
Prospects for Development and Commercialisation of Allogeneic CAR-Based Therapies for Autoimmune Disease



Simple Summary
Abstract
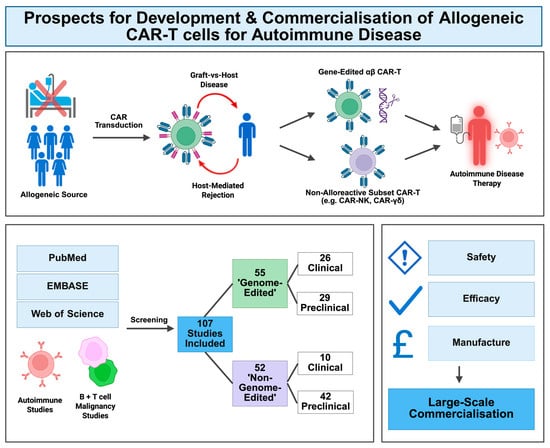
1. Introduction
1.1. Chimeric Antigen Receptors
1.2. Autoimmune Disease and CAR-T
1.3. Production of Allogeneic CAR-T
2. Results
2.1. Genome-Edited Database Search
2.2. Non-Genome-Edited Database Search
2.3. Data Analysis
- Complete Responses (CR). These were designated by the authors in autoimmunity, using relevant rheumatological measures of disease activity. For malignancy, this refers to the proportion of complete responses/complete responses with incomplete haematological recovery (CRi), with negative minimal residual disease (MRD), at day (D)28 of the study (or time period indicated).
- Incidence of Graft-vs.-Host Disease (GVHD), at any point during the study period.
- Incidence of Prolonged Cytopenia, persisting at D28 of the study (or time period indicated). Any specific reduced-cell-type data used, e.g., neutropenia, was highlighted in the tables. If the incidence of prolonged cytopenia was unavailable, with only overall cytopenia incidence or cytopenia data at an unclear time point being available, this was indicated by ‘†’.
- Incidence of cytokine release syndrome (CRS) at any point in the study period.
- Incidence of Immune-Effector Cell-Associated Neurotoxicity Syndrome (ICANS), or other CAR-T-induced neurotoxicity, reported at any point in the study period.
- Incidence of Opportunistic Infections or Infectious Reactivations, at any point in the study period.
- Fatalities, clearly stated as related to the interventional treatment administered, as well as the overall number of deaths reported across the study period in parentheses. As per the ‘Common Terminology Criteria for Adverse Events’ [17] guidance, any ‘grade 5’ adverse events (AE), indicating ‘death related to AE’, were recorded as a fatality.
- Any severe, stated as grade 3 or above (≥grade 3) [17], adverse reactions recorded were represented using bold and underlined text.
- If no data were available for an outcome, this was recorded as ND (no data).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Cell Design | Study Design | CR | GvHD/ Cytopenia | CRS/ ICANS | Infection | Fatalities 1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| Autoimmunity | ||||||||
| TyU19: An anti-CD19 CAR-T for necrotising myositis and systemic sclerosis | Healthy donor PBMC, lentiviral transduction, CRISPR Cas-9 KO of HLA-A, HLA-B, CIITA, TRAC and PD-1 | DCSSc (n = 2), Necrotising myositis (n = 1) Flu/Cy LD | 3/3 | 0/3 3/3 lymphopenia 2 | 0/3 0/3 | ND | 0/3 | [18] |
| YTS109: An anti-CD19 targeting chimeric TCR (STAR) | Healthy donor PBMC, AAV transduction, CRISPR Cas-9 KO of TRAC, PD1, HLA-A, HLA-B and CIITA | SLE with nephritis (n = 5) Flu/Cy LD | 2/5 3 | 0/5 5/5 leukocytopaenia) 4 | 2/5 0/5 | 2/5 | 0/5 | [19] |
| BRL-303: An anti-CD19 CAR-T for SLE | Healthy donor T cells, Multiplex genome-edited (ND method, no genes specified), Transduction (ND method) | r/r SLE (n = 4) Flu/Cy LD (n = 3) No LD (n = 1) | ND 5 | 0/4 1/4 (leukopaenia at 1 month) | 4/4 0/4 | ND | 0/4 | [16] |
| Cancer | ||||||||
| BE-CAR7: An anti-CD7 CAR-T, for relapsed T-ALL | Healthy donor PBMC, lentiviral transduction, Base editing disruption of TRBC1 + TRBC2, CD52 and CD7 | r/r T-ALL (n = 3)Flu/Cy/Alem. LD | 2/3 | 1/3 ND | 2/3 2/3 | 3/3 | 1/3 | [20] |
| CTA101: A CD19/CD22 dual targeting CAR-T, for r/r B-ALL | CD3+ T cells isolated, lentiviral transduction, CRISPR Cas-9 KO of TRAC and CD52 | r/r B-ALL (n = 6) Flu/Cy LD | 5/6 | 0/6 3/6 | 6/6 (1/6) 0/6 | 3/6 | (1/6 overall) | [21] |
| TT52CAR19: An anti-CD19 CAR-T, for paediatric r/r B-ALL | Healthy donor peripheral blood lymphocytes, lentiviral transduction (incorporating self-duplicating CRISPR sgRNA), Next generation CRISPR Cas-9 KO of TRAC and CD52 | r/r B-ALL (n = 6) Flu/Cy/Alem. LD | 3/6 | 1/6 6/6 (6/6) neutropenia | 6/6 2/6 (1/6) | 6/6 | (4/6 overall) | [22] |
| UCART19: An anti-CD19 CAR-T, for adult B-ALL (CALM) | Healthy donor PBMCs, lentiviral transduction, TALEN editing of TRAC and CD52 | r/r B-ALL (n = 25) Flu/Cy +/− Alem. LD | 9/25 | 2/25 10/25 | 20/25 (6/25) 7/25 (1/25) | 12/25 (7/25) | 4/25 (14/25 overall) | [23] |
| U-CAR-T19 + nU-CAR-T19: Anti-CD19 CAR-Ts, for r/r B-ALL | PBMCs, lentiviral transduction, CRISPR Cas-9 (1) U-CAR-T19: B2M and TRAC KO (2) nU-CAR-T19: HLA-A, HLA-B and TRAC KO | (1) U-CAR-T19: DLBCL (n = 3), B-ALL (n = 3) (2) nU-CAR-T19: r/r B-ALL (n = 3) Flu/Cy/Etoposide LD | (1) 0/6 (2) 3/3 (D14 MRD neg. 6) | 0/9 5/9 (D14) | 9/9 0/9 | 2/9 | 0/9 | [24] |
| CD5 CAR-T: A CD5 targeting CAR-T for r/r T-ALL | PBMCs (previous transplant donor or healthy matched donors), lentiviral transduction, (EGFRt incorporated), CRISPR Cas-9 (sgRNA) KO of CD5 | r/r T-ALL: previous transplant donor (n = 11), healthy donor (n = 5) Flu/Cy LD | 16/16 | 11/16 16/16 (15/16 at 1 month) | 12/16 4/16 | 9/16 (7/16) | (8/16 overall) | [25] |
| WU-CART-007: A CD7 targeting CAR-T, for r/r T-ALL/T-LBL | Healthy Donor T cells, transduction (ND method), CRISPR Cas-9 editing to KO CD7 and TRAC | T-ALL (n = 18), T-LBL (n = 8) -phase 1 (n = 13); Flu/Cy LD -phase 2 (n = 13); enhanced LD | 12/23 (7/10 MRD neg. 7) | 1/26 5/26 (neutropenia) † | 23/26 (5/26) 2/26 | 7/26 8 | 1/26 (3/26 overall) | [26] |
| UCART19: An anti-CD19 CAR-T, for infant B-ALL | Healthy Donor T cells, lentiviral transduction (incorporated RQR8 epitope tag), TALEN-mediated KO of TRAC and CD52 | Infant B-ALL (n = 2) -followed by alloHSCT Flu/Cy/Alem. LD | 2/2 | 2/2 1/2 | 0/2 1/2 | ND | 0/2 | [27] |
| UCD7-CAR: An anti-CD7 CAR-T, for paediatric R/R T-ALL/LBL | Healthy Donor T cell, transduction (ND method), CRISPR Cas-9 editing to KO CD7 and TCR | r/r T-LBL (n = 1), r/r T-ALL (n = 4) Flu/Cy LD | 4/5 | 0/5 ND | 4/5 0/5 | 1/5 | 0/5 | [28] |
| CTX110: An anti-CD19 CAR T, for adult r/r LBCL | Allogeneic source T cells, transduction (ND method), CRISPR Cas-9 editing to disrupt TRAC and B2M | r/r LBCL (n = 32) -2nd dose (n = 8) Flu/Cy LD | 11/32 | 0/32 ND | 18/32 3/32 (2/32) | 4/32 | (1/32 overall) | [29] |
| CB-010: An anti-CD19 CAR-T, for r/r B-NHL | Healthy Donor T cells, chRDNA CAR insertion into TRAC locus, chRDNA TRAC and PD-1 KO | LBCL (n = 10), MCL (n = 3), FL (n = 2), MZL (n = 1) -3 DL Flu/Cy LD | 11/16 | 0/16 9/16 (neutropenia) † | 7/16 4/16 (2/16) | 1/16 | ND | [30] |
| ET-901: An anti-CD19 CAR-T, for B-NHL | Allogeneic T cell source, transduction (ND method), CRISPR KO of undisclosed gene (Gene X) and TCR | r/r LBCL (n = 4), FL (n = 2) Flu/Cy LD | 4/6 | 1/6 6/6 † | 6/6 2/6 | 2/6 | ND | [31] |
| UCART22 P2: An anti-CD22 CAR-T for r/r B-ALL | Donor-derived T cells, lentiviral transduction, TALEN-mediated KO of TRAC and CD52 | r/r B-ALL (n = 3) Flu/Cy/Alem. LD | 1/3 9 | ND ND | 2/3 0/3 | 1/3 | 1/3 | [32] |
| UCART20x22: An anti-CD20/CD22 dual-targeted CAR-T, for r/r B-NHL | Healthy donor T cells, lentiviral transduction, TALEN-mediated KO of TRAC and CD52 | r/r DLBCL (n = 2), THL (n = 1) Flu/Cy/Alem. LD | 2/3 | 0/3 ND | 3/3 0/3 | 1/3 | ND | [33] |
| FT819: An iPSC-derived anti-CD19 CAR-T, for r/r B cell leukaemia/lymphoma | iPSCs, transduction into TRAC locus (ND method), Gene-edited bi-allelic KO of TCR (ND tech.) | r/r BCL (n = 12) -regimens A: 1 dose (n = 9), B: 3 split doses (n = 3) Flu/Cy LD | ND | 0/12 ND | 3/12 0/12 | ND | ND | [34] |
