
Ajuforrestin A Inhibits Tumor Proliferation and Migration by Targeting the STAT3/FAK Signaling Pathways and VEGFR-2

Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Assessment of Cell Viability In Vitro
2.3. Flow Cytometric Analysis of Apoptosis
2.4. Cell Cycle Distribution Analysis
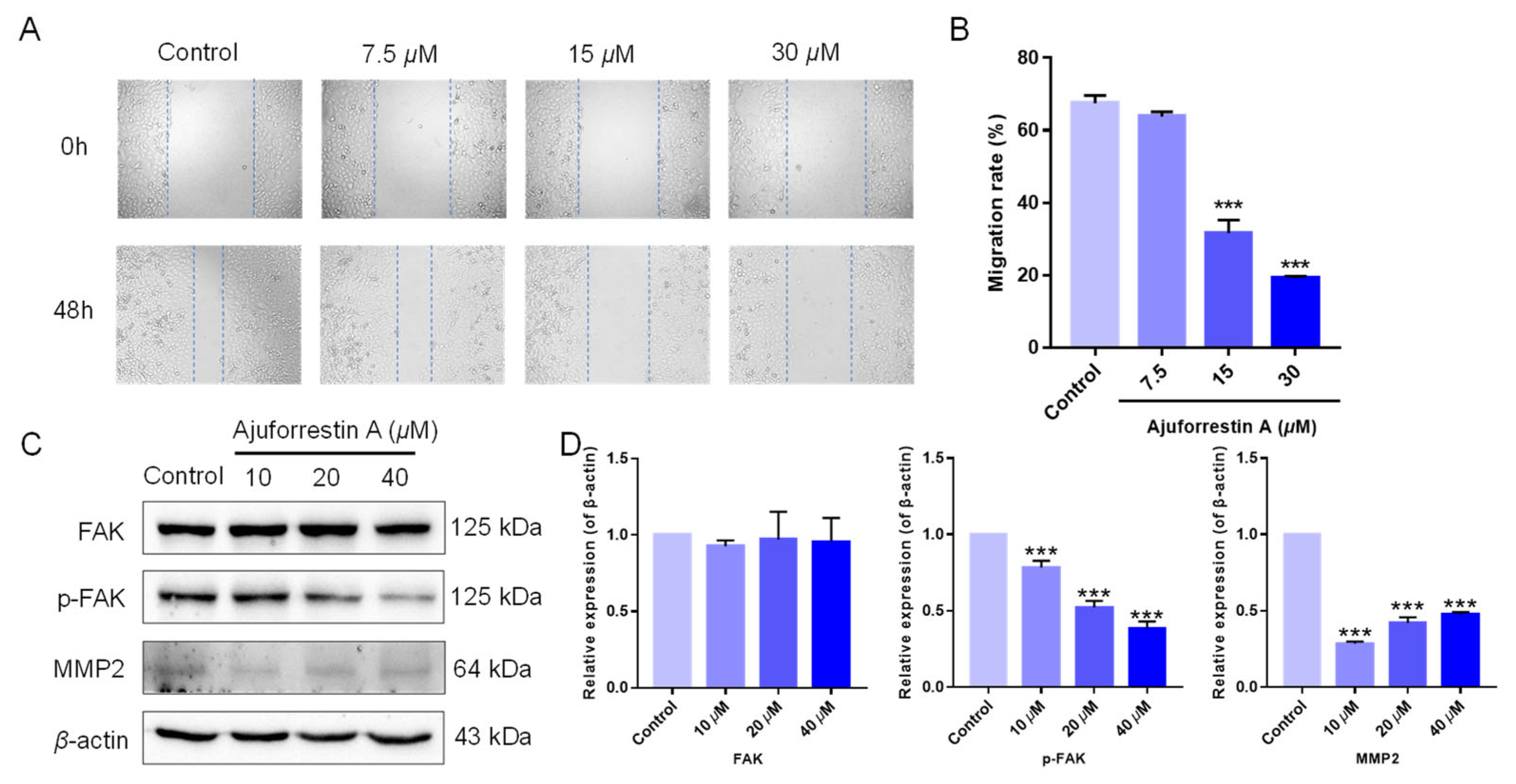
2.5. Cell Migration Analysis via Wound-Healing Assay
2.6. Western Blotting Analysis
2.7. Zebrafish Husbandry and Care
2.8. In Vivo Anti-Angiogenetic Activity of Ajuforrestin A in Zebrafish
2.9. Surface Plasmon Resonance (SPR) Assay
2.10. Molecular Docking Simulation
2.11. In Vivo Antitumor Activity of Ajuforrestin A in Zebrafish
2.12. Statistical Analysis
3. Results
3.1. In Vitro Antiproliferative Activity
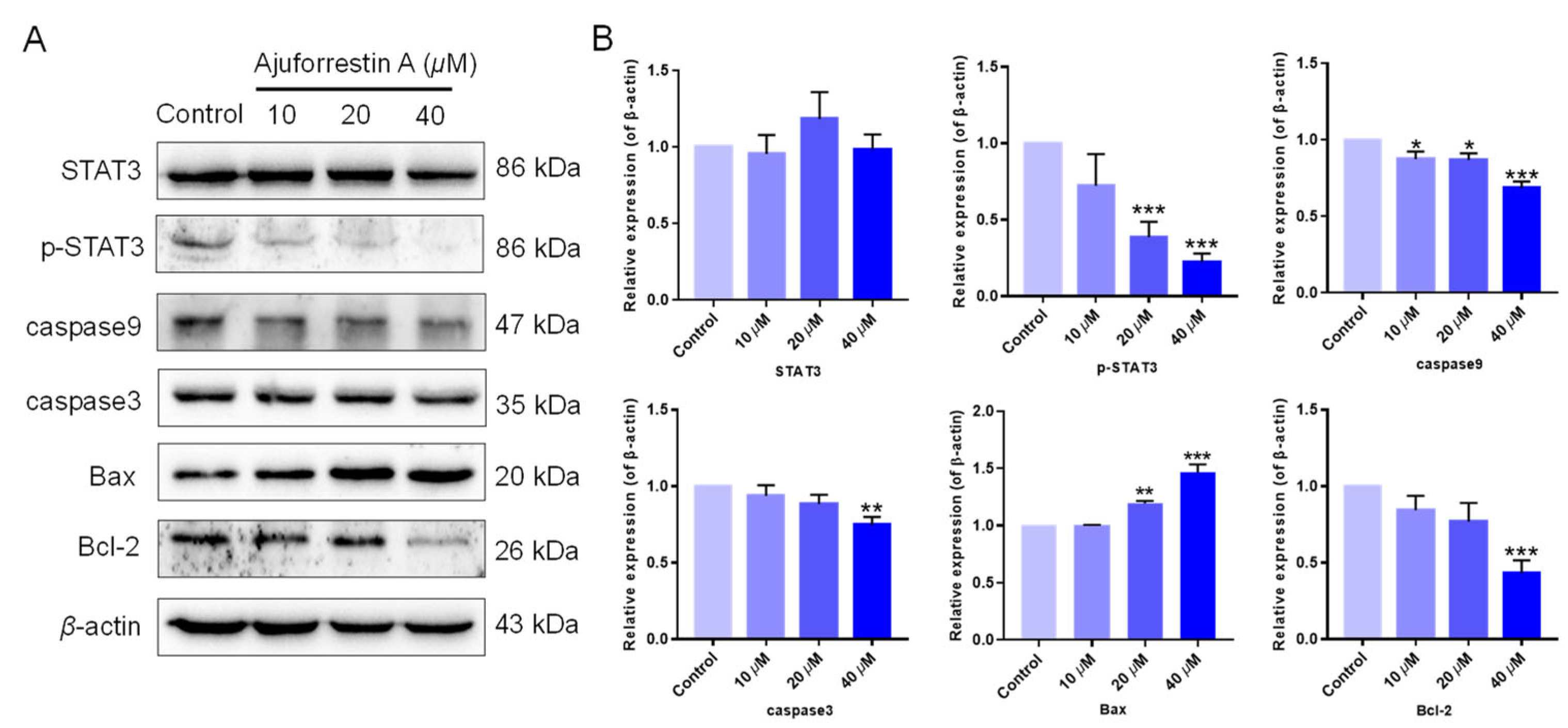
3.2. Ajuforrestin A Triggered Apoptosis in A549 Cells
3.3. Ajuforrestin A Induced G0/G1 Cell Cycle Arrest in A549 Cells
3.4. Ajuforrestin A Modulated the STAT3-Associated Signaling Pathway
3.5. Ajuforrestin A Inhibited Cell Migration via Regulation of the FAK Signaling Pathway
3.6. In Vivo Anti-Angiogenic Efficacy of Ajuforrestin A
3.7. Ajuforrestin A Interacted with VEGFR-2 as Determined by SPR
3.8. Molecular Docking Simulation Insights
3.9. In Vivo Antitumor Efficacy of Ajuforrestin A in Zebrafish Xenografts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Murphy, J.M.; Rodriguez, Y.A.R.; Jeong, K.; Ahn, E.-Y.E.; Lim, S.-T.S. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med. 2020, 52, 877–886. [Google Scholar] [CrossRef]
- Modi, S.J.; Kulkarni, V.M. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: Medicinal chemistry perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Naeem, A.; Hu, P.; Yang, M.; Zhang, J.; Liu, Y.; Zhu, W.; Zheng, Q. Natural products as anticancer agents: Current status and future perspectives. Molecules 2022, 27, 8367. [Google Scholar] [CrossRef]
- Yang, Y.; Li, N.; Wang, T.-M.; Di, L. Natural products with activity against lung cancer: A review focusing on the tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 10827. [Google Scholar] [CrossRef]
- Qing, X.; Yan, H.-M.; Ni, Z.-Y.; Vavricka, C.J.; Zhang, M.-L.; Shi, Q.-W.; Gu, Y.-C.; Kiyota, H. Chemical and pharmacological research on the plants from genus Ajuga. Heterocycl. Commun. 2017, 23, 245–268. [Google Scholar] [CrossRef]
