Simple Summary
Non-alcoholic fatty liver disease (NAFLD) is an unmet medical need due to its increasingly high incidence, severe clinical consequences, and the absence of feasible diagnostic tools and effective drugs. This review summarizes the preclinical and clinical data on adipokines, cytokine-like hormones secreted by adipose tissue, and NAFLD. The aim is to establish the potential of adipokines as diagnostic and prognostic biomarkers, as well as their potential as therapeutic targets for NAFLD. The limitations of current research are also discussed, and future perspectives are outlined.
Abstract
Non-alcoholic fatty liver disease (NAFLD) has become the major cause of chronic hepatic illness and the leading indication for liver transplantation in the future decades. NAFLD is also commonly associated with other high-incident non-communicable diseases, such as cardiovascular complications, type 2 diabetes, and chronic kidney disease. Aggravating the socio-economic impact of this complex pathology, routinely feasible diagnostic methodologies and effective drugs for NAFLD management are unavailable. The pathophysiology of NAFLD, recently defined as metabolic associated fatty liver disease (MAFLD), is correlated with abnormal adipose tissue–liver axis communication because obesity-associated white adipose tissue (WAT) inflammation and metabolic dysfunction prompt hepatic insulin resistance (IR), lipid accumulation (steatosis), non-alcoholic steatohepatitis (NASH), and fibrosis. Accumulating evidence links adipokines, cytokine-like hormones secreted by adipose tissue that have immunometabolic activity, with NAFLD pathogenesis and progression; however, much uncertainty still exists. Here, the current knowledge on the roles of leptin, adiponectin, ghrelin, resistin, retinol-binding protein 4 (RBP4), visfatin, chemerin, and adipocyte fatty-acid-binding protein (AFABP) in NAFLD, taken from preclinical to clinical studies, is overviewed. The effect of therapeutic interventions on adipokines’ circulating levels are also covered. Finally, future directions to address the potential of adipokines as therapeutic targets and disease biomarkers for NAFLD are discussed.
Keywords:
adipokines; NAFLD; liver; steatosis; inflammation; fibrosis; biomarkers; therapy; clinical trials; preclinical studies 1. Introduction
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of liver abnormalities characterized by increased hepatic fat content (>5%)—steatosis—in the absence of secondary causes, namely excessive alcohol consumption (>20 g/day for women and >30 g/day in men), medications, viral hepatitis, or certain hereditable conditions. Histologically, NAFLD can be categorized into non-alcoholic fatty liver (NAFL), when there is only evidence of hepatic steatosis, and non-alcoholic steatohepatitis (NASH), when, apart from steatosis, lobular inflammation and hepatocyte ballooning with or without perisinusoidal fibrosis can be observed. NASH, especially in the fibrotic state, often presents worse prognoses and frequently progresses to more severe conditions, such as cirrhosis and hepatocellular carcinoma [1].
NAFLD has become the major cause of chronic hepatic illness in children and adults, as well as the leading indication for liver transplantation in the future decades, replacing chronic hepatitis C. This high-incident pathology is also associated with extra-hepatic complications, such as chronic kidney disease and cardiovascular disease, which is the leading cause of death among NAFLD patients [2]. Mainly asymptomatic, NAFLD is considered a major societal, clinical, and research challenge due to its increasingly high prevalence, difficulties in its diagnosis, the lack of approved therapies, and its complex pathophysiology.
The development and progression of NAFLD is induced by multiple factors in a “multiple parallel-hit” model, where numerous genetic and environmental determinants (“hits”) interplay on an individual basis. These factors encompass, but are not limited to, genetic alterations, inflammation, gut dysbiosis, and metabolic abnormalities. Indeed, almost 90% of NAFLD patients present at least one of the metabolic syndrome features (abdominal obesity, hypertriglyceridemia, low HDL-cholesterol, hypertension, and high fasting glucose), and about 33% fulfill the criteria for diagnosing metabolic syndrome [3]. Therefore, metabolic-associated fatty liver disease (MAFLD) was recently proposed as a more appropriate overarching term that recognizes the metabolic risk profile of patients as the master criterion for diagnosis and considers the disease activity grade to be a continuum, which better describes disease pathophysiology [4]. Therefore, it is not surprising that the worldwide prevalence of NAFLD is rising in parallel with obesity [2], the main driver of metabolic abnormalities. Stepping up the strict correlation between obesity and NAFLD, there is a dose-response between weight loss and histological disease improvement; a 7% weight loss reverts NASH in 65–90% of patients, and a ≥10% weight loss causes fibrosis relapse in 45% of patients [3].
In the last 10 years, researchers have gained substantial insights into the role of adipose tissue in NAFLD. Initially seen as a simple energy storage tissue, adipose tissue is now recognized as an active endocrine organ, the dysfunction of which impacts either the initial stages of liver steatosis or the progression to NASH and fibrosis [5]. Adipokines, cytokine-like hormones secreted by adipose tissue, have emerged as cornerstone players in the regulation of energy metabolism, inflammation, and fibrosis [6,7], making them potential therapeutic targets for NAFLD. Furthermore, a vast number of clinical trials have established the serum profiles of classical (leptin and adiponectin) and emergent adipokines, such as ghrelin, resistin, retinol binding protein 4 (RBP4), visfatin, chemerin, and adipocyte fatty acid-binding protein (AFABP), in NAFLD patients [8].
In this review, we briefly summarize the main aspects of the adipose tissue–liver communication driving NAFLD pathophysiology. Following this, we overview the current knowledge on the role of adipokines in the development and progression of NAFLD and present data from clinical studies on adipokine profile in NAFLD patients. Moreover, we cover the effect of therapeutic interventions on circulating adipokines in the context of the management of NAFLD-associated metabolic complications. Finally, we discuss future directions to address the potential of adipokines as therapeutic targets and disease biomarkers for NAFLD.
2. NAFLD, an Unmet Medical Need
Over the last century, dramatic changes in lifestyle behaviors have evoked a growing incidence of noncommunicable diseases. The Global Burden of Disease Study 2017 estimated the prevalence of NAFLD for the first time, reflecting that the emerged epidemic in chronic liver disease is related to the burden of this pathology [9]. Parallel to the worldwide augment of obesity, the global prevalence of NAFLD is currently estimated to be 24%, with South America and the Middle East having the highest rates, followed by Asia, the USA, and Europe [2]. The clinical consequences of NAFLD, particularly its histological phenotype NASH, which likely progresses to cirrhosis and hepatocellular carcinoma, are the second-most common indication of liver transplantation in the USA [2]. NAFLD also potentially contributes to the burden of extra-hepatic chronic complications, namely cardiovascular disease, type 2 diabetes (T2DM), and chronic kidney disease [2]. Aggravating the health and economic consequences of NAFLD, weight-management difficulties, together with unavailability of routinely feasible diagnostic procedures and effective pharmacotherapeutic approaches, challenged the increasing need of the management of this complex and frequently asymptomatic pathology.
The “gold standard” method for diagnosing NASH (and differentiation from NAFL) is liver biopsy, even though it has limitations related to invasiveness, patient discomfort, risk of adverse events, sampling variability, pathologist experience, and unsuitability for large-scale screening, as well as its debatable cost-effectiveness needs, since approved NASH-specific therapies are not presently available [3]. A non-invasive multistep approach for NAFLD diagnosis and follow up, which considers patients clinical history (patients with obesity and/or metabolic syndrome or T2DM are at high risk of NAFL/NASH), imaging techniques (such as ultrasonography and transient elastography), biomarkers, and scoring systems to estimate steatosis or fibrosis (NAFLD Activity Score or NAS, Fibrosis-4 index or FIB-4, NAFLD Fibrosis Score or NFS, and Aspartate aminotransferase to Platelet Ratio Index or APRI), is a useful tool to guide disease management. However, these predictive approaches have decreased sensitivity and specificity to the earlier and more moderate stages of fibrosis and have broad diagnostic grey zones [10]. Therefore, liver biopsy remains the only reliable diagnostic tool for NAFL/NASH identification and assessing fibrosis severity.