| RD13-01: An anti-CD7 CAR-T, for T-ALL + T-LBL | Healthy donor-derived T cells, transduction (incorporated NK32 inhibitory ligand) (ND method), CRISPR Cas-9 edited (no genes specified) | T-ALL (n = 7), T-LBL (n = 3) Flu/Cy/Etoposide LD | 7/10 | ND ND | 10/10 (1/10) 1/10 | 0/10 | (4/10 overall) | [35] |
| GC027: An anti-CD7 CAR-T, for r/r T cell malignancy | Healthy donor T cells isolated, lentiviral transduction, CRISPR Cas-9 KO of TRAC and CD7 | r/r T-ALL (n = 11), T-LBL (n = 1) Flu/Cy + melphalan, etoposide, or pred. LD | 11/12 | 1/12 (post HSCT) ND | 10/12 (8/12) 0/12 | 1/12 10 | (2/12 overall) | [36] |
| UCART123v1.2: An anti-CD123 CAR-T, for AML | Healthy Donor T cells, lentiviral transduction, TALEN-mediated KO of TRAC and CD52 | r/r AML (n = 17) Flu/Cy +/− Alem. LD | 1/16 | ND ND | 17/17 (4/17) 1/17 | ND | (2/16 overall) | [37] |
| CTX130: An anti-CD70 CAR-T, for r/r TCL | Healthy Donor T cells, AAV transduction into TRAC locus, CRISPR Cas-9 KO of TRAC, B2M and CD70 | PTCL (n = 22), Mycosis Fungoides or Sézary Syndrome (n = 17) Flu/Cy LD | 6/39 | 0/39 15/39 (14/39 neutropenia) † | 26/39 (3/39) 4/39 | 10/39 | 0/39 (16/39 overall) | [38] |
| ALLO-501: An anti-CD19 CAR-T, for r/r B-NHL | Allogeneic source T cells, transduction (ND method), TALEN-mediated KO of TRAC and CD52 | B-NHL (n = 46) -2nd dose (n = 7) Flu/Cy/ALLO-647 LD | 18/36 (Auto CAR-T naïve) | 0/46 38/46 † | 11/46 (1/46) 0/46 | 11/46 | 1/46 (5/46 overall) | [39] |
| ALLO-715: An anti-BCMA CAR-T, for r/r MM | Healthy donor PBMCs, lentiviral transduction, TALEN-mediated KO of TRAC and CD52 | r/r MM (n = 43) Cy/ALLO-647 +/− Flu LD | 6/43 | 0/43 8/43 (D56) | 24/43, 1/43 6/43 | 23/43, 10/43 | (10/43 overall) | [40] |
| ALLO-501A: An anti-CD19 CAR-T, for LBCL | Allogeneic source T cells, transduction (ND method), TALEN-mediated KO of TRAC and CD52 | LBCL (n = 15) -Con: 2nd consolidation dose (n = 9) Flu/Cy/ALLO-647 LD | 6/12 | Con: 0/9 72% † | Con: 0/9 Con: 0/9 | Con: 0/9 | ND | [41] |
| UCART19: An anti-CD19 CAR-T, for paediatric B-ALL (continued trial [31]) | Healthy Donor T cells, lentiviral transduction (incorporated RQR8), TALEN-mediated KO of TRAC and CD52 | r/r B-ALL (n = 5) -additional participants from original study [27] Flu/Cy/Alem. LD | 3/5 | 1/5 2/5 (neutropenia) | 5/5 (1/5) ND | 4/5 | (3/5 overall) | [42] |
| Description | Cell Design | Study Design | CR | GvHD/ Cytopenia | CRS/ ICANS | Infection | Fatalities | Ref. |
|---|---|---|---|---|---|---|---|---|
| Autoimmunity | ||||||||
| QN-139b: CD19 BCMA dual CAR targeted iPSC-derived NK cells | iPSC engineered using CRISPR Cas-9 to express CD19 and BCMA CARs in addition to KO by cytosine base editing of B2M, CIITA, CD16 and over-expression of HLA-E, HLA-G, IL-2 receptor fusion and EGFRt. Differentiated thereafter into NK cells | DCSSc (n = 1) Flu/Cy LD | PR 1 | 0/1 1/1 2 | 0/1 0/1 | 1/1 | 0/1 | [43] |
| Allogeneic CD19 CAR-NK: An anti-CD19 CAR-NK for SLE | Allogeneic Source, CAR-NK (ND methods) | r/r SLE (n = 24) Flu/Cy LD | 8/12 (DORIS at 12 months) 3 | 0/24 ND | 2/24 4 0/24 | ND | ND | [44] |
| Cancer | ||||||||
| CYAD-211: An anti-BCMA CAR-T (engineered using miRNA-based shRNA), for r/r MM | Healthy donor PBMCs, retroviral transduction (encoding miRNA-based shRNA against CD3ζ) | r/r MM (n = 12) Flu/Cy LD | 0/12 (3/12 PR) | 0/12 2/12 (neutropenia) † | 1/12 0/12 (CRES) | 5/12 (2/12) | (1/12 overall) | [45] |
| FT596: An iPSC-derived anti-CD19 CAR-NK for BCL | iPSCs, lentiviral transduction (incorporated hnCD16 + IL-15-RF and NK-optimised CD19 CAR with NKG2D transmembrane domain, 2B4 co-stimulatory domain and CD3ζ activation domain), differentiated into NK cells | BCL (n = 86) regimen A (without rituximab, n = 18) or B (with rituximab, n = 68) Flu/Cy LD | 25/68 (efficacy reported for B alone) | 0/86 17/81 | 10/86 0/86 | 17/86 | 0/86 (43/86 overall) | [46] |
| CD33 CAR-NK: An anti-CD33 CAR-NK for r/r AML | NK cells from healthy donor UCB, lentiviral transduction | r/r AML (n = 10) Flu/Cy LD | 6/10 | 0/10 10/10 (5/10) (neutropenia) † | 1/10 0/10 | 3/10 (1/10) | (9/10 overall) | [47] |
| CNTY-101: An anti-CD19 iPSC-derived CAR-NK for B-NHL | iPSC, CRISPR MAD-7 mediated KO of B2M and CIITA, CRISPR mediated HLA-E expression, transduction (incorporated secreted IL-15 and EGFR safety switch) (ND method), differentiated into CAR-NK [48] 5 | DLBCL (n = 5), FL (n = 1), MZL (n = 1) Chemo. LD | 2/7 | 0/7 ND | 2/7 0/7 | ND | ND | [49] |
| NKX101: An anti-NKG2D ligand CAR-NK for r/r AML | Healthy donor NK cells, transduction (incorporated membrane-bound IL-15) (ND method) | r/r AML (n = 6) Flu/Ara-C LD | 3/6 | 0/6 3/6 (neutropenia) † | 0/6 0/6 | 3/6 | ND | [50] |
| ADI-001: An anti-CD20 CAR-γδ for BCL | Allogeneic γδ T cells, transduction (ND method) | LBCL (n = 8), MCL (n = 1) Flu/Cy LD | 7/9 | 0/9 ND | 2/9 1/9 | 1/9 | 0/9 (1/9 overall) | [51] |
| CD19-CAR NKT (ANCHOR): An anti-CD19 CAR- iNKT for B-NHL + B-ALL | NK T cells from leukapheresis product, transduction (incorporated IL-15, and encoding shRNA targeting B2M and CD74) (ND method) | B-NHL (n = 4), B-ALL (n = 1) Flu/Cy LD | 2/5 | ND ND | 1/5 ND | ND | ND | [52] |
| 19-28z CAR EBV-CTLs: An anti-CD19 CAR-EBV-CTL for r/r B cell malignancy | EBV-CTLs, from primary HSCT donors (n = 4) or 3rd Party donors (n = 6), transduction (ND method) | B-ALL (n = 5), Burkitt’s (n = 1), CLL (n = 1), PMBCL (n = 2), DLBCL (n = 1) ND on LD | 7/10 | 1/10 ND | 0/10 0/10 | ND | ND | [53] |
3. Discussion
3.1. A Summary of the Findings
3.2. Genome-Editing to Address Immunological Barriers Imposed by Allogeneic T Cells
3.3. Genome-Editing Technologies
3.4. Non-Genome-Edited Strategies Using Unconventional Cell Types
3.5. Safety Considerations
3.6. Lymphodepletion
3.7. Enhancing Efficacy and Persistence
3.8. Manufacture of Allogeneic CAR-Engineered Products
3.9. CAR Design and Transduction
3.10. Genome-Edited vs. Non-Genome-Edited Allogeneic CAR Therapies
3.11. Focus on Autoimmune Disease Application
3.12. Limitations of the Study
4. Clinical Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Halim, L.; Maher, J. CAR T-cell immunotherapy of B-cell malignancy: The story so far. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520927164. [Google Scholar] [CrossRef]
- Li, Y.-R.; Lyu, Z.; Chen, Y.; Fang, Y.; Yang, L. Frontiers in CAR-T cell therapy for autoimmune diseases. Trends Pharmacol. Sci. 2024, 45, 839–857. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Taubmann, J.; Boltz, S.; Hagen, M.; Wirsching, A.; Muller, F.; Volkl, S.; Kharboutli, S.; Sporl, S.; Garantziotis, P.; Aigner, M.; et al. Long-term follow up of efficiency and safety of CD19-CAR T-cell treatment of systemic lupus erythematosus. Z. Rheumatol. 2025, 84, 612–620. [Google Scholar] [CrossRef]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-Cell Therapy in Autoimmune Disease—A Case Series with Follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Qasim, W. Genome-edited allogeneic donor “universal” chimeric antigen receptor T cells. Blood 2023, 141, 835–845. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhu, Y.; Chen, Y.; Yang, L. The clinical landscape of CAR-engineered unconventional T cells. Trends Cancer 2025, 11, 520–539. [Google Scholar] [CrossRef]
- What Is PICO? Available online: https://www.cochranelibrary.com/about-pico (accessed on 12 December 2024).