- Kwak, A.; Lee, J.; Lee, S.; Seo, J.; Park, J.W.; Choi, Y.H.; Cho, S.; Yoon, G.; Lee, M.; Shim, J. Echinatin induces reactive oxygen species-mediated apoptosis via JNK/p38 MAPK signaling pathway in colorectal cancer cells. Phytother. Res. 2023, 37, 563–577. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Y.; He, X.; Su, Y.; Hu, F.; Wei, X.; Pan, M.; Zhou, Q.; Yang, W. Lutein inhibits tumor progression through the ATR/Chk1/p53 signaling pathway in non-small cell lung cancer. Phytother. Res. 2023, 37, 1260–1273. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Peng, M.; Ma, J.; Zhang, Q.; Guo, Y.; Xu, J. Modification of a natural diterpene and its antitumor mechanism: Promoting apoptosis, suppressing migration, and inhibiting angiogenesis. Arab. J. Chem. 2024, 17, 105603. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X.; Li, Y.; Liu, Y.; Hou, J.; Guo, Y. Preparation and photothermal therapy of gold nanorods modified by Belamcanda chinensis (L.) DC polysaccharide. Int. J. Biol. Macromol. 2024, 255, 127854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Che, D.; Zeng, L.; Zhang, Y.; Nan, K.; Zhang, X.; Zhang, H.; Guo, Z. 6-Methoxydihydrosanguinarine induces apoptosis and autophagy in breast cancer MCF-7 cells by accumulating ROS to suppress the PI3K/AKT/mTOR signaling pathway. Phytother. Res. 2023, 37, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Chien, P.-Y.; Lan, Y.-H.; Wu, I.-T.; Huang, Y.-P.; Hung, C.-C. Mosloflavone from Fissistigma petelotii ameliorates oncogenic multidrug resistance by STAT3 signaling modulation and P-glycoprotein blockade. Phytomedicine 2024, 123, 155210. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, Y.; Zheng, X.; Liang, G. Reveal the interaction mechanism of five old drugs targeting VEGFR2 through computational simulations. J. Mol. Graph. Model. 2020, 96, 107538. [Google Scholar] [CrossRef]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.-L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.S.; Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Wu, X.-L.; Liu, L.; Wang, Q.-C.; Wang, H.-F.; Zhao, X.-R.; Lin, X.-B.; Lv, W.-J.; Niu, Y.-B.; Lu, T.-L.; Mei, Q.-B. Antitumor Activity and Mechanism Study of Riluzole and Its Derivatives. Iran J. Pharm. Res. 2018, 19, e124401. [Google Scholar] [CrossRef]