Despite the increasingly high incidence of NAFLD worldwide, its associated morbidity and mortality, and the intensive research on the field, there is no specific drug approved for its treatment, and lifestyle change remains the cornerstone of NAFLD management, as indicated by clinical guidelines [11]. If exercise and diet fail to achieve the targets in NAFLD patients, bariatric surgery or pharmacotherapy to manage the underlying metabolic complications, namely obesity, T2DM, dyslipidemia and cardiovascular disease, is recommended [12]. The current and emerging pharmaceutical therapeutic strategies for NASH aim to improve metabolic function, reduce steatosis, decrease inflammation, and halt or reverse the progression of fibrosis. Among the extensive list of potential pharmacotherapies are those targeting oxidative stress (vitamin E), insulin resistance (IR) or high glucose blood levels (thiazolidinediones, metformin and sodium-glucose cotransporter-2 inhibitors), and dyslipidemia/lipotoxicity (statins). Nevertheless, to date, no molecule studied for the treatment of NAFLD has demonstrated convincing effects on halting or reverting liver disease [13].
3. Adipose Tissue-Liver Axis in NAFLD Pathophysiology
Adipose tissue (AT) and the liver play a critical role in the regulation of whole-body energy homeostasis. Triglycerides (TG) and excess carbohydrates from the diet are converted into fatty acids (FA) in the liver. In particular, dietary TG are emulsified by bile acids and hydrolyzed by pancreatic lipase, yielding FA, while excess carbohydrates are converted into FA by de novo lipogenesis (DNL). Hepatic FA are then esterified and stored in cytoplasmatic lipid droplets or secreted into bloodstream. Circulating FA are uptaken and used as energy sources by brown adipose tissue and muscle or re-esterified to TG and stored in white adipose tissue (WAT). In WAT, insulin-stimulated FA uptake and TG synthesis inhibits the intracellular lipolysis of cytosolic TG, thereby promoting adipocyte TG storage [14].
In overweight and obese subjects, the capacity for WAT expansion reaches its limit; therefore, energy cannot be stored in WAT, and ectopic lipid accumulation occurs in other tissues, including the liver. Resistance to insulin also promotes lipolysis in adipocytes with a further increase of circulating free FA. Consequently, intrahepatic fat accumulation (steatosis) develops, which can be further amplified by FA synthesis from excess carbohydrates through DNL. At this stage, TGs-derived toxic metabolites can cause hepatic lipotoxicity, this results in cellular dysfunction, lipoapoptosis, IR, and the activation of hepatic innate immune cells, including dendritic cells, Kupffer cells, and hepatic stellate cells (HSCs); all of these are essential features of NASH [15].
Beyond its energy-storage functions, WAT is a recognized active endocrine and an immunological organ constituted by mature and developing adipocytes, endothelial cells, fibroblasts, and immune cells that maintain tissue homeostasis, such as AT macrophages, eosinophils, mast cells, neutrophils, and T and B cells [16]. In the WAT of lean subjects, eosinophils and T regulatory cells (Treg) secrete anti-inflammatory cytokines, such as interleukin (IL)-4 and IL-10, that polarize AT macrophages towards the M2 phenotype (anti-inflammatory phenotype), thus preserving a tolerogenic environment [17]. Adipocyte expansion derived from TG accumulation is accompanied by adipocyte hypoxia, cell stress, and apoptosis, able to promote chemoattractant molecules’ expression and immune-cell infiltration. The immune profile of WAT is then modified; T cells become activated, the presence of T reg is reduced, and the macrophage’s phenotype switches from M2 to M1 (pro-inflammatory phenotype), which accumulates around necrotic adipocytes forming ‘crown-like structures’ and producing pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and IL-6 [16]. Of relevance, this pro-inflammatory profile is correlated with local and systemic IR [18].
In the WAT of obese subjects, a deregulated production of cytokine-like hormones, known as adipokines, is observed. This family of low-molecular-weight, bioactive peptides has a proven pleiotropic function as hormones and cytokines with both pro- and anti-inflammatory activities. Namely, adipokines show a fundamental role in energy metabolism by communicating the nutrient status of the body through the induction of anorexigenic signals and the suppression of orexigenic factors at the hypothalamus [19]. Adipokines have also been revealed to be critical regulators of hepatic lipogenesis and insulin sensitivity; for example, adiponectin has been shown to augment insulin sensitivity, maintain healthy AT expansion, and rescue the body from ectopic lipid accumulation [20]. Moreover, adipokines were highlighted as cornerstone modulators of both the innate and adaptive immune systems. For instance, leptin, the first adipokine to be discovered, modulates both the innate immune response, by increasing natural killer cells cytotoxicity and modulating granulocytes, macrophages, or dendritic cell activation, and the adaptive immunity, by increasing naïve T and B cell proliferation, promoting a switch towards a pro-inflammatory Th1 phenotype, and reducing Treg proliferation [6]. The deregulated production of adipokines thus contributed to obesity-associated systemic chronic low-grade inflammation, and the liver is progressively infiltrated by immune cells, such as monocytes, neutrophils, and T cells. When inflammation perpetuates, fibrogenesis starts with the progression to NASH with fibrosis, the most severe grade of the disease [21].
In summary, the adipose tissue–liver axis is essential for the regulation of glucose and lipid metabolism, as well as immunological homeostasis, and a dysfunctional secretion of adipokines from WAT could (i) determine the fluxes of lipid to the liver and, thus, hepatic steatosis; and (ii) promote hepatic inflammation (characteristic of NASH) that, if not managed in a timely fashion, can progress to hepatic fibrosis (Figure 1). In this context, adipokines (overviewed in Table 1) have been identified as potential therapeutic targets and investigated as diagnostic and prognostic biomarkers of NAFLD. The following sections review the current knowledge on adipokines in NAFLD, focusing on their activity on NAFLD pathophysiological mechanisms, namely IR, hepatic lipid accumulation, inflammation, and fibrosis. Moreover, clinical studies establishing the serum profiles of adipokines in NAFLD and therapeutic approaches used in the management of NAFLD and affecting adipokines levels in humans were described. Since very recent reviews summarized the action of leptin [22,23] and adiponectin [24,25] in NAFLD in detail, here we will focus on the preclinical and clinical data of less-studied adipokines (ghrelin, resistin, RBP4, visfatin, chemerin, and AFABP) in NAFLD, briefly overviewing the activity of leptin and adiponectin.
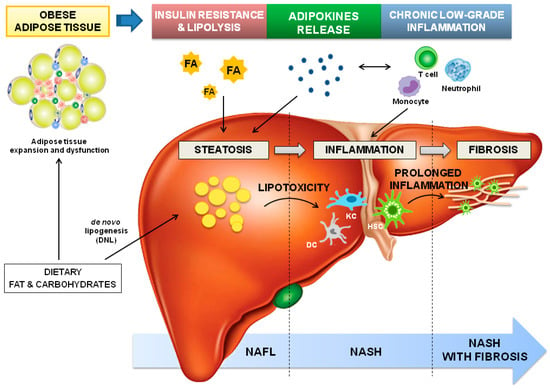
Figure 1.