- Autoimmune Condition: NCI Dictionary of Cancer Terms. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/autoimmune-condition (accessed on 12 December 2024).
- Deguine, J.; Xavier, R.J. B cell tolerance and autoimmunity: Lessons from repertoires. J. Exp. Med. 2024, 221, e20231314. [Google Scholar] [CrossRef]
- Sanber, K.; Savani, B.; Jain, T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: The diametric opposition of T cells. Br. J. Haematol. 2021, 195, 660–668. [Google Scholar] [CrossRef]
- Jørgensen, L.V.; Christensen, E.B.; Barnkob, M.B.; Barington, T. The clinical landscape of CAR NK cells. Exp. Hematol. Oncol. 2025, 14, 46. [Google Scholar] [CrossRef]
- Mahne, A.; Rodriguez, R.; Wang, J.; Anaya, D.; Cheng, J.K.; Kwong, B.; Banuelos, J.; Starokadomskyy, P.; Park, S.; Gibson, C.; et al. Preclinical Development and Manufacturability of KYV-201, an Investigational Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cell for the Treatment of Autoimmune Disease. In Proceedings of the Arthritis & Rheumatology 2024, Washington, DC, USA, 14–19 November 2024; Volume 76 (Suppl. S9). [Google Scholar]
- Xiang, J.; Devenport, J.M.; Carter, A.J.; Staser, K.W.; Kim, M.Y.; O’ Neal, J.; Ritchey, J.K.; Rettig, M.P.; Gao, F.; Rettig, G.; et al. An “off-the-shelf” CD2 universal CAR-T therapy for T-cell malignancies. Leukemia 2023, 37, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sun, C.; Tan, B.; Hu, C.; Wan, L.; Xie, C.; Tan, Q.; Liu, M.; Lin, J.; Du, B.; et al. Allogenic anti-CD19 CAR-T cells induce remission in refractory systemic lupus erythematosus. In Proceedings of the Arthritis & Rheumatology 2025, Chicago, IL, USA, 24–29 October 2025; Volume 77 (Suppl. S9). [Google Scholar]
- National Cancer Insititute. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0; U.S. Department of Health and Human Services: Washington, DC, USA, 2017.
- Wang, X.; Wu, X.; Tan, B.; Zhu, L.; Zhang, Y.; Lin, L.; Xiao, Y.; Sun, A.; Wan, X.; Liu, S.; et al. Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis. Cell 2024, 187, 4890–4904.e4899. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Wang, H.; Wu, X.; He, C.; Lin, S.; Pang, K.; Li, Y.; Chen, Y.; Tang, X.; et al. Allogeneic CD19-targeting T cells for treatment-refractory systemic lupus erythematosus: A phase 1 trial. Nat. Med. 2025, 31, 3713–3724. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, R.; Georgiadis, C.; Syed, F.; Zhan, H.; Etuk, A.; Gkazi, S.A.; Preece, R.; Ottaviano, G.; Braybrook, T.; Chu, J.; et al. Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2023, 389, 899–910. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, Y.; Zhang, M.; Ge, W.; Li, Y.; Yang, L.; Wei, G.; Han, L.; Wang, H.; Yu, S.; et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2021, 27, 2764–2772. [Google Scholar] [CrossRef]
- Ottaviano, G.; Georgiadis, C.; Gkazi, S.A.; Syed, F.; Zhan, H.; Etuk, A.; Preece, R.; Chu, J.; Kubat, A.; Adams, S.; et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci. Transl. Med. 2022, 14, eabq3010. [Google Scholar] [CrossRef]
- Benjamin, R.; Jain, N.; Maus, M.V.; Boissel, N.; Graham, C.; Jozwik, A.; Yallop, D.; Konopleva, M.; Frigault, M.J.; Teshima, T.; et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): A phase 1, dose-escalation trial. Lancet Haematol. 2022, 9, e833–e843. [Google Scholar] [CrossRef]
- Chen, X.; Tan, B.; Xing, H.; Zhao, X.; Ping, Y.; Zhang, Z.; Huang, J.; Shi, X.; Zhang, N.; Lin, B.; et al. Allogeneic CAR-T cells with of HLA-A/B and TRAC disruption exhibit promising antitumor capacity against B cell malignancies. Cancer Immunol. Immunother. 2024, 73, 13. [Google Scholar] [CrossRef]
- Pan, J.; Tan, Y.; Shan, L.; Seery, S.; Deng, B.; Ling, Z.; Xu, J.; Duan, J.; Wang, Z.; Wang, K.; et al. Allogeneic CD5-specific CAR-T therapy for relapsed/refractory T-ALL: A phase 1 trial. Nat. Med. 2024, 31, 126–136. [Google Scholar] [CrossRef]
- Ghobadi, A.; Aldoss, I.; Maude, S.; Bhojwani, D.; Wayne, A.; Bajel, A.; Dholaria, B.; Faramand, R.; Mattison, R.; Rijneveld, A.; et al. Anti-CD7 allogeneic WU-CART-007 in patients with relapsed/refractory T-cell acute lymphoblastic leukemia/lymphoma: A phase 1/2 trial. Res. Sq. 2024, 1–27. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Guo, Y.; Yang, W.; Wang, Y.; Zhu, X. Gene-Edited Universal CD7 Chimeric Antigen Receptor T Cells for Pediatric T-Cell Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma. Blood 2023, 142, 2109. [Google Scholar] [CrossRef]
- McGuirk, J.P.; Tam, C.S.; Kröger, N.; Riedell, P.A.; Murthy, H.S.; Ho, P.J.; Maakaron, J.E.; Waller, E.K.; Awan, F.T.; Shaughnessy, P.J.; et al. CTX110 Allogeneic CRISPR-Cas9-Engineered CAR T Cells in Patients (Pts) with Relapsed or Refractory (R/R) Large B-Cell Lymphoma (LBCL): Results from the Phase 1 Dose Escalation Carbon Study. Blood 2022, 140, 10303–10306. [Google Scholar] [CrossRef]
- Hu, B.; Nastoupil, L.J.; Holmes, H.; Hamdan, A.; Kanate, A.; Farooq, U.; Cherry, M.; Brem, E.; Pinter-Brown, L.C.; Ermann, D.A.; et al. A CRISPR-edited allogeneic anti-CD19 CAR-T cell therapy with a PD-1 knockout (CB-010) in patients with relapsed/refractory B cell non-Hodgkin lymphoma (r/r B-NHL): Updated phase 1 results from the ANTLER trial. J. Clin. Oncol. 2024, 42, 7025. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, J.; Yang, Q.; Wang, C.; Liang, F.; Wu, Z.; Lamao, Q.; Yang, J.; Qiu, Y.; Tang, W.; et al. The interim analysis of a first-in-human phase 1 trial of ET-901, a CRISPR edited allogeneic immune-cloaked anti-CD19 CAR-T cell therapy in patients with r/r B-NHL. J. Clin. Oncol. 2024, 42, e19010. [Google Scholar] [CrossRef]
- Jain, N.; Chevallier, P.; Liu, H.; Schiller, G.J.; Méar, J.-B.; DeAngelo, D.J.; Curran, K.J.; Grupp, S.; Baruchel, A.; Balsat, M.; et al. Updated Results of the Phase I BALLI-01 Trial of UCART22 Process 2 (P2), an Anti-CD22 Allogeneic CAR-T Cell Product Manufactured By Cellectis Biologics, in Patients with Relapsed or Refractory (R/R) CD22+ B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2023, 142, 4847. [Google Scholar] [CrossRef]
- Abramson, J.S.; Ramakrishnan, A.; Pierola, A.A.; Braunschweig, I.; Cartron, G.; Thieblemont, C.; Pérez-Simón, J.A.; Barba, P.; Riedell, P.A.; Solano, C.; et al. Preliminary Results of Nathali-01: A First-in-Human Phase I/IIa Study of UCART20x22, a Dual Allogeneic CAR-T Cell Product Targeting CD20 and CD22, in Relapsed or Refractory (R/R) Non-Hodgkin Lymphoma (NHL). Blood 2023, 142, 2110. [Google Scholar] [CrossRef]
- Mehta, A.; Farooq, U.; Chen, A.; McGuirk, J.P.; Ly, T.; Wong, L.; Cooley, S.; Valamehr, B.; Elstrom, R.; Chu, Y.-W.; et al. Interim Phase I Clinical Data of FT819-101, a Study of the First-Ever, Off-the-Shelf, iPSC-Derived TCR-Less CD19 CAR T-Cell Therapy for Patients with Relapsed/Refractory B-Cell Malignancies. Blood 2022, 140, 4577–4578. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Y.; Yang, J.; Li, J.; Qiu, L.; Ge, W.; Pei, B.; Chen, J.; Han, L.; Ren, J.; et al. A Novel Universal CD7-Targeted CAR-T Cell Therapy for Relapsed or Refractory T-Cell Acute Lymphoblastic Leukemia and T-Cell Lymphoblastic Lymphoma. Blood 2022, 140, 4566–4567. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Liu, L.; Liu, J.; Rao, J.; Yuan, Z.; Gao, L.; Li, Y.; Luo, L.; Li, G.; et al. CD7 targeted “off-the-shelf” CAR-T demonstrates robust in vivo expansion and high efficacy in the treatment of patients with relapsed and refractory T cell malignancies. Leukemia 2023, 37, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; De Angelo, D.J.; Pemmaraju, N.; Dinner, S.; Gill, S.; Olin, R.L.; Wang, E.S.; Stark, E.; Korngold, A.; Figliola, C.; et al. AMELI-01: A Phase I Trial of UCART123v1.2, an Anti-CD123 Allogeneic CAR-T Cell Product, in Adult Patients with Relapsed or Refractory (R/R) CD123+ Acute Myeloid Leukemia (AML). Mol. Ther. 2023, 31, 51. [Google Scholar] [CrossRef]
