHO-FCH aemodynamics (NS) Survival | Jaw pain Flushing Headaches Catheter related sepsis |
| BREATHE-1 [69] (bosentan) | None | 213 | 12 | 6MWD | Borg dyspnoea index WHO-FC TTCW | Abnormal hepatic function |
| BREATHE-2 [70] (bosentan) | Epoprostenol | 33 | 16 | TPR (NS) | CI (NS) PVR (NS) 6MWD (NS) WHO-FC (NS) | Mainly related to epoprostenol therapy |
| COMPASS-2 [71] (bosentan) | Sildenafil | 334 | 38 months | Time to first morbidity or mortality event (NS) | 6MWD WHO-FC (NS) PAH-related admissions (NS) | Abnormal hepatic function |
| EARLY [72] (bosentan) | None or Sildenafil | 185 | 24 | 6MWD (NS) PVR | TTCW WHO-FC Quality of life | Abnormal liver function test |
| FREEDOM-C [73] (treprostinil) | ERA and/or PDE-5i | 350 | 16 | 6MWD (NS) | Clinical worsening (NS) WHO-FC (NS) Borg dyspnoea score (NS) | Headache Nausea and vomiting Diarrhoea Flushing Jaw pain |
| FREEDOM-C2 [74] (treprostinil) | ERA and/or PDE-5i | 310 | 16 | 6MWD (NS) | Clinical worsening (NS) WHO-FC (NS) | Headache Nausea and vomiting Diarrhoea Flushing Jaw pain |
| GRIPHON [75] (selexipag) | ERA and/or PDE-5i | 1156 | 71 | Event point event | 6MWD WHO-FC | Headache Jaw pain Flushing Diarrhoea |
| PACES [76] (sildenafil) | Epoprostenol | 267 | 16 | 6MWD | TTCW WHO-FC | Headache Dyspepsia |
| PATENT-1 [77] (riociguat) | None or ERA or PCA | 443 | 12 | 6MWD | PVR Borg dyspnoea score WHO-FC | Headache Dyspepsia Hypotension |
| PHIRST [78] (Tadalafil) | None or Bosentan | 405 | 16 | 6MWD | WHO-FC (NS) TTCW Quality of life | Headache Myalgia Flushing |
| Rubin and colleagues [79] (epoprostenol) | None | 23 | 12 | 6MWD | Haemodynamics | Diarrhoea Jaw pain Photosensitivity |
| SERAPHIN [80] (macitentan) | None or PDE-5i or PCA | 742 | 100 | Time to first event | 6MWD WHO-FC PAH related admissions | Nasopharyngitis Headache Anaemia |
| Simonneau and colleagues [81] (treprostinil) | None | 470 | 12 | 6MWD | Symptoms Borg dyspnoea score Haemodynamics | Infusion site pain Jaw pain Diarrhoea |
| SUPER [82] (sildenafil) | None | 278 | 12 | 6MWD | WHO-FC TTCW (NS) Haemodynamics | Flushing Dyspepsia Diarrhoea |
| TRIUMPH [83] (treprostinil) | ERA or PDE-5i | 235 | 12 | 6MWD | Quality of life TTCW (NS) Symptoms (NS) | Cough Headache Flushing |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, N.S.H.; Massam, B.D.; Kulkarni, S.S.; Lang, C.C. Pulmonary Arterial Hypertension: Pathophysiology and Treatment. Diseases 2018, 6, 38. https://doi.org/10.3390/diseases6020038
Lan NSH, Massam BD, Kulkarni SS, Lang CC. Pulmonary Arterial Hypertension: Pathophysiology and Treatment. Diseases. 2018; 6(2):38. https://doi.org/10.3390/diseases6020038
Chicago/Turabian StyleLan, Norris S. H., Benjamin D. Massam, Sandeep S. Kulkarni, and Chim C. Lang. 2018. "Pulmonary Arterial Hypertension: Pathophysiology and Treatment" Diseases 6, no. 2: 38. https://doi.org/10.3390/diseases6020038
APA StyleLan, N. S. H., Massam, B. D., Kulkarni, S. S., & Lang, C. C. (2018). Pulmonary Arterial Hypertension: Pathophysiology and Treatment. Diseases, 6(2), 38. https://doi.org/10.3390/diseases6020038