Dysfunctional adipose tissue-driven pathophysiology of NAFLD. The expansion of white adipose tissue in obesity induces adipocyte dysfunction and insulin resistance leading to lipolysis. As a consequence, circulating fatty acid (FA) and adipokine imbalance towards pro-steatogenic ones contribute to intrahepatic fat accumulation (steatosis), which is amplified by the high dietary fat and carbohydrate levels (commonly observed in obesity), the latter augmenting de novo lipogenesis (DNL). The ectopic accumulation of triglycerides (TGs)-derived toxic metabolites induces lipotoxicity and the consequent cellular dysfunction (endoplasmic reticulum and oxidative stress, and mitochondrial defects), lipoapoptosis and activation of inflammatory pathways. The expansion of adipocytes also elevates the expression of chemoattractants, and immune cells infiltration, and promotes deregulation of adipokines secretion; altogether, contributing to systemic chronic low-grade inflammation. Additionally, in the liver, dendritic cells (DC), Kupffer cells (KC), and hepatic stellate cells (HSCs) are activated, and hepatic infiltration by circulating immune cells, including neutrophils, monocytes, and T-lymphocytes, occurs. When inflammation perpetuates, fibrogenesis starts, with HSCs as key players. Abbreviations: DC-Dendritic cells; DNL-de novo lipogenesis; FA-Fatty acids; HSC-Hepatic stellate cells; KC-Kupffer cells; NAFL-Non-alcoholic fatty liver; NASH-Non-alcoholic steatohepatitis; TGs- triglycerides.

Table 1.
Overview of adipokines.
4. Adipokines as Potential Therapeutic Targets for NAFLD
4.1. Leptin and Adiponectin
Leptin, the forerunner and best-characterized member of the adipokine family, plays a pivotal role in appetite and body-weight homeostasis by augmenting anorexigenic neuropeptides and suppressing orexigenic factors in the central nervous system [6]. Likewise, leptin has been described as modulating several physiological processes, such as lipid and glucose metabolism, as well as both innate and adaptive immunity [6]. Most of the current knowledge about leptin’s action arose from leptin-deficient ob/ob mice and leptin-receptor-deficient db/db mice. These murine models exhibited marked hepatic alterations, such as IR, accumulation of TG and lipids, and steatosis, which were partially reverted following leptin administration to ob/ob mice [22,23]. Although leptin acts as an anti-steatotic hormone preventing the accumulation of and promoting the mobilization of hepatic lipids, leptin resistance may cause the leptin inability to alleviate hepatic steatosis [22,23]. Furthermore, high leptin plasma levels derived from obese adipose tissue are associated with hepatic inflammatory and fibrogenic mechanisms and therefore, with NAFLD development [22,23].
On the contrary, adiponectin, an adipocyte-secreted hormone inversely correlated with obesity and with determining roles in insulin sensitivity, glucose levels, and lipid metabolism [25], has been reported to protect the liver from steatosis, inflammation, and fibrosis [24]. Adiponectin augments insulin’s capacity to suppress glucose production, prevents hepatic DNL, suppresses FA synthesis in hepatocytes, and enhances FA β-oxidation [24], overall protecting the liver from steatosis. Adiponectin was also described as decreasing the production of inflammatory cytokines, such as IL-6 and TNF-α, through the modulation of toll-like receptor 4 (TLR4) signaling. By boosting the beneficial effects of adiponectin in NAFLD development, this adipokine was described to possess anti-fibrotic effects by preventing leptin profibrogenic signaling [24]. Therefore, strategies aiming to rescue the leptin–adiponectin balance, i.e., reverting the obesity-associated increased leptin levels and reduced adipokines levels, are of relevance for NAFLD treatment [24]; this will be further explored in Section 5.
4.2. Ghrelin
Ghrelin stands out as one of the few peripheral peptide hormones with an orexigenic effect. Initially identified as a stomach-derived hormone, ghrelin is an endogenous ligand for the growth hormone secretagogue receptor 1a (GHSR1a). It stimulates food intake and adiposity and acts as a regulator of glucose metabolism, reward behavior, gut motility, or even hepatic lipid metabolism and the immune system [26,27]. This hormone circulates in blood in two forms, an acylated (AG) form and an unacylated form (UAG, also named DAG from desacyl ghrelin). This post-transcriptional modification is catalyzed by ghrelin O-acyltransferase (GOAT), and the producing AG is the active form that triggers the signaling of the cognate receptor GHSR1a. Initially thought to be an inactive form, it has been suggested that UAG to antagonizes AG activity on glucose metabolism and lipolysis and reduces food consumption and body weight [27,34]. Given its regulatory activity on metabolism and immune system, there is a growing interest on the role of ghrelin-GOAT system in the development and progression of NAFLD.
In murine models, the administration of ghrelin during or after diet-induced NAFLD development counteracts dysregulated hepatic lipid metabolism, oxidative stress, apoptosis, and inflammation [35,36]. Furthermore, the action mechanisms involved in the beneficial effects of ghrelin on NAFLD were investigated. The amelioration of liver injury by ghrelin was accompanied by the reestablishment of phosphoinositide 3-kinase (PI3K)/Akt and liver kinase B1 (LKB1)/AMP-activated protein kinase (AMPK) pathways [35]. Ghrelin also attenuates lipotoxicity by upregulating autophagy via AMPK/mTOR restoration and inhibiting nuclear factor kappa B (NF-κB) [36]. Recently, ghrelin was demonstrated to block the progression of NASH induced by lipopolysaccharide (LPS) in mice fed with a high-fat diet through the reduction of Kupffer cells’ M1 polarization, which is mediated by GHSR1a [37]. In addition to these results demonstrating the beneficial effects of ghrelin, the genetic deletion of ghrelin in mice also significantly reduced age-associated hepatic steatosis, partly by downregulating diacylglycerol O-acyltransferase-1, one of the key enzymes of TG synthesis [38].
Concerning the effects of the ghrelin’s isoforms, it was verified that plasma UAG levels were reduced after a sleeve gastrectomy and a Roux-en-Y gastric bypass in Wistar rats, whereas the AG:UAG ratio was augmented. Concomitantly, both surgeries diminished obesity-associated hepatic steatosis, inflammation, mitochondrial dysfunction, and endoplasmic reticulum stress. Moreover, AG has a similar effect in palmitate-treated hepatocytes, which suggests that the increased AG:UAG ratio after bariatric surgery might ameliorate obesity-associated NAFLD [39]. Possible underlying mechanisms are the reduction of lipogenesis and stimulation of mitochondrial FA β-oxidation, as well as hepatic autophagy, by relatively increased AG levels [40]. Nevertheless, in lean rats, the administration of exogenous AG induced hepatic IR and lipid accumulation, while the co-administration of UAG prevented the AG-induced effects [41]. Thus, further evaluation of the ghrelin-GOAT system and the effects of AG and UAG isoforms on NAFLD development is needed.
4.3. Resistin
Resistin (named for its ability to induce “resistance to insulin”) is the founding member of resistin-like molecules (RELMs), a family of small, secreted cysteine-rich peptides with hormone-like and pro-inflammatory activities. It is mainly secreted by adipose tissue and inflammatory cells, and its action is thought to be mediated by the TLR4 receptor, although the receptors tyrosine kinase-like orphan receptor (ROR)-1, insulin-like growth factor type 1 receptor (IGF-1R), and adenylyl cyclase-associated protein 1 (CAP1) have also emerged as potential candidate receptors [28]. Resistin has been shown to have pleiotropic effects, including the regulation of blood–glucose levels and lipid metabolism, as well as the induction of pro-inflammatory cytokines secretion or monocyte differentiation into macrophages [28]. In fact, resistin administration to C57BL/6J mice increased blood glucose, i.e., impaired glucose tolerance, due to the decreased insulin sensitivity, which was rescued after the administration of an anti-resistin antibody [42]. Similarly, the high-fat diet (HFD)-fed mice presented increased plasma resistin levels and severe IR, which is completely reversed after treatment with a resistin antisense oligonucleotide [43]. More recently, the mechanisms of action of resistin in a HFD-induced NAFLD model were disclosed [44]. Acute elevated resistin altered mitochondrial morphology and content, increased lipid accumulation, and up-regulated pro-inflammatory mediators in HFD-fed mice and palmitate-treated HepG2 cells. Furthermore, steatosis aggravation induced by resistin in mice is mediated by the AMPK/peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) pathway [44]. It was also reported that resistin treatment augments the suppressor of cytokine signaling 3 (SOCS3) expression, a suppressor of insulin signaling, in adipocytes [45].