- Iyer, S.P.; Sica, R.A.; Ho, P.J.; Prica, A.; Zain, J.; Foss, F.M.; Hu, B.; Beitinjaneh, A.; Weng, W.-K.; Kim, Y.H.; et al. Safety and activity of CTX130, a CD70-targeted allogeneic CRISPR-Cas9-engineered CAR T-cell therapy, in patients with relapsed or refractory T-cell malignancies (COBALT-LYM): A single-arm, open-label, phase 1, dose-escalation study. Lancet Oncol. 2025, 26, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Nath, R.; Munoz, J.; Tees, M.; Miklos, D.B.; Frank, M.J.; Malik, S.A.; Stevens, D.; Shin, C.R.; Balakumaran, A.; et al. ALPHA Study: ALLO-501 Produced Deep and Durable Responses in Patients with Relapsed/Refractory Non-Hodgkin’s Lymphoma Comparable to Autologous CAR T. Blood 2021, 138, 3878. [Google Scholar] [CrossRef]
- Mailankody, S.; Matous, J.V.; Chhabra, S.; Liedtke, M.; Sidana, S.; Oluwole, O.O.; Malik, S.; Nath, R.; Anwer, F.; Cruz, J.C.; et al. Allogeneic BCMA-targeting CAR T cells in relapsed/refractory multiple myeloma: Phase 1 UNIVERSAL trial interim results. Nat. Med. 2023, 29, 422–429. [Google Scholar] [CrossRef]
- Lekakis, L.J.; Locke, F.L.; Tees, M.; Neelapu, S.S.; Malik, S.A.; Hamadani, M.; Frank, M.J.; Popplewell, L.L.; Abramson, J.S.; de Vos, S.; et al. ALPHA2 Study: ALLO-501A Allogeneic CAR T in LBCL, Updated Results Continue to Show Encouraging Safety and Efficacy with Consolidation Dosing. Blood 2021, 138, 649. [Google Scholar] [CrossRef]
- Waseem, Q.; Oana, C.; Stuart, A.; Sarah, I.; Claire, M.; Christine, R.; Gary, W.; Giovanna, L.; Juliana, S.; Kanchan, R.; et al. Gene-edited allogeneic CAR19 T cells (UCART19) induce molecular remission ahead of allo-SCT in high risk pediatric patients with CD19+ relapsed/refractory B-cell acute lymphoblastic leukemia (The 44th Annual Meeting of the European Society for Blood and Marrow Transplantation: Physicians Oral Session). Bone Marrow Transplant. 2019, 53, 27–28. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Jin, Y.; Dai, L.; Yue, Y.; Hu, J.; Liu, X.; Pang, K.; Ye, S.; Chen, Y.; et al. An iPSC-derived CD19/BCMA CAR-NK therapy in a patient with systemic sclerosis. Cell 2025, 188, 4225–4238.e4212. [Google Scholar] [CrossRef]
- Gao, J.; Yu, Y.; Kong, R.; Xu, X.; Liu, S.; Chen, Q.; Li, X.; Wu, Y.; Xu, E.; Sun, M.; et al. Lb0009 allogenic cd19 car nk cell therapy in refractory systemic lupus erythematosus-a case series study. Ann. Rheum. Dis. 2025, 84, 321. [Google Scholar] [CrossRef]
- Lonez, C.; Bolsée, J.; Huberty, F.; Nguyen, T.; Jacques-Hespel, C.; Anguille, S.; Flament, A.; Breman, E. Clinical Proof-of-Concept of a Non-Gene Editing Technology Using miRNA-Based shRNA to Engineer Allogeneic CAR T-Cells. Int. J. Mol. Sci. 2025, 26, 1658. [Google Scholar] [CrossRef]
- Ghobadi, A.; Bachanova, V.; Patel, K.; Park, J.H.; Flinn, I.; Riedell, P.A.; Bachier, C.; Diefenbach, C.S.; Wong, C.; Bickers, C.; et al. Induced pluripotent stem-cell-derived CD19-directed chimeric antigen receptor natural killer cells in B-cell lymphoma: A phase 1, first-in-human trial. Lancet 2025, 405, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, X.; Yan, H.; Tan, X.; Ma, Y.; Wang, M.; Han, X.; Liu, J.; Gao, L.; Gao, L.; et al. Safety and efficacy of CD33-targeted CAR-NK cell therapy for relapsed/refractory AML: Preclinical evaluation and phase I trial. Exp. Hematol. Oncol. 2025, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, M.; Idank, K.; Budd, C.; Lebid, A.; Perry, D.; Jessup, H.; Gurung, B.; Morse, B.; Carton, J.; Campion, L.; et al. Characterization of CNTY-101, an Allogeneic Anti-CD19 iPSC-Derived NK Product, for the Treatment of B Cell-Driven Autoimmune Diseases. In Proceedings of the ASGCT 27th Annual Meeting, Baltimore, MD, USA, 7–11 May 2024. [Google Scholar]
- Patel, K.; Namburi, S.; Latif, T.; Oluwole, O.O.; Cross, S.J.; Simmons, G.; Iragavarapu, C.; Hu, B.; Jih, G.; Bullaughey, K.; et al. Interim results from the ELiPSE-1 study: A phase 1, multicenter, open-label study of CNTY-101 in subjects with relapsed or refractory CD19-positive B-cell malignancies. J. Clin. Oncol. 2024, 42, 7023. [Google Scholar] [CrossRef]
- Sauter, C.S.; Borthakur, G.; Mountjoy, L.; Rotta, M.; Liu, H.; Murthy, H.S.; Lin, M.; Trager, J.; Chang, C.; Kothari, N.; et al. A Phase 1 Study of NKX101, a Chimeric Antigen Receptor Natural Killer (CAR-NK) Cell Therapy, with Fludarabine and Cytarabine in Patients with Acute Myeloid Leukemia. Blood 2023, 142, 2097. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Stevens, D.A.; Hamadani, M.; Frank, M.J.; Holmes, H.; Jacobovits, A.; Hinkle, J.; Kennedy-Wilde, J.; Maller, O.; Weinstein, B.; et al. A Phase 1 Study of ADI-001: Anti-CD20 CAR-Engineered Allogeneic Gamma Delta1 (γδ) T Cells in Adults with B-Cell Malignancies. Blood 2022, 140, 4617–4619. [Google Scholar] [CrossRef]
- Ramos, C.A.; Courtney, A.N.; Robinson, S.N.; Dakhova, O.; Lulla, P.D.; Kamble, R.; Carrum, G.; Wang, T.; Zhang, C.; Di Pierro, E.; et al. Allogeneic NKT Cells Expressing a CD19-Specific CAR in Patients with Relapsed or Refractory B-Cell Malignancies: An Interim Analysis. Blood 2021, 138, 2819. [Google Scholar] [CrossRef]
- Curran, K.J.; Sauter, C.S.; Kernan, N.A.; Prockop, S.E.; Boulad, F.; Perales, M.; Giralt, S.A.; Riviere, I.; Wang, X.; Boelens, J.-J.; et al. Durable Remission Following “Off-the-Shelf” Chimeric Antigen Receptor (CAR) T-Cells in Patients with Relapse/Refractory (R/R) B-Cell Malignancies. Biol. Blood Marrow Transplant. 2020, 26, S89. [Google Scholar] [CrossRef]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Li, W.; Zhu, X.; Xu, Y.; Chen, J.; Zhang, H.; Yang, Z.; Qi, Y.; Hong, J.; Li, Y.; Wang, G.; et al. Simultaneous editing of TCR, HLA-I/II and HLA-E resulted in enhanced universal CAR-T resistance to allo-rejection. Front. Immunol. 2022, 13, 1052717. [Google Scholar] [CrossRef]
- Degagné, E.; Donohoue, P.D.; Roy, S.; Scherer, J.; Fowler, T.W.; Davis, R.T.; Reyes, G.A.; Kwong, G.; Stanaway, M.; Larroca Vicena, V.; et al. High-Specificity CRISPR-Mediated Genome Engineering in Anti-BCMA Allogeneic CAR T Cells Suppresses Allograft Rejection in Preclinical Models. Cancer Immunol. Res. 2024, 12, 462–477. [Google Scholar] [CrossRef]
- Zhu, S.; Zuo, S.; Li, C.; You, X.; Jiang, E.; Feng, X.; Luo, Y. LLT1 overexpression renders allogeneic-NK resistance and facilitates the generation of enhanced universal CAR-T cells. J. Exp. Clin. Cancer Res. 2025, 44, 25. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Manner, K.; DeJesus, R.; White, K.; Gattis, C.; Ngo, P.; Bandoro, C.; Tham, E.; Chu, E.Y.; Young, C.; et al. Hypoimmune anti-CD19 chimeric antigen receptor T cells provide lasting tumor control in fully immunocompetent allogeneic humanized mice. Nat. Commun. 2023, 14, 2020. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Wright, H.; Hu, X.; Kinder, J.; van Hoeven, N.; Liang, O.; Granger, B.; Duback, T.; Baldeviano, C.; Chandra, S.; et al. Hypoimmune, Allogeneic CD22-Directed CAR T Cells That Evade Innate and Adaptive Immune Rejection for the Treatment of Large B Cell Lymphoma Patients That Are Relapsed/Refractory to CD19-Directed CAR T Cell Therapy. Blood 2023, 142, 3437. [Google Scholar] [CrossRef]
- Johnson, A.; Wright, H.; Hu, X.; van Hoeven, P.N.; Granger, B.; Liang, O.; Moreno, J.; Lamba, M.; Young, C.; McAlister, A.; et al. A Dual-Antigen Targeting, Hypoimmune Allogeneic CAR T to Evade Innate and Adaptive Immune Rejection and Overcome Antigen Escape. Blood 2022, 140, 4552–4553. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, D.; Li, H.; Zhu, Z.; Zhang, Z.; Chen, Y.; Yang, N.; Li, J.; Wang, Z.; Niu, T.; et al. Delivery of CD47-SIRPα checkpoint blocker by BCMA-directed UCAR-T cells enhances antitumor efficacy in multiple myeloma. Cancer Lett. 2024, 585, 216660. [Google Scholar] [CrossRef]
- PD-1: NCI Dictionary of Cancer Terms. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/pd-1 (accessed on 12 December 2024).