- Choisy-Rossi, C.; Yonish-Rouach, E. Apoptosis and the cell cycle: The p53 connection. Cell Death Differ. 1998, 5, 129–131. [Google Scholar] [CrossRef]
- Mohrherr, J.; Uras, I.Z.; Moll, H.P.; Casanova, E. STAT3: Versatile functions in non-small cell lung cancer. Cancers 2020, 12, 1107. [Google Scholar] [CrossRef]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Gamble, J.; Elson, D.; Greenwood, J.; Tanguay, R.; Kolluri, S. The zebrafish xenograft models for investigating cancer and cancer therapeutics. Biology 2021, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Yesudasu, V.; Pradhan, H.S.; Pandya, R.J. Recent progress in surface plasmon resonance based sensors: A comprehensive review. Heliyon 2021, 7, e06321. [Google Scholar] [CrossRef]
- Alsehli, M.; Ali, A.A.S.; Nafie, M.S.; Bardaweel, S.; Aljuhani, A.; Darwish, K.M.; Alraqa, S.Y.; Rezki, N.; Aouad, M.R. Discovery of novel tris-1,2,3-triazole-based hybrids as VEGFR2 inhibitors with potent anti-proliferative and cytotoxicity through apoptosis induction. Bioorganic Chem. 2025, 155, 108131. [Google Scholar] [CrossRef]
- Moradi, M.; Mousavi, A.; Emamgholipour, Z.; Giovannini, J.; Moghimi, S.; Peytam, F.; Honarmand, A.; Bach, S.; Foroumadi, A. Quinazoline-based VEGFR-2 inhibitors as potential anti-angiogenic agents: A contemporary perspective of SAR and molecular docking studies. Eur. J. Med. Chem. 2023, 259, 115626. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Liu, W.; Hou, J.; Xu, J.; Guo, Y.; Hu, P. Cratoxylumxanthone C, a natural xanthone, inhibits lung cancer proliferation and metastasis by regulating STAT3 and FAK signal pathways. Front. Pharmacol. 2022, 13, 920422. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Li, Q.; Wang, Y.; Wang, L.; Li, X.; Ge, N.; Wang, Y.; Guo, C. Niclosamide Induces Cell Cycle Arrest in G1 Phase in Head and Neck Squamous Cell Carcinoma Through Let-7d/CDC34 Axis. Front. Pharmacol. 2019, 9, 1544. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Li, Y.; Li, Y.; Zhang, H.; Song, Z.; Xu, J.; Guo, Y. A natural xanthone suppresses lung cancer growth and metastasis by targeting STAT3 and FAK signaling pathways. Phytomedicine 2022, 102, 154118. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Jung Kim, M.; Ho Kang, K.; Kim, C.H.; Choi, S.Y. Real-time imaging of mitochondria in transgenic zebrafish expressing mitochondrially targeted GFP. BioTechniques 2008, 45, 331–334. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.; Li, Z.; Liu, W.; Zhang, H.; Ohizumi, Y.; Nakajima, A.; Xu, J.; Guo, Y. Structure, anti-tumor activity, and potential anti-tumor mechanism of a fungus polysaccharide from Fomes officinalis. Carbohydr. Polym. 2022, 295, 119794. [Google Scholar] [CrossRef]