Resistin-deficient mice demonstrated reduced hepatic glucose production and, consequently, their blood–glucose levels after fasting were low [46]. In addition, ob/ob mice and diet-induced obese mice, both lacking resistin, had reduced hepatic steatosis, since the expression of genes involved in hepatic lipogenesis and the secretion of very-low-density lipoprotein (VLDL) were decreased [47].
At the cellular level, resistin hampered glycogen synthase kinase 3β (GSK3β) phosphorylation in primary rat hepatocytes under hyperinsulinemic and hyperglycemic conditions, with further reduction of glycogen synthesis and hepatic insulin action [48]. Resistin also exerts pro-inflammatory activity by inducing the expression of cell adhesion molecules and pro-inflammatory cytokines in macrophages and mononuclear cells, contributing to the recruitment of leukocytes to inflammation sites [28]. Moreover, the secretion of resistin by infiltrated monocytes/macrophages is enhanced by pro-inflammatory mediators [28]. Of note, hepatic myeloid cells and T-lymphocytes from NAFLD patients showed a decreased response to resistin, which is associated with a failure to maintain redox homeostasis, which would be a risk factor for NAFLD severity [49]. It was also verified that resistin increased hepatic inflammation through mitogen-activated protein kinase (MAPK) signaling and the activation of a coagulation cascade in animal models [50]. Finally, resistin demonstrated profibrogenic effects by activating HSCs, which release IL8/CXCL8 and monocyte chemoattractant protein (MCP)-1/CCL2 via NF-κB, and increasing the transforming growth-factor beta (TGFβ) and collagen type I production in Kupffer cells [51,52].
4.4. Retinol Binding Protein 4 (RBP4)
Distinct mouse models have been used to elucidate the RBP4 activity in metabolic diseases. In general, elevated circulating and adipose tissue RBP4 levels have been correlated with IR, dyslipidemia, and T2DM [29]. The possible RBP4-dependent mechanisms contributing to IR include impaired insulin signaling, the down-regulation of GLUT-4 translocation, and the induction of inflammatory and lipolytic pathways in adipose tissue, as well as the induction of phosphoenolpyruvate carboxykinase in the liver, thereby increasing glucose production [29].
In genetic and dietary mouse models of NAFLD, the results of hepatic expression of RBP4 are controversial. Liu et al. observed an abnormal hepatic RBP4 expression in apoE−/− mice fed with a high-fat and high-cholesterol (HFC) diet [53]. However, Saeed et al. described reduced hepatic RBP4 levels in C57BL/6J mice fed with a HFC diet and in ob/ob mice, while serum RBP4 levels were increased in both models [54]. Apart from that, transgenic mice overexpressing human RBP4 had increased hepatic lipid accumulation, cellular ballooning, and inflammation, which were exacerbated when mice were challenged with a high-fat diet, likely through RBP4-induced mitochondrial dysfunction [53,55]. Of note, recently, novel RBP4 antagonists were reported to reduce hepatic steatosis in transgenic mice with adipocyte-specific overexpression of RBP4 [56].
4.5. Visfatin
Nicotinamide phosphoribosiltransferase (NAMPT), also called pre-B cell colony-enhancing factor (PBEF) or visfatin, functions as an intracellular enzyme (iNAMPT) mediating the synthesis of nicotinamide adenine dinucleotide (NAD+) and as a cytokine-like soluble factor secreted into extracellular space (eNAMPT) [31]. Intracellular NAMPT regulates mitochondrial biogenesis, cellular metabolism, and survival, as well as the adaptive response to cell stress; it was described as modulating pancreatic β-cell function, likely regulating glucose homeostasis and IR [30]. On the other hand, extracellular NAMPT acts mainly as an inductor of pro-inflammatory cytokine production [31]. The NAMPT extracellular form has been associated with metabolic and inflammatory disorders, but its pathophysiological mechanisms are still ill-defined [31].
Administration of NAMPT to a methionine-choline-deficient (MCD)-diet-fed mouse model of NAFLD aggravated hepatic steatosis, increased inflammatory cell infiltration and inflammatory cytokines levels and exacerbated the expression of fibrotic markers in the liver, together with the induction of endoplasmic reticulum and oxidative stress [57]. In hepatocytes, NAMPT also induced the expression of inflammatory cytokines and diminished insulin signaling through a signal transducer and activator of transcription 3 (STAT3) and NF-κB activation [58]. These results support the adverse effects of NAMPT in hepatic steatosis, inflammation, and fibrosis. However, the pharmacologic inhibition or genetic ablation of NAMPT also showed deleterious effects. The intracellular NAMPT inhibitor FK866 promoted liver steatosis in HFD-fed mice and hepatic lipid accumulation in vitro via the sirtuin 1 (SIRT1)/sterol regulatory element-binding protein 1 (SREBP1)/fatty acid synthase (FASN) pathway [59]. Similarly, the knockdown of NAMPT in hepatocyte cells led to TG accumulation through the regulation of DNL via SIRT1/AMPK pathway [60]. Accordingly, the overexpression of NAMPT in hepatocyte cell lines mitigated lipid accumulation [59]. However, hepatocyte-specific NAMPT knockout mice on low-methionine and choline-free high-fat diet showed less TG accumulation than wild-type controls but had augmented histological scores for hepatic inflammation, necrosis, and fibrosis [61]. Of note, NAMPT KO mice on a control diet also showed liver injury, since they had decreased mitochondrial proteins and respiratory capacity and increased fibrosis due to low NAD+ levels [61]. Taken together, the present data suggest that the opposing activity of NAMPT in NAFLD pathophysiology could be derived from the different roles of extracellular and intracellular NAMPT, but further studies are needed.
4.6. Chemerin
Chemerin, also named tazarotene-induced gene 2 (TIG2) and retinoic acid responder 2 (RARRES2), is secreted as inactive precursor, which is activated by proteases of the coagulation cascade, neutrophil-derived proteases (elastase and cathepsin G), bacterial proteases, and mast cell products (tryptase) [32]. This chemotactic adipokine binds to the G protein-coupled receptor chemokine-like receptor 1 (CMKLR1), which is expressed in dendritic cells, macrophages, and natural killer (NK) cells and may serve as bridge between innate and adaptive immunity [62]. Although the C-C chemokine receptor-like 2 (CCRL2) and the G protein-coupled receptor 1 (GPR1)/CMKLR2 were also described as chemerin receptors, their physiological activity is still uncertain. Chemerin and its receptor CMKLR1 are both expressed in adipose tissue [63], and they have been reported to be augmented in obesity and IR states (T2DM), decreasing after weight loss [64]. This adipokine also seems to regulate adipocyte differentiation, glucose and lipid homeostasis, and insulin sensitivity [32].
In addition to its ability to regulate glucose metabolism, IR, and inflammation, the role of chemerin in NAFLD is still unclear. The administration of recombinant chemerin ameliorate HFD-induced NASH in mice, as well as IR, leptin resistance, and liver lesions, by alleviating oxidative stress and promoting autophagy, at least in part, due to chemerin/CMKLR1-dependent activation of janus kinase 2 (JAK2)-STAT3 pathway [65]. On the contrary, the administration of a chemerin-derived C15 peptide did not affect hepatic TG accumulation, inflammation, or fibrotic gene expression in atherogenic diet-induced murine NASH [66]. Moreover, PI3K inhibition mitigated liver steatosis and KC-mediated inflammation due to the down-regulation of the chemerin receptor CMKLR1 in the liver [66]. Nevertheless, whole-body Cmklr1-gene abrogation in mice did not affect either the hepatic lipid accumulation, inflammation, fibrotic gene expression, NAS, or the IR [67]. Inconsistencies in the current data could be related to the differential modulation of hepatic chemerin in distinct murine models of NAFLD [68].