- Mishra, A.K.; Qasim, W. Genome editing approaches for universal chimeric antigen receptor T cells. EJC Paediatr. Oncol. 2024, 3, 100149. [Google Scholar] [CrossRef]
- Asmamaw, M.; Zawdie, B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics 2021, 15, 353–361. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Nain, V. TALENs—An indispensable tool in the era of CRISPR: A mini review. J. Genet. Eng. Biotechnol. 2021, 19, 125. [Google Scholar] [CrossRef]
- Madison, B.B.; Patil, D.; Richter, M.; Li, X.; Tong, M.; Cranert, S.; Wang, X.; Martin, R.; Xi, H.; Tan, Y.; et al. Cas-CLOVER is a novel high-fidelity nuclease for safe and robust generation of TSCM-enriched allogeneic CAR-T cells. Mol. Ther. Nucleic Acids 2022, 29, 979–995. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- Diorio, C.; Murray, R.; Naniong, M.; Barrera, L.; Camblin, A.; Chukinas, J.; Coholan, L.; Edwards, A.; Fuller, T.; Gonzales, C.; et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood 2022, 140, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, R.; Parekh, F.; Lamb, K.; Mekkaoui, L.; Allen, C.; Smetanova, K.; Huang, J.; Williams, A.; Toledo, G.S.; Lilova, K.; et al. Large-scale manufacturing of base-edited chimeric antigen receptor T cells. Mol. Ther. Methods Clin. Dev. 2023, 31, 101123. [Google Scholar] [CrossRef] [PubMed]
- Lonez, C.; Breman, E. Allogeneic CAR-T Therapy Technologies: Has the Promise Been Met? Cells 2024, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Moscarelli, J.; Zahavi, D.; Maynard, R.; Weiner, L.M. The Next Generation of Cellular Immunotherapy: Chimeric Antigen Receptor-Natural Killer Cells. Transpl. Cell Ther. 2022, 28, 650–656. [Google Scholar] [CrossRef]
- Taylor, N.P. Nkarta’s Blood Cancer CAR Stalls Again, with Falling Response Rate Forcing It Out of the Race. Available online: https://www.fiercebiotech.com/biotech/nkartas-blood-cancer-car-stalls-again-falling-response-rate-forcing-it-out-race# (accessed on 12 December 2024).
- O’Neal, J.; Cooper, M.L.; Ritchey, J.K.; Gladney, S.; Niswonger, J.; Gonzalez, L.S.; Street, E.; Haas, G.J.; Carter, A.; Amayta, P.N.; et al. Anti-myeloma efficacy of CAR-iNKT is enhanced with a long-acting IL-7, rhIL-7-hyFc. Blood Adv. 2023, 7, 6009–6022. [Google Scholar] [CrossRef]
- Nishimoto, K.P.; Barca, T.; Azameera, A.; Makkouk, A.; Romero, J.M.; Bai, L.; Brodey, M.M.; Kennedy-Wilde, J.; Shao, H.; Papaioannou, S.; et al. Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models. Clin. Transl. Immunol. 2022, 11, e1373. [Google Scholar] [CrossRef]
- Perko, R.; Kang, G.; Sunkara, A.; Leung, W.; Thomas, P.G.; Dallas, M.H. Gamma delta T cell reconstitution is associated with fewer infections and improved event-free survival after hematopoietic stem cell transplantation for pediatric leukemia. Biol. Blood Marrow Transpl. 2015, 21, 130–136. [Google Scholar] [CrossRef]
- Sanchez Martinez, D.; Tirado, N.; Mensurado, S.; Martinez-Moreno, A.; Romecin, P.; Gutierrez Aguera, F.; Correia, D.V.; Silva-Santos, B.; Menendez, P. Generation and proof-of-concept for allogeneic CD123 CAR-Delta One T (DOT) cells in acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e005400. [Google Scholar] [CrossRef]
- Almeida, A.R.; Correia, D.V.; Fernandes-Platzgummer, A.; da Silva, C.L.; da Silva, M.G.; Anjos, D.R.; Silva-Santos, B. Delta One T Cells for Immunotherapy of Chronic Lymphocytic Leukemia: Clinical-Grade Expansion/Differentiation and Preclinical Proof of Concept. Clin. Cancer Res. 2016, 22, 5795–5804. [Google Scholar] [CrossRef]
- Wu, Y.; Biswas, D.; Usaite, I.; Angelova, M.; Boeing, S.; Karasaki, T.; Veeriah, S.; Czyzewska-Khan, J.; Morton, C.; Joseph, M.; et al. A local human Vdelta1 T cell population is associated with survival in nonsmall-cell lung cancer. Nat. Cancer 2022, 3, 696–709. [Google Scholar] [CrossRef]
- Luoma, A.M.; Castro, C.D.; Adams, E.J. gammadelta T cell surveillance via CD1 molecules. Trends Immunol. 2014, 35, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.T.; von Borstel, A.; Chevour, P.; Awad, W.; Howson, L.J.; Littler, D.R.; Gherardin, N.A.; Le Nours, J.; Giles, E.M.; Berry, R.; et al. Recognition of the antigen-presenting molecule MR1 by a Vdelta3+ gammadelta T cell receptor. Proc. Natl. Acad. Sci. USA 2021, 118, e2110288118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lyu, H.; Guo, R.; Cao, X.; Feng, J.; Jin, X.; Lu, W.; Zhao, M. Epstein–Barr virus–associated cellular immunotherapy. Cytotherapy 2023, 25, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Houen, G.; Trier, N.H. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front. Immunol. 2021, 11, 587380. [Google Scholar] [CrossRef]
- Moradi, V.; Omidkhoda, A.; Ahmadbeigi, N. The paths and challenges of “off-the-shelf” CAR-T cell therapy: An overview of clinical trials. Biomed. Pharmacother. 2023, 169, 115888. [Google Scholar] [CrossRef]
- Kinoshita, S.; Ishii, M.; Furukawa, Y.; Sato, S.; Ando, J.; Nakauchi, H.; Ando, M. iPSC-Derived Dual Antigen Receptor T Cells Targeting GD2 and LMP2 Antigens for Extranodal NK/T-Cell Lymphoma, Nasal Type. Blood 2023, 142, 2079. [Google Scholar] [CrossRef]
- Oelsner, S.; Wagner, J.; Friede, M.E.; Pfirrmann, V.; Genßler, S.; Rettinger, E.; Buchholz, C.J.; Pfeifer, H.; Schubert, R.; Ottmann, O.G.; et al. Chimeric antigen receptor-engineered cytokine-induced killer cells overcome treatment resistance of pre-B-cell acute lymphoblastic leukemia and enhance survival. Int. J. Cancer 2016, 139, 1799–1809. [Google Scholar] [CrossRef]
- Fang, K.K.-L.; Lee, J.; Khatri, I.; Na, Y.; Zhang, L. Targeting T-cell malignancies using allogeneic double-negative CD4-CAR-T cells. J. Immunother. Cancer 2023, 11, e007277. [Google Scholar] [CrossRef]
- Chan, W.K.; Suwannasaen, D.; Throm, R.E.; Li, Y.; Eldridge, P.W.; Houston, J.; Gray, J.T.; Pui, C.H.; Leung, W. Chimeric antigen receptor-redirected CD45RA-negative T cells have potent antileukemia and pathogen memory response without graft-versus-host activity. Leukemia 2015, 29, 387–395. [Google Scholar] [CrossRef]
- Li, Y.R.; Fang, Y.; Niu, S.; Zhu, Y.; Chen, Y.; Lyu, Z.; Zhu, E.; Tian, Y.; Huang, J.; Rezek, V.; et al. Allogeneic CD33-directed CAR-NKT cells for the treatment of bone marrow-resident myeloid malignancies. Nat. Commun. 2025, 16, 1248. [Google Scholar] [CrossRef]
- Lee, J.B.; Kang, H.; Fang, L.; D’Souza, C.; Adeyi, O.; Zhang, L. Developing Allogeneic Double-Negative T Cells as a Novel Off-the-Shelf Adoptive Cellular Therapy for Cancer. Clin. Cancer Res. 2019, 25, 2241–2253. [Google Scholar] [CrossRef] [PubMed]
- Hammer, Q.; Perica, K.; Mbofung, R.M.; van Ooijen, H.; Martin, K.E.; Momayyezi, P.; Varady, E.; Pan, Y.; Jelcic, M.; Groff, B.; et al. Genetic ablation of adhesion ligands mitigates rejection of allogeneic cellular immunotherapies. Cell Stem Cell 2024, 31, 1376–1386.e1378. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Moon, B.K.; Han, M.; Lee, T.-W.; Lee, J.; Kang, K.-S. Genetically stable multi-gene edited iPSCs-derived NK cells for enhanced cancer immunotherapy. Mol. Ther. Oncol. 2024, 32, 200885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xia, C.; Weng, Q.; Zhang, L.; Wang, Y.; Liu, Y.; Zheng, X.; Lin, Y.; Chen, Y.; Shen, Y.; et al. Hypoimmunogenic CD19 CAR-NK cells derived from embryonic stem cells suppress the progression of human B-cell malignancies in xenograft animals. Front. Immunol. 2024, 15, 1504459. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, H.; Lu, Q.; Zhang, Z.; Li, J.; Wang, Z.; Yang, N.; Yu, Z.; Yang, C.; Chen, Y.; et al. mRNA-Engineered CD5-CAR-γδTCD5- Cells for the Immunotherapy of T-Cell Acute Lymphoblastic Leukemia. Adv. Sci. 2024, 11, 2400024. [Google Scholar] [CrossRef]