- Lv, D.; Xu, J.; Qi, M.; Wang, D.; Xu, W.; Qiu, L.; Li, Y.; Cao, Y. A strategy of screening and binding analysis of bi-oactive components from traditional Chinese medicine based on surface plasmon resonance biosensor. J. Pharma-Ceutical Anal. 2022, 12, 500–508. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, W.; Guan, X.; Guo, S.; Li, C.; Niu, R.; Gao, J.; Jiang, M.; Bai, L.; Leung, E.L.; et al. 20(S)-Protopanaxatriol promotes the binding of P53 and DNA to regulate the antitumor network via multiomic analysis. Acta Pharm. Sin. B 2020, 10, 1020–1035. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, J.; Qiu, L. Synergistic effects of A-B-C-type amphiphilic copolymer on reversal of drug resistance in MCF-7/ADR breast carcinoma. Int. J. Nanomed. 2016, 11, 5205–5220. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) 1 |
|---|---|
| Ajuforrestin A | 9.0 ± 1.1 |
| Etoposide | 33.8 ± 1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Li, Y.; Rong, M.; Li, Y.; Lu, Y.; Li, S.; Lee, D.; Xu, J.; Guo, Y. Ajuforrestin A Inhibits Tumor Proliferation and Migration by Targeting the STAT3/FAK Signaling Pathways and VEGFR-2. Biology 2025, 14, 908. https://doi.org/10.3390/biology14080908
Wang S, Li Y, Rong M, Li Y, Lu Y, Li S, Lee D, Xu J, Guo Y. Ajuforrestin A Inhibits Tumor Proliferation and Migration by Targeting the STAT3/FAK Signaling Pathways and VEGFR-2. Biology. 2025; 14(8):908. https://doi.org/10.3390/biology14080908
Chicago/Turabian StyleWang, Sibei, Yeling Li, Mingming Rong, Yuejun Li, Yaxin Lu, Shen Li, Dongho Lee, Jing Xu, and Yuanqiang Guo. 2025. "Ajuforrestin A Inhibits Tumor Proliferation and Migration by Targeting the STAT3/FAK Signaling Pathways and VEGFR-2" Biology 14, no. 8: 908. https://doi.org/10.3390/biology14080908
APA StyleWang, S., Li, Y., Rong, M., Li, Y., Lu, Y., Li, S., Lee, D., Xu, J., & Guo, Y. (2025). Ajuforrestin A Inhibits Tumor Proliferation and Migration by Targeting the STAT3/FAK Signaling Pathways and VEGFR-2. Biology, 14(8), 908. https://doi.org/10.3390/biology14080908