4.7. Adipocyte Fatty Acid-Binding Protein (AFABP)
There is strong evidence correlating elevated AFABP with IR and adipose tissue lipolysis in obesity and metabolic syndrome [33]. Interestingly, a recent review pointed out that AFABP as a metabolic/functional marker regulating macrophage functions likely having a determining role in pathophysiology [69]. Concerning to NAFLD, AFABP expression was elevated in Kupffer cells in both LPS-induced acute liver injury and diet-induced NAFLD [70]. In these NAFLD mice models, the pharmacological inhibition of AFABP ameliorated hepatic steatosis, macrophage infiltration, and hepatocellular ballooning [70]. Genetic ablation and the pharmacological inhibition of AFABP also attenuated bile-duct-ligation- and carbon-tetrachloride-induced liver fibrosis in mice through the reduction of collagen accumulation, liver sinusoidal endothelial cells (LSEC) capillarization, and HSC activation [71]. Mechanistically, elevated AFABP promotes LSEC capillarization, an early event of NAFLD pathogenesis, and LSEC-derived AFABP activate HSCs that augments TGFβ production and further extracellular matrix accumulation and fibrosis [71]. Furthermore, AFABP could promote hepatic inflammation through Kupffer cell activation [70]. Altogether, these findings suggest pharmacological inhibition of AFABP as a promising therapeutic strategy for NAFLD.
5. Adipokines in NAFLD: Evidence from Clinical Studies
5.1. Ghrelin
Clinical studies have demonstrated that obese patients with IR or metabolic syndrome had lower UAG and total ghrelin levels, while the AG:UAG ratio was elevated [72,73]. Moreover, the AG:UAG ratio was positively correlated with IR in both obese children and adults [72,74]. After the proof about the influence of ghrelin on insulin sensitivity, some research focused on ghrelin’s role in NAFLD, but the evidence is scarce, and not all studies take into consideration the concentrations of total ghrelin and its isoforms, AG and UAG.
Ghrelin levels were associated negatively with body mass index (BMI) in obese NAFLD patients diagnosed by ultrasonography [75] or biopsy [76]. Compared to matched healthy controls, patients with NAFLD showed reduced ghrelin levels, which correlated with IR [77]. However, in a study with NAFLD biopsy-proven patients that underwent bariatric surgery, UAG levels were increased in NASH patients compared to non-NASH subjects, while similar levels of AG were observed. Additionally, higher levels of AG, but not UAG, were observed in higher stages of fibrosis [78]. Further evidencing the role of ghrelin in NAFLD, recent case-control retrospective studies of biopsy-proven NAFLD patients suggested that the Leu72Met (rs696217 G > T) polymorphism and the “GG” genotype of rs26802 variant in the ghrelin gene have a protective effect against NAFLD [79,80].
5.2. Resistin
As described above, the positive correlation between resistin and IR, steatosis, and inflammation is well stablished in murine and cellular models, but data in human NAFLD are conflicting. A systematic review of Bekaert et al. found 12 studies reporting resistin levels in biopsy-proven NAFLD patients, of which 6 reported statistically significant results [81]. Circulating resistin levels were positively correlated with hepatic steatosis, portal inflammation, and NAFLD ACTIVITY SCORES in non-diabetic NAFLD patients [82,83,84,85]. Of relevance, Aller et al. found that resistin association with the steatosis grade was lost when the homeostatic model assessment of insulin resistance (HOMA-IR) parameter was included in the multivariate logistic analysis, indicating that resistin is a surrogate marker of IR [83]. Supporting the relevance of resistin in NAFLD, a predicting diagnostic biomarker panel for histological NASH in obese subjects included the serum levels of resistin together with adiponectin and cytokeratin 18 (marker of cell death) [86]. However, resistin was not included in the predicting algorithm for NASH or NASH-related fibrosis in a more recent study by the same group [87]. In contrast, a study described a negative correlation between circulating resistin levels and the steatosis grade in severely obese NAFLD patients [88]. The remaining studies included in the cited meta-analysis did not demonstrate an association of resistin with liver histological parameters in obese and non-obese NAFLD patients [89,90,91,92]. It is worth mentioning that, in this meta-analysis, 4 out of the 12 studies did not adjust their results for confounding factors, and the potential association between resistin and IR was conflicting among the studies [81]. More recently, the determination of serum resistin levels in severe obese NAFLD patients found no correlation with steatohepatitis or fibrosis severity [93]. Similarly, resistin circulating levels did not associate with steatosis grade, NASH diagnosis, hepatic ballooning, or lobular inflammation grade, but they did correlate with fibrosis stage in obese NAFLD patients [94].
Although there are ambiguous data on circulating resistin levels in NAFLD patients, the existing results on its hepatic expression are more consistent. Augmented hepatic resistin mRNA levels were reported in patients with NASH compared to steatosis or control subjects and in steatosis patients compared to control individuals [95]. Moreover, a positive association between hepatic resistin mRNA levels and hepatic steatosis, inflammation, or fibrosis was reported in several studies [95,96]. Additionally, a positive correlation between hepatic resistin protein expression and NAS, aspartate aminotransferase (AST), alanine aminotransferase (ALT), BMI, glucose, insulin, HOMA-IR, gamma-glutamyl transferase (GGT), lactate dehydrogenase (LDH), TG, and glycated haemoglobin was verified in obese NAFLD patients [97]. The resistin mRNA expression in subcutaneous adipose tissue was also verified to be increased in non-obese NAFLD patients, but no correlation with liver histological parameters was observed [84]. Interestingly, the immuno–histological assessment of hepatic samples of NAFLD patients revealed a resistin distribution predominantly in perisinusoidal cells (Kupffer cells and HSCs) [95], histiocytes of inflammatory infiltrate, and histiocytes surrounding the hepatocytes with steatosis [97]. Finally, a significant association was observed between resistin rs1862513 polymorphism and NAFLD [98], which supports the significance of resistin in the development of NAFLD.
5.3. Retinol Binding Protein 4 (RBP4)
Several clinical studies showed that elevated circulating RBP4 levels were associated with insulin-resistance states, namely obesity and T2DM [29]. In addition, decreased levels of RBP4 were correlated with recovering insulin sensitivity after weight loss or lifestyle intervention in obese adult or children populations [99,100]. Given the close association of NAFLD pathogenesis and IR, NAFLD is assumed to be correlated with increased levels of serum RBP4; however, inconsistent findings were observed. In studies without histological confirmation, serum RBP4 levels seemed to be positively correlated with liver fat [101] and were found to be higher in NAFLD patients than controls, in adult and pediatric subjects [102,103]. Nevertheless, a systematic review reported that only three out of seven studies verified a positive correlation between serum RBP4 levels and liver histology among patients with biopsy-proven NAFLD [81]. Similarly, a meta-analysis research did not find any significant differences between NAFLD, NASH, or SS patients compared to controls, neither between NASH nor SS patients [104]. The authors highlighted the heterogeneity across patient populations or the lack of adjustment for confounding factors in the analyzed research, which challenges comparisons between studies and limits the conclusions that can be drawn about the associations between adipokines levels and NAFLD. More recently, a 3-year follow-up study in a Chinese cohort of NAFLD patients diagnosed by abdominal ultrasonography verified that baseline serum RBP4 concentrations are positively associated with NAFLD development and inversely correlated with NAFLD regression [105]. Moreover, higher serum RBP4 levels were associated with an increased risk for prediabetes and metabolic syndrome in obese patients with NAFLD [106].