- Reiser, J.; Chan, S.R.; Mathavan, K.; Sillitti, D.; Mottershead, C.; Mattson, B.; Pache, M.; Gutierrez, A.; Scoon, W.; Zhu, Y.; et al. FT555: Off-the-Shelf CAR-NK Cell Therapy Co-Targeting GPRC5D and CD38 for the Treatment of Multiple Myeloma. Blood 2022, 140, 4560–4561. [Google Scholar] [CrossRef]
- Wang, C.Q.; Lim, P.Y.; Tan, A.H. Gamma/delta T cells as cellular vehicles for anti-tumor immunity. Front. Immunol. 2023, 14, 1282758. [Google Scholar] [CrossRef]
- Boucher, J.C.; Austin, A.L.; Kostenko, E.; Reid, K.; Nagy, M.Z.; Davila, M.L.; Guevara-Patino, J.A.; Bejanyan, N. CD33 Targeted gd T cells for Treatment of Acute Myeloid Leukemia. In Proceedings of the 27th Annual Meeting of the American Society of Gene & Cell Therapy Abstracts, Baltimore, MD, USA, 7–11 May 2024; pp. 633–634. [Google Scholar]
- Guercio, M.; Manni, S.; Boffa, I.; Caruso, S.; Di Cecca, S.; Sinibaldi, M.; Abbaszadeh, Z.; Camera, A.; Ciccone, R.; Polito, V.A.; et al. Inclusion of the Inducible Caspase 9 Suicide Gene in CAR Construct Increases Safety of CAR.CD19 T Cell Therapy in B-Cell Malignancies. Front. Immunol. 2021, 12, 755639. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Oelsner, S.; Waldmann, A.; Billmeier, A.; Röder, J.; Lindner, A.; Ullrich, E.; Marschalek, R.; Dotti, G.; Jung, G.; Große-Hovest, L.; et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int. J. Cancer 2019, 145, 1935–1945. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Savoldo, B.; Lau, W.; Rubinos, C.; Grover, N.; Armistead, P.; Coghill, J.; Hagan, R.S.; Morrison, K.; Buchanan, F.B.; et al. Utility of a safety switch to abrogate CD19.CAR T-cell–associated neurotoxicity. Blood 2021, 137, 3306–3309. [Google Scholar] [CrossRef] [PubMed]
- Bouquet, L.; Bôle-Richard, E.; Warda, W.; Neto Da Rocha, M.; Trad, R.; Nicod, C.; Haderbache, R.; Genin, D.; Ferrand, C.; Deschamps, M. RapaCaspase-9-based suicide gene applied to the safety of IL-1RAP CAR-T cells. Gene Ther. 2023, 30, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef]
- Cichocki, F.; Goodridge, J.P.; Bjordahl, R.; Mahmood, S.; Davis, Z.B.; Gaidarova, S.; Abujarour, R.; Groff, B.; Witty, A.; Wang, H.; et al. Dual antigen-targeted off-the-shelf NK cells show durable response and prevent antigen escape in lymphoma and leukemia. Blood 2022, 140, 2451–2462. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Goodridge, J.P.; Mahmood, S.; Gaidarova, S.; Abujarour, R.; Davis, Z.B.; Merino, A.; Tuininga, K.; Wang, H.; et al. Quadruple gene-engineered natural killer cells enable multi-antigen targeting for durable antitumor activity against multiple myeloma. Nat. Commun. 2022, 13, 7341. [Google Scholar] [CrossRef]
- Frankel, N.W.; Deng, H.; Yucel, G.; Gainer, M.; Leemans, N.; Lam, A.; Li, Y.; Hung, M.; Lee, D.; Lee, C.-T.; et al. Precision off-the-shelf natural killer cell therapies for oncology with logic-gated gene circuits. Cell Rep. 2024, 43, 114145. [Google Scholar] [CrossRef]
- Lickefett, B.; Chu, L.; Ortiz-Maldonado, V.; Warmuth, L.; Barba, P.; Doglio, M.; Henderson, D.; Hudecek, M.; Kremer, A.; Markman, J.; et al. Lymphodepletion—An essential but undervalued part of the chimeric antigen receptor T-cell therapy cycle. Front. Immunol. 2023, 14, 1303935. [Google Scholar] [CrossRef]
- Li, Z.; Richards, S.; Surks, H.K.; Jacobs, A.; Panzara, M.A. Clinical pharmacology of alemtuzumab, an anti-CD52 immunomodulator, in multiple sclerosis. Clin. Exp. Immunol. 2018, 194, 295–314. [Google Scholar] [CrossRef]
- Baden, L.R.; Swaminathan, S.; Almyroudis, N.G.; Angarone, M.; Baluch, A.; Barros, N.; Buss, B.; Cohen, S.; Cooper, B.; Chiang, A.D.; et al. Prevention and Treatment of Cancer-Related Infections, Version 3.2024, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2024, 22, 617–644. [Google Scholar] [CrossRef]
- Kilgour, M.K.; Bastin, D.J.; Lee, S.H.; Ardolino, M.; McComb, S.; Visram, A. Advancements in CAR-NK therapy: Lessons to be learned from CAR-T therapy. Front. Immunol. 2023, 14, 1166038. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Ren, Q.; Zhang, J.; Su, H.; Lu, Q.; Song, Y.; Zhou, J. Targeting CD5 chimeric antigen receptor-engineered natural killer cells against T-cell malignancies. Exp. Hematol. Oncol. 2024, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Lei, W.; Jin, X.; Liu, H.; Wang, J.Q.; Deng, W.; Qian, W. CD70-specific CAR NK cells expressing IL-15 for the treatment of CD19-negative B-cell malignancy. Blood Adv. 2024, 8, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Zu, Y.; Su, H.; Lu, Q.; Xiang, B.; Luo, Y.; Zhang, J.; Song, Y. Single VHH-directed BCMA CAR-NK cells for multiple myeloma. Exp. Hematol. Oncol. 2023, 12, 98. [Google Scholar] [CrossRef]
- Mansour, A.G.; Teng, K.-Y.; Li, Z.; Zhu, Z.; Chen, H.; Tian, L.; Ali, A.; Zhang, J.; Lu, T.; Ma, S.; et al. Off-the-shelf CAR–engineered natural killer cells targeting FLT3 enhance killing of acute myeloid leukemia. Blood Adv. 2023, 7, 6225–6239. [Google Scholar] [CrossRef]
- Silvestre, R.N.; Eitler, J.; de Azevedo, J.T.C.; Tirapelle, M.C.; Fantacini, D.M.C.; de Souza, L.E.B.; Swiech, K.; Covas, D.T.; Calado, R.T.; Montero, P.O.; et al. Engineering NK-CAR.19 cells with the IL-15/IL-15Rα complex improved proliferation and anti-tumor effect in vivo. Front. Immunol. 2023, 14, 1226518. [Google Scholar] [CrossRef]
- Ngai, H.; Tian, G.; Courtney, A.N.; Ravari, S.B.; Guo, L.; Liu, B.; Jin, J.; Shen, E.T.; Di Pierro, E.J.; Metelitsa, L.S. IL-21 Selectively Protects CD62L(+) NKT Cells and Enhances Their Effector Functions for Adoptive Immunotherapy. J. Immunol. 2018, 201, 2141–2153. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, R.; Wang, Z.; Xiong, J.; Wang, X.; Zhang, X. Facing challenges with hope: Universal immune cells for hematologic malignancies. Cancer Biol. Med. 2023, 20, 229–247. [Google Scholar] [CrossRef]
- Tian, G.; Courtney, A.N.; Yu, H.; Bhar, S.; Xu, X.; Barragan, G.A.; Martinez Amador, C.; Ghatwai, N.; Wood, M.S.; Schady, D.; et al. Hyperleukocytosis in a neuroblastoma patient after treatment with natural killer T cells expressing a GD2-specific chimeric antigen receptor and IL-15. J. Immunother. Cancer 2025, 13, e010156. [Google Scholar] [CrossRef]
- Sedloev, D.; Chen, Q.; Unglaub, J.M.; Schanda, N.; Hao, Y.; Besiridou, E.; Neuber, B.; Schmitt, A.; Raffel, S.; Liu, Y.; et al. Proteasome inhibition enhances the anti-leukemic efficacy of chimeric antigen receptor (CAR) expressing NK cells against acute myeloid leukemia. J. Hematol. Oncol. 2024, 17, 85. [Google Scholar] [CrossRef]
- Verbrugge, S.E.; Scheper, R.J.; Lems, W.F.; de Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther. 2015, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Hiepe, F.; Alexander, T.; Peukert, R.; Rubbert, A.; Rech, J.; Braun, T.; Schott, G.; Wiesener, M.; Eckardt, K.U.; Baeuerle, M.; et al. Refractory SLE patients respond to the proteasome inhibitor bortezomib. Ann. Rheum. Dis. 2012, 71, A15–A16. [Google Scholar] [CrossRef]