5.4. Visfatin
Several studies have evaluated the levels of visfatin in histologically confirmed NAFLD patients as well as the possible correlations with hepatic steatosis, inflammation, and fibrosis; but, current data are limited and inconclusive, as verified by two systematic reviews [81,107]. Most data reported similar serum visfatin levels in NAFLD [108], simple steatosis (SS) [109], or NASH patients [109,110] compared to control subjects, as well as in NASH compared to SS patients [109,111]. Likewise, similar hepatic visfatin expression was found in NASH and SS patients [112]. However, some studies also verified the augmented levels of serum visfatin in NAFLD compared to controls [113] or the decreased visfatin levels in NAFLD [114], SS, or NASH patients versus controls [86,111]. Of note, increased serum visfatin levels were associated with a reduced hepatic DNL index in women with ultrasound-diagnosed NAFLD, while in men it was correlated with augmented hepatic fat but not with DNL index, which suggests a sex-dependent interpretation for the serum visfatin levels in NAFLD prognosis [115].
Concerning histological parameters, most data did not report any correlation between serum visfatin and hepatic steatosis, inflammation, or fibrosis [108,109]. However, Aller et al. reported that circulating visfatin levels may predict portal inflammation, but not steatosis or fibrosis, in non-diabetic obese NAFLD patients [116]. In addition, Kukla et al. found a positive correlation between hepatic visfatin expression and the fibrosis stage but not hepatic steatosis and inflammation in morbidly obese NAFLD patients [112], while Gaddipati et al. reported a positive correlation between visfatin expression in visceral adipose tissue and steatosis degree in non-diabetic NAFLD patients [114].
Interestingly, visfatin was recently proposed as a potential serum biomarker related to the degree of hepatic steatosis and fibrosis among pediatric obese patients diagnosed by non-invasive methods (abdominal ultrasound and transient elastography with liver stiffness and controlled attenuation parameter) [117]. Moreover, a 10-year follow-up study verified no association between serum visfatin levels and leukocyte infiltration in fatty liver at the baseline, but visfatin serum levels were significantly increased during the follow-up, likely due to the combined effects of augmented BMI and diabetes prevalence [118]. However, this study had certain limitations, in particular, the use of ultrasonography as the NAFLD diagnosis method, without confirmation by hepatic biopsy, and the measurement of visfatin levels in all samples (basal and 10-years after) at the end of the study, which likely affects the visfatin concentrations due to different storage times.
Thus, the different methodological strategies used to study visfatin levels in human NAFLD likely determine the inconsistencies among the current data, and future research is still needed.
5.5. Chemerin
Since chemerin regulates insulin signaling and chronic inflammation, it is reasonable to hypothesize that this adipokine may be related to NAFLD development. In fact, elevated serum chemerin levels were identified as a risk factor for NAFLD development in T2DM patients [119] and were pointed out as a novel non-invasive serum marker predicting liver steatosis in obese children [120]. A recent meta-analysis [121] further explored the correlations between serum chemerin levels and NAFLD (steatosis and/or NASH) and its specific hepatic histologic lesions (liver steatosis, lobular and portal inflammation, and fibrosis). Overall, circulating chemerin levels were consistently higher in patients with NAFLD and steatosis compared to controls, although no significant difference was verified between NASH patients and controls. Moreover, data on serum chemerin levels and specific liver lesions are inconsistent, and no correlations were verified [81,121].
It was found that chemerin expression in visceral adipose tissue was negatively correlated with the steatosis score and NAFLD ACTIVITY SCORES of obese NAFLD patients, likely through the modulation of IR and, thus, NAFLD [122]. Data on hepatic chemerin mRNA expression are contradictory; its levels were found to be negatively associated with inflammation, fibrosis, and NAS, but not with steatosis, in non-obese NAFLD patients [123], while other studies verified an increased hepatic chemerin mRNA expression, as well as hepatic CMKLR1 expression, that correlated with hepatic steatosis, hepatocyte ballooning, and the NAFLD activity score in obese NAFLD patients [124,125]. Of note, the hepatic expression of both chemerin and CMKLR1 was associated with obesity [125], which can partially explain the inconsistency of the results.
5.6. Adipocyte Fatty Acid-Binding Protein (AFABP)
In ultrasound-diagnosed NAFLD patients, serum AFABP levels were higher in NAFLD patients compared with non-NAFLD group [126,127]. The same was observed in biopsy-proven NAFLD patients, where serum AFABP had an independent positive correlation with lobular inflammation and hepatocyte ballooning, even after adjusting for confounding factors [128,129]. Milner et al. also reported higher serum AFABP levels in NASH patients compared with SS and correlated AFABP with IR, adiposity, and the fibrosis stage [128]. Nevertheless, other studies did not verify an association between AFABP and fibrosis, or that this adipokine was able to distinguish NASH from non-NASH patients [110]. In summary, serum AFABP levels are elevated in NAFLD, but its correlation with NASH, and particular fibrosis, is still unclear.
In addition to uncertainties, the general accepted roles of adipokines on NAFLD development are summarized in Figure 2.
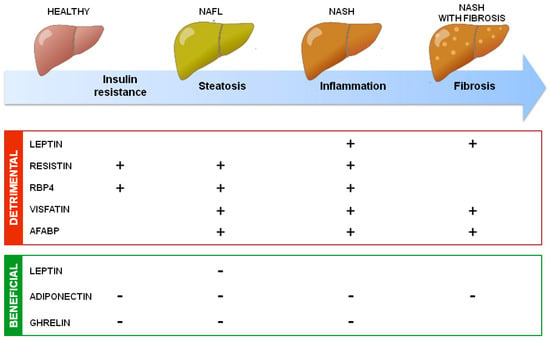
Figure 2.
Summary of adipokines’ action in NAFLD development. Adipokines secreted by adipose tissue could have detrimental or beneficial effects on NAFLD development through, respectively, increasing (+) or decreasing (−) insulin resistance, hepatic steatosis, inflammation, and fibrosis.
6. Therapeutic Interventions and Modulation of Adipokines’ Levels
As described above, clinical studies bring out much uncertainty regarding the correlation between adipokine levels and NAFLD. However, small interventional studies have revealed that lifestyle modifications and pharmacologic agents could affect the circulating levels of adipokines in NAFLD patients. Although only a few of these studies have adipokines as their main endpoint and most of them are characterized by a reduced sample size and the absence of control groups, they highlight the importance of adipokines in NAFLD amelioration and so are also summarized here.
Weight loss following bariatric surgery or lifestyle modification, namely healthy eating habits and physical exercise, in the NAFLD population, diminishes circulating leptin [8,22,130,131,132,133], resistin [131], RBP4 [8], visfatin [131], and chemerin [8], while elevating circulating adiponectin [8,131,133,134]. Nevertheless, the direct correlations between hepatic steatosis, weight loss, and adipokine levels have barely been investigated. Recently, the DIRECT PLUS randomized clinical trial verified that a green Mediterranean diet, enriched in specific green polyphenols (Mankai, green tea, and walnuts) and restricted in red and processed meat, led to weight loss, a decline of NAFLD prevalence, and a significant reduction in intrahepatic fat assessed by magnetic resonance, which was associated with a decline in leptin and chemerin plasma levels [135]. Furthermore, a randomized single-center study investigating the effects of guided lifestyle changes with endurance activity and nutrition advising or meal replacement with soy protein-based preparation in NASH patients demonstrated the positive correlation of leptin changes with body weight, fat mass, and reduction in waist circumference and total abdominal fat, while adiponectin changes were inversely correlated with these parameters [136]. More importantly, adiponectin increased with a reduction of intrahepatic lipid content, assessed by magnetic resonance imaging, while leptin declined as the intrahepatic lipid content decreased [136].
Thiazolidinediones (TZDs) are a family of drugs used in the treatment of T2DM [137]. As activators of the peroxisome proliferator-activated receptor γ (PPAR-γ), these drugs improve insulin sensitivity, and, therefore, some clinical trials have evaluated the effect of approved TZDs (pioglitazone and rosiglitazone) on NASH [138,139,140]. Both pioglitazone (30 mg/day for 22 months) [140] and rosiglitazone (8 mg/day for 3 years) [139] clearly reduced hepatic steatosis, whereas pioglitazone further reduced lobular inflammation in non-diabetic NAFLD patients [140], as well as fibrosis in biopsy-proven NAFLD patients with T2DM [141]. Rosiglitazone had no beneficial effects on hepatic lobular inflammation or fibrosis [139]. Additionally, a systematic review found an inverse association between the increase of adiponectin levels and histological steatosis, IR, and liver function markers (ALT and AST) after TZDs treatment [142]. So, TZDs increase adiponectin levels in addition to any weight gain, commonly seen with these drugs and usually associated with reduced adiponectin levels and adverse effects in NAFLD, likely through the induction of adiponectin production and secretion, which counterbalances the increase of body weight induced by TZDs [142]. A recent randomized controlled trial also correlated the amelioration of hepatic steatosis and necroinflammation in NASH patients with the enhancement of adiponectin levels and the decrease of visceral-to-subcutaneous fat ratio after pioglitazone (45 mg/d for 6-months) treatment [143]. Altogether, these results reinforce the role of adiponectin on NAFLD. Regarding the leptin levels, it has been demonstrated that TZDs reduced the leptin expression in adipose tissue, which could counteract the enhancement of leptin levels due to the TDZs-induced adipose tissue increase, with a neutral effect of TZDs on circulating leptin levels in NASH patients [8]. Of relevance, preliminary data revealed that the beneficial effects of pioglitazone on adiponectin levels and hepatic histology in NAFLD patients were reversed after therapy discontinuation, suggesting that long-term therapy with TZDs may be required [144]. However, treatment with rosiglitazone has been restricted due to the increased risk of myocardial infarction [145], whereas the administration of pioglitazone has been halted in some European countries due to its association with bladder-cancer risk after long-term use in diabetic patients [146]. Therefore, TZDs have demonstrated promising results in NAFLD amelioration, mainly through the reduction of hepatic steatosis correlated with increased adiponectin levels, but their clinical use could be limited by poor activity on hepatic fibrosis and long-term deleterious effects. The newer selective PPAR-γ modulators (SPPARMs) have been identified as promising agents that could provide better outcomes in NAFLD patients, but long term placebo-controlled randomized trials are still needed [147].
Another drug extensively used as a first-line therapy in T2DM is metformin, because of its outstanding glucose-lowering properties and its ability to improve insulin sensitivity [148]. So, it is not surprising that the therapeutic activity of metformin in NAFLD has been investigated. Preclinical data found that metformin prevented NAFLD development through the suppression of hepatic TG accumulation [149]. However, the data of clinical trials on metformin activity in NAFLD patients are not so promising. Biochemical parameters, including fasting plasma glucose, postprandial glucose, HOMA-IR, hemoglobin A1c, AST, ALT, GGT, TG, and total cholesterol, as well as body weight, BMI, and waist-to-hip ratio, all significantly improved with metformin therapy comparing to the control group; however, liver histologic scores, namely steatosis, ballooning, NAFLD activity score, and fibrosis, did not significantly change after metformin treatment, and lobular inflammation even became worse after therapy [150,151,152,153]. The beneficial effects of metformin on biochemical parameters and body weight in NAFLD patients were parallel to an increase in adiponectin levels [152,153] and a reduction in chemerin levels [151]. Interestingly, the one-year treatment with metformin decreased the central aortic augmentation index, which increased circulating adiponectin levels, an independent predictor of arterial stiffness improvement, which indicates that metformin therapy could ameliorate vascular dysfunction in NAFLD patients [154].
In the last few years, sodium-glucose cotransporter-2 (SGLT2) inhibitors, namely, empagliflozin, dapagliflozin, ipragliflozin, luseogliflozin, canagliflozin, and tofogliflozin, have demonstrated beneficial effects in T2DM patients with NAFLD. These blood–glucose-lowering drugs can significantly reduce hepatic enzymes and hepatic fat and improve metabolic and fibrosis indexes without increasing adverse effects [155,156]. The effect of SGLT2 on adipokine levels was poorly explored in clinical trials with NAFLD patients. A study verified no significant differences in adiponectin and leptin levels in the dapagliflozin-treated group compared to the placebo group [157], whereas the lower serum level of high-molecular-weight adiponectin was correlated with an improved response to dapagliflozin in T2DM patients with NAFLD [156]. Altogether, SGLT2 inhibitors seem to be an interesting therapeutic option for T2DM patients with NAFLD, but clinicians need to consider their efficacy and safety when individualizing the treatment [156].
Despite the modulation of adipokine levels by therapeutic approaches used in the management of NAFLD and its comorbidities T2DM and obesity, the effect of direct adipokine targeting in NAFLD has been poorly investigated. As described above, increased circulating levels of adiponectin seem to have beneficial effects in NAFLD patients, and, so, its administration could be seen as an appealing strategy for NAFLD. However, adiponectin is subjected to extensive post-translational modifications and multimerization, which challenges the production of functionally active recombinant adiponectin [158]. This is why the discovery of adiponectin analogues, such as osmotin, a plant antifungal protein, might result in alternative therapies for NAFLD [159,160]. Another alternative therapeutic approach for NAFLD in patients with remaining functional adipose tissue could be the up-regulation of endogenous adiponectin expression and/or its secretion induced by sustained weight loss or pharmacological agents, such as TZDs or SPPARMs, as described above [5]. The only “adipokine drug” approved by the United States Food and Drug Administration (FDA) is recombinant human leptin—metreleptin, used to treat complications of leptin deficiency in patients with lipodystrophy [161]. Data from small interventional studies verified a reduction in hepatic steatosis and improvements in hepatocellular ballooning and NAS, but not fibrosis, in NASH patients with a relative leptin deficiency or partial lipodystrophy [162,163,164]. So, although large well-controlled studies are needed, it seems that leptin treatment could become a therapeutic tool for hypoleptinemic NAFLD patients. Moreover, the discovery of leptin analogues with anti-steatotic action but lacking the inflammatory and fibrogenic leptin activity, such as 7i [165], will be of relevance in the development of adipokine-based therapies for NAFLD [22].
7. Future Perspectives and Conclusions
The Global Burden of Disease Study 2017 evidenced the alarming trend of NAFL/NASH to become the major cause of chronic liver disease in children and adults and to potentially contribute to the burden of non-communicable diseases, such as cardiovascular disease [2]. Therefore, the early screening of NAFLD patients at highest risk for liver-related complications and the development of successful management strategies are mandatory. The emerging roles of adipokines as metabolic and inflammatory players, together with encouraging preclinical data, have identified adipokines as promising biomarkers and therapeutic targets for steatosis and NASH. Nevertheless, the available clinical data are inconsistent.
Every clinical study has a unique set of inclusion and exclusion criteria for participants. A considerable number of clinical trials used magnetic-resonance-imaging-based NAFLD diagnosis without liver biopsy confirmation or histological NAFLD grade evaluation, which led to highly heterogeneous groups and limited the conclusions of these studies. NAFL and NASH classifications are still widely used in clinical practice, but the recent definition of MAFLD will allow a better characterization of patients’ profiles, excluding those with steatosis unrelated to metabolic dysfunction and, thus, contributing to more homogeneous cohorts [4]. Moreover, the establishment of diagnosis and follow-up guidelines [1,11,166] as effective tools to identify and stratify NAFLD patients is helpful. Moreover, the pathogenic factors associated with NAFLD are multifactorial, including lifestyle (high-calorie diet and sedentarism), genetic alterations, or the presence of comorbidities [21], and the analysis of possible confounding factors are critical. In this context, future clinical trials with highly-phenotyped patient cohorts, similarly to the European NAFLD Registry [167], are demanding.