- Karasiewicz, K.; He, S.; Ng, M.; Tess, K.; Ling, W.; Kaufmann, G.F.; Zeldis, J.B.; Ji, H.; Hariri, R.; Zhang, X. Preclinical Evaluation of Human Placental-Derived Allogeneic CD19 CAR-T Cells Against B Cell Malignancies. Blood 2019, 134, 3222. [Google Scholar] [CrossRef]
- Georgiadis, C.; Nickolay, L.; Syed, F.; Zhan, H.; Gkazi, S.A.; Etuk, A.; Abramowski-Mock, U.; Preece, R.; Cuber, P.; Adams, S.; et al. Umbilical cord blood T cells can be isolated and enriched by CD62L selection for use in ‘off the shelf’ chimeric antigen receptor T-cell therapies to widen transplant options. Haematologica 2024, 109, 3941–3951. [Google Scholar] [CrossRef]
- Bachiller, M.; Perez-Amill, L.; Battram, A.M.; Carné, S.C.; Najjar, A.; Verhoeyen, E.; Juan, M.; Urbano-Ispizua, A.; Martin-Antonio, B. NK cells enhance CAR-T cell antitumor efficacy by enhancing immune/tumor cells cluster formation and improving CAR-T cell fitness. J. Immunother. Cancer 2021, 9, e002866. [Google Scholar] [CrossRef]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef]
- Stikvoort, A.; van der Schans, J.; Sarkar, S.; Poels, R.; Ruiter, R.; Naik, J.; Yuan, H.; de Bruijn, J.D.; van de Donk, N.; Zweegman, S.; et al. CD38-specific Chimeric Antigen Receptor Expressing Natural Killer KHYG-1 Cells: A Proof of Concept for an “Off the Shelf” Therapy for Multiple Myeloma. Hemasphere 2021, 5, e596. [Google Scholar] [CrossRef]
- Suck, G.; Branch, D.R.; Smyth, M.J.; Miller, R.G.; Vergidis, J.; Fahim, S.; Keating, A. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp. Hematol. 2005, 33, 1160–1171. [Google Scholar] [CrossRef]
- Mark, C.; Czerwinski, T.; Roessner, S.; Mainka, A.; Hörsch, F.; Heublein, L.; Winterl, A.; Sanokowski, S.; Richter, S.; Bauer, N.; et al. Cryopreservation impairs 3-D migration and cytotoxicity of natural killer cells. Nat. Commun. 2020, 11, 5224. [Google Scholar] [CrossRef]
- Saultz, J.N.; Otegbeye, F. Optimizing the cryopreservation and post-thaw recovery of natural killer cells is critical for the success of off-the-shelf platforms. Front. Immunol. 2023, 14, 1304689. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Adami, A.; Metoudi, M.; Beatson, R.; George, M.S.; Achkova, D.; Williams, E.; Arif, S.; Reid, F.; Elstad, M.; et al. Intratumoral pan-ErbB targeted CAR-T for head and neck squamous cell carcinoma: Interim analysis of the T4 immunotherapy study. J. Immunother. Cancer 2023, 11, e007162. [Google Scholar] [CrossRef]
- Lu, Q.; Li, H.; Wu, Z.; Zhu, Z.; Zhang, Z.; Yang, D.; Tong, A. BCMA/CD47-directed universal CAR-T cells exhibit excellent antitumor activity in multiple myeloma. J. Nanobiotechnol. 2024, 22, 279. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.; Lee, E.; Park, H.J.; Lee, H.H.; Yun, S.H.; Kim, H.S. A chimeric antigen receptor tailored to integrate complementary activation signals potentiates the antitumor activity of NK cells. J. Exp. Clin. Cancer Res. 2025, 44, 86. [Google Scholar] [CrossRef] [PubMed]
- Raikar, S.S.; Fleischer, L.C.; Moot, R.; Fedanov, A.; Paik, N.Y.; Knight, K.A.; Doering, C.B.; Spencer, H.T. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology 2018, 7, e1407898. [Google Scholar] [CrossRef]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef]
- Ren, J.; Liao, X.; Lewis, J.M.; Chang, J.; Qu, R.; Carlson, K.R.; Foss, F.; Girardi, M. Generation and optimization of off-the-shelf immunotherapeutics targeting TCR-Vβ2+ T cell malignancy. Nat. Commun. 2024, 15, 519. [Google Scholar] [CrossRef]
- Tipanee, J.; Samara-Kuko, E.; Gevaert, T.; Chuah, M.K.; VandenDriessche, T. Universal allogeneic CAR T cells engineered with Sleeping Beauty transposons and CRISPR-CAS9 for cancer immunotherapy. Mol. Ther. 2022, 30, 3155–3175. [Google Scholar] [CrossRef]
- Calviño, C.; Ceballos, C.; Alfonso, A.; Jauregui, P.; Calleja-Cervantes, M.E.; San Martin-Uriz, P.; Rodriguez-Marquez, P.; Martin-Mallo, A.; Iglesias, E.; Abizanda, G.; et al. Optimization of universal allogeneic CAR-T cells combining CRISPR and transposon-based technologies for treatment of acute myeloid leukemia. Front. Immunol. 2023, 14, 1270843. [Google Scholar] [CrossRef]
- Micklethwaite, K.P.; Gowrishankar, K.; Gloss, B.S.; Li, Z.; Street, J.A.; Moezzi, L.; Mach, M.A.; Sutrave, G.; Clancy, L.E.; Bishop, D.C.; et al. Investigation of product-derived lymphoma following infusion of piggyBac-modified CD19 chimeric antigen receptor T cells. Blood 2021, 138, 1391–1405. [Google Scholar] [CrossRef]
- Schomer, N.T.; Jiang, Z.K.; Lloyd, M.I.; Klingemann, H.; Boissel, L. CCR7 expression in CD19 chimeric antigen receptor-engineered natural killer cells improves migration toward CCL19-expressing lymphoma cells and increases tumor control in mice with human lymphoma. Cytotherapy 2022, 24, 827–834. [Google Scholar] [CrossRef]
- Egli, L.; Kaulfuss, M.; Mietz, J.; Picozzi, A.; Verhoeyen, E.; Munz, C.; Chijioke, O. CAR T cells outperform CAR NK cells in CAR-mediated effector functions in head-to-head comparison. Exp. Hematol. Oncol. 2024, 13, 51. [Google Scholar] [CrossRef]
- Khairallah, C.; Chu, T.H.; Sheridan, B.S. Tissue Adaptations of Memory and Tissue-Resident Gamma Delta T Cells. Front. Immunol. 2018, 9, 2636. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Beauchesne, P.; Wang, C.; Wong, A.; Deuse, T.; Schrepfer, S. Hypoimmune CD19 CAR T cells evade allorejection in patients with cancer and autoimmune disease. Cell Stem Cell 2025, 32, 1356–1368.e1354. [Google Scholar] [CrossRef] [PubMed]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef]
- Xie, L.; Gu, R.; Yang, X.; Qiu, S.; Xu, Y.; Mou, J.; Wang, Y.; Xing, H.; Tang, K.; Tian, Z.; et al. Universal Anti-CD7 CAR-T Cells Targeting T-ALL and Functional Analysis of CD7 Antigen on T/CAR-T Cells. Hum. Gene Ther. 2023, 34, 1257–1272. [Google Scholar] [CrossRef]
- Maldonado-Pérez, N.; Tristán-Manzano, M.; Justicia-Lirio, P.; Martínez-Planes, E.; Muñoz, P.; Pavlovic, K.; Cortijo-Gutiérrez, M.; Blanco-Benítez, C.; Castella, M.; Juan, M.; et al. Efficacy and safety of universal (TCRKO) ARI-0001 CAR-T cells for the treatment of B-cell lymphoma. Front. Immunol. 2022, 13, 1011858. [Google Scholar] [CrossRef]
- Sugita, M.; Galetto, R.; Zong, H.; Ewing-Crystal, N.; Trujillo-Alonso, V.; Mencia-Trinchant, N.; Yip, W.; Filipe, S.; Lebuhotel, C.; Gouble, A.; et al. Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia. Nat. Commun. 2022, 13, 2227. [Google Scholar] [CrossRef]
- Georgiadis, C.; Preece, R.; Nickolay, L.; Etuk, A.; Petrova, A.; Ladon, D.; Danyi, A.; Humphryes-Kirilov, N.; Ajetunmobi, A.; Kim, D.; et al. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-leukemic Effects. Mol. Ther. 2018, 26, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Gouble, A.; Black, K.L.; Skwarska, A.; Naqvi, A.S.; Taylor, D.; Zhao, M.; Yuan, Q.; Sugita, M.; Zhang, Q.; et al. Targeting CD123 in blastic plasmacytoid dendritic cell neoplasm using allogeneic anti-CD123 CAR T cells. Nat. Commun. 2022, 13, 2228. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Boldajipour, B.; Kuo, T.C.; Bentley, T.; Sutton, J.; Chen, A.; Geng, T.; Dong, H.; Galetto, R.; Valton, J.; et al. Preclinical Evaluation of Allogeneic CAR T Cells Targeting BCMA for the Treatment of Multiple Myeloma. Mol. Ther. 2019, 27, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Kloos, A.; Georgiadis, C.; Etuk, A.; Gkazi, S.A.; Syed, F.; Braybrook, T.; Zhan, H.; Kadirkamanathan, R.; Kattre, N.; Görlich, K.; et al. Single and Combinational Multiplex Base-Edited ‘Universal’ CAR T Cells in a Humanised Model of Primary CD7+CD33+ AML. Blood 2022, 140, 7425–7426. [Google Scholar] [CrossRef]
- Korst, C.L.B.M.; Bruins, W.S.C.; Cosovic, M.; Verkleij, C.P.M.; Twickler, I.; Le Clerre, D.; Chion-Sotinel, I.; Zweegman, S.; Galetto, R.; Mutis, T.; et al. Preclinical Activity of Allogeneic CS1-Specific CAR T-Cells (UCARTCS1) in Multiple Myeloma. Blood 2022, 140, 4215–4216. [Google Scholar] [CrossRef]
- Oumzil, I.; Guerrero, V.; Keerthipati, P.S.; Colangelo, J.; Lawler, B.; Singson, V.; Khedkar, A.; Singh, S.; Terrett, J.; Hostetter, D. 367 CRISPR/Cas9 gene-edited, allogeneic anti-CD83 CAR-T cells demonstrate potent activity in GvHD and AML tumor models. J. Immunother. Cancer 2022, 10, A386. [Google Scholar] [CrossRef]
- Klaihmon, P.; Samart, P.; Rojanasakul, Y.; Issaragrisil, S.; Luanpitpong, S. Anti-TIM3 chimeric antigen receptor-natural killer cells preferentially target primitive acute myeloid leukemia cells with minimal fratricide and exhaustion. Exp. Hematol. Oncol. 2024, 13, 67. [Google Scholar] [CrossRef]
- Rowan, A.G.; Ponnusamy, K.; Ren, H.; Taylor, G.P.; Cook, L.B.M.; Karadimitris, A. CAR-iNKT cells targeting clonal TCRVβ chains as a precise strategy to treat T cell lymphoma. Front. Immunol. 2023, 14, 1118681. [Google Scholar] [CrossRef]
- Caruso, S.; De Angelis, B.; Del Bufalo, F.; Ciccone, R.; Donsante, S.; Volpe, G.; Manni, S.; Guercio, M.; Pezzella, M.; Iaffaldano, L.; et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J. Hematol. Oncol. 2022, 15, 163. [Google Scholar] [CrossRef]
- Quintarelli, C.; Sivori, S.; Caruso, S.; Carlomagno, S.; Falco, M.; Boffa, I.; Orlando, D.; Guercio, M.; Abbaszadeh, Z.; Sinibaldi, M.; et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia 2020, 34, 1102–1115. [Google Scholar] [CrossRef]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef]
- Boonchalermvichian, C.; Yan, H.; Baker, J.; Bubb, Q.R.; Gupta, B.; Czechowicz, A.; Negrin, R.S. Engineered Invariant Natural Killer T Cells (iNKT cells) Targeting CD 117 As a Novel Treatment for Acute Myeloid Leukemia. Transplant. Cell. Ther. 2025, 31, S201–S202. [Google Scholar] [CrossRef]
- Lin, P.; Reyes Silva, F.C.; Lin, P.; Gilbert, A.L.; Acharya, S.; Nunez Cortes, A.K.; Banerjee, P.; Fang, D.; Melo Garcia, L.; Daher, M.; et al. CD70 CAR NK Cells in the Treatment of Multiple Myeloma. Blood 2023, 142, 3463. [Google Scholar] [CrossRef]
- Zhou, X. Preclinical Development of Non-Edited CD7 CAR-Modified Invariant NKT Cell Therapy for T-Cell Malignancies and Acute Myeloid Leukemia. Blood 2022, 140, 7417–7418. [Google Scholar] [CrossRef]







| ClinicalTrials.gov ID | CAR-T | Conditions |
|---|---|---|
| NCT05859997 | BRL-301 | SLE, Sjogren’s Syndrome, Systemic Sclerosis, Inflammatory Myopathy, ANCA-Associated Systemic Vasculitis, Antiphospholipid Syndrome |
| NCT06978738 | CD19/BCMA UCAR | Inflammatory Myopathy, Autoimmune Haemolytic Anaemia, SLE, Systemic Sclerosis, ANCA-Associated Systemic Vasculitis, IgG4-RD, Myasthenia Gravis |
| NCT05988216 | BRL-301 | SLE |
| NCT06886919 | IC19 | Refractory SLE |
| NCT06939166 | CD19/BCMA UCAR | Autoimmune Encephalitis, Multiple Sclerosis, Chronic Inflammatory Demyelinating Polyradiculoneuropathy, Myasthenia Gravis, Neuromyelitis Optica Spectrum Disorders |
| NCT06828042 | CD19 CAR-γδT | Inflammatory Myopathies, SLE, Systemic Sclerosis, ANCA-Associated Systemic Vasculitis, Antiphospholipid Syndrome, Sjogren Syndrome |
| NCT06485232 | Universal BCMA + CD19 CAR-T | Neuromyelitis Optica Spectrum Disorders, Myasthenia Gravis, Multiple Sclerosis, Chronic Inflammatory Demyelinating Polyradiculoneuropathy |
| NCT07085104 | ALLO-329 | Idiopathic Inflammatory Myopathy, Lupus Nephritis, SLE, Systemic Sclerosis |
| NCT06941129 | CD19 + BCMA UCAR | SLE, Systemic Sclerosis, Inflammatory Myopathy, ANCA-Associated Systemic Vasculitis |
| NCT06920433 | CD19/BCMA UCAR | SLE |
| NCT07115745 | BMS-986515 | Refractory Autoimmune Disease |
| NCT07155369 | CD19/BCMA UCAR | SLE, Systemic Sclerosis |
| NCT06946485 | CHT101 | SLE |
| NCT06821659 | UWD-CD19 | Inflammatory Myopathies, SLE, Systemic Sclerosis, ANCA Associated Systemic Vasculitis, Sjogren Syndrome |
| NCT06691152 | CD19 UCAR | SLE |
| NCT06633042 | BCMA Universal CAR-T | AQP4 Antibody Positive Neuromyelitis Optica Spectrum Disease |
| NCT06340490 | RJMty19 | SLE |
| NCT06106893 | CD19 γδ T | SLE |
| NCT06212154 | ThisCART19A | Autoimmune Haemolytic Anaemia |
| NCT06752876 | CB-010 | SLE, Lupus Nephritis |
| NCT06980597 | OL-108 | Idiopathic Inflammatory Myositis, SLE, Systemic Sclerosis, ANCA-Associated Systemic Vasculitis |
| NCT06294236 | SC291 | SLE, ANCA-Associated Systemic Vasculitis, Granulomatous Polyangiitis, Microscopic Polyangiitis |
| NCT06733610 | Universal anti-CD19/BCMA CAR T | Autoimmune Haemolytic Anaemia |
| NCT06983964 | CD19 CART | Autoimmune Diseases |
| NCT06920446 | CD19/BCMA UCAR | Autoimmune Haemolytic Anaemia |
| NCT07105735 | RN1201 | Autoimmune Disease |
| NCT07129642 | Allogeneic Anti-CD19 CAR-T | Graves’ Disease |
| NCT07142161 | RD13-02 | Type 1 Diabetes |
| NCT06375993 | ADI-001 | Lupus Nephritis, SLE, Systemic Sclerosis, ANCA-Associated Systemic Vasculitis, Idiopathic Inflammatory Myopathies, Stiff Person Syndrome |
| NCT06925542 | CTX112 | Lupus Nephritis, SLE, Systemic Sclerosis, Idiopathic Inflammatory Myopathies |
| NCT07031713 | CT1192 | SLE |
| NCT06933563 | CD19/BCMA UCAR | Myasthenia Gravis |
| NCT07072247 | RN1201 | Immune-mediated Platelet Transfusion Refractoriness, Immune Thrombocytopenia |
| NCT06986018 | RD06-04 | ANCA-Associated Systemic Vasculitis, Idiopathic Inflammatory Myopathies |
| NCT06308978 | FT819 | Idiopathic Inflammatory Myositis, SLE, Systemic Sclerosis, ANCA-Associated Systemic Vasculitis |
| NCT06686524 | Anti-CD19 UCAR-T | Refractory Juvenile Dermatomyositis |
| NCT06681337 | BCMA + CD19 CAR-T | Lupus Nephritis |
| NCT07100873 | ADI-001 | Rheumatoid Arthritis |
| NCT07033299 | CT1192 | ANCA-Associated Systemic Vasculitis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osborne, M.; Maher, J. Prospects for Development and Commercialisation of Allogeneic CAR-Based Therapies for Autoimmune Disease. Biology 2025, 14, 1790. https://doi.org/10.3390/biology14121790
Osborne M, Maher J. Prospects for Development and Commercialisation of Allogeneic CAR-Based Therapies for Autoimmune Disease. Biology. 2025; 14(12):1790. https://doi.org/10.3390/biology14121790
Chicago/Turabian StyleOsborne, Madeleine, and John Maher. 2025. "Prospects for Development and Commercialisation of Allogeneic CAR-Based Therapies for Autoimmune Disease" Biology 14, no. 12: 1790. https://doi.org/10.3390/biology14121790
APA StyleOsborne, M., & Maher, J. (2025). Prospects for Development and Commercialisation of Allogeneic CAR-Based Therapies for Autoimmune Disease. Biology, 14(12), 1790. https://doi.org/10.3390/biology14121790