The existing clinical studies on adipokines and NAFLD are mostly case-control or cross-sectional studies, which limited the conclusions regarding causalities and possible associations. Prospective cohort studies with paired biopsies, long-term follow-ups and carefully matched controls can offer a higher level of evidence on adipokines’ pathophysiological role in NAFLD and on their diagnosis and prognosis power. In this regard, it will be important to define standardized methodology for high-sensitivity, rapid, single-analysis, and, most importantly, the reliable identification and quantification of adipokines [168], without forgetting that some of them have multiple forms, such as adiponectin, resistin, and ghrelin [26,42,158], or can be found in the circulation as free or protein-bounded forms, such as leptin [169]. Moreover, the levels of endogenous antagonists of adipokines [170] or inhibitors of their receptors should not be ignored. Finally, increasingly important topics are proper clinical study design, analysis, reporting, and sharing of data, to ensure accurate and robust findings as well as appropriate interpretation [171,172].
As described in this review, the potential role of adipokines in NAFLD development has been broadly explored using in vitro and in vivo approaches. While cellular models lack systemic perspective and cross-organ communication, current NAFL/NASH in vivo models have limited predictive value to the full spectrum of human NAFLD [173]. However, nutritional models or combined models of genetic modifications and nutritional challenges presenting metabolic comorbidities (IR, dyslipidemia and obesity) [173] are likely to be the best-suited models with a higher translatable potential for the study of adipokines’ pathophysiological role in NAFLD. Furthermore, recent advances on 3D cell cultures and biochip-based culture systems [174] could allow a better assessment of cellular responses to adipokines. Altogether, preclinical models are useful tools to unravel pathophysiological links and signaling pathways, which could be very valuable in disclosing uncertainties on the role of adipokines in NAFLD. Curiously, there are limited studies either on the hepatic functionality of adipokines receptors or on the activity of endogenous adipokines antagonists in NAFLD, which could add other (dys)regulatory levels to the intricate communication between adipose tissue and the liver. Furthermore, several emerging adipokines, such as vaspin [175,176,177], adipsin [178], apelin [179,180], obestatin [181], and omentin [176,177], have been associated with NAFLD, but their roles and mechanisms of action remain unclear.
Future insights into the pathophysiologic role of adipokines in NAFLD will be of great relevance for the development of new therapeutic approaches. In particular, the control of bioactive adipokines levels using high-affinity binding molecules, miRNAs targeting specific adipokines, and antagonists or monoclonal humanized antibodies against adipokines receptors are likely to be feasible options [182]. Accordingly, as mentioned above, recombinant leptin was described to improve steatosis and NAFLD activity score in hypoleptinemic NAFLD patients [162,163,164]. However, there are some concerns to take into account when targeting adipokines or their receptors. Anti-drug antibodies, developed after adipokine administration, could cross-react with endogenous adipokine and limit their therapeutic efficacy [183]. Furthermore, given the pleiotropic activity of adipokines, existence of compensatory mechanisms, and interplay among detrimental and beneficial adipokines, a systematic approach might be discouraged; instead, strategies targeting specific receptor isoforms or adipokines activity precisely in specific cell populations could be viable therapeutic options [6,8]. The development of adipokines analogues without deleterious effects, such as the leptin analogue 7i [165] are also an attractive strategy. Above all, adipokines are immunometabolic players [7] acting in a dose-dependent and continuous dynamic cross-talk way, and, thus, therapies aiming to reestablish a healthy adipokine network and adipose tissue function are likely to be more effective than single-adipokine targeting.
In conclusion, much uncertainty still exists regarding the roles of adipokines in NAFLD, mainly due to complex NAFLD pathophysiology and intricate adipokines network. Long-term prospective studies of well-characterized cohorts presenting paired liver biopsies and suitable matched controls, together with improved preclinical models, are demanding to accurately evaluate adipokines as NAFLD biomarkers as well as adipokine-based therapies for NAFLD. Through the path, strict collaboration between clinicians and basic scientists will certainly elicit cross-fertilization of translational research and accelerate the transfer of the results to the clinical practice. Nevertheless, we should keep in mind that the more effective approach to counteract adipokine imbalance and NAFLD is to prevent and/or to revert excessive fat.
Author Contributions
Conceptualization, V.F., M.J.S., J.T.R. and O.G.; investigation, V.F., P.M., M.C. and D.A.E.; data curation, V.F.; writing—original draft preparation, V.F.; writing—review and editing, V.F., M.J.S., J.T.R., P.M. and O.G.; project administration, V.F.; funding acquisition, V.F., M.J.S., J.T.R. and O.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Instituto de Salud Carlos III (ISCIII) and co-funded by the European Social Fund Plus (ESF+) through a Miguel Servet Program contract to Vera Francisco (CP21/00025). This work was also supported by the Spanish Ministry of Science and Innovation: (grant number PID2020-120336RB-I00), the European Regional Development Fund (FEDER), and the Generalitat Valenciana (PROMETEO/2019/032). Patrice Marques was funded by postdoctoral grant from the Generalitat Valenciana (PROMETEO/2019/032). O.G. is Staff Personnel (I3SNS stable Researchers) of Xunta de Galicia (Servizo Galego de Saude, SERGAS) through a research–staff contract (Instituto de Salud Carlos III (ISCIII) /SERGAS). O.G. is a member of the RICORS Program, RD21/0002/0025 via ISCIII and FEDER. The work of O.G. (PI20/00902) is funded by ISCIII and FEDER. O.G. is a beneficiary of a project funded by the Research Executive Agency of the European Union under the framework of the MSCA-RISE Action of the H2020 Program (project number 734899). O.G. is beneficiary of a grant funded by Xunta de Galicia, Consellería de Educación, Universidade e Formación Profesional, and Consellería de Economía, Emprego e Industria (GAIN) (GPC IN607B-2019/10). The funders were not involved in study design, data collection and analysis, decision to publish, or manuscript preparation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Leoni, S.; Tovoli, F.; Napoli, L.; Serio, I.; Ferri, S.; Bolondi, L. Current guidelines for the management of non-alcoholic fatty liver disease: A systematic review with comparative analysis. World J. Gastroenterol. 2018, 24, 3361–3373. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Perakakis, N.; Mantzoros, C.S. Fatty liver in lipodystrophy: A review with a focus on therapeutic perspectives of adiponectin and/or leptin replacement. Metabolism 2019, 96, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, fat mass and immune system: Role for leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; González-Gay, M.Á.; Lago, F.; Karppinen, J.; Tervonen, O.; Mobasheri, A.; Gualillo, O. A new immunometabolic perspective of intervertebral disc degeneration. Nat. Rev. Rheumatol. 2022, 18, 47–60. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 Diseases and Injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Papatheodoridi, M.; Cholongitas, E. Diagnosis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Concepts. Curr. Pharm. Des. 2018, 24, 4574–4586. [Google Scholar] [CrossRef]
- Aller, R.; Fernández-Rodríguez, C.; lo Iacono, O.; Bañares, R.; Abad, J.; Carrión, J.A.; García-Monzón, C.; Caballería, J.; Berenguer, M.; Rodríguez-Perálvarez, M.; et al. Consensus document. Management of non-alcoholic fatty liver disease (NAFLD). Clinical practice guideline. Gastroenterol. Hepatol. 2018, 41, 328–349. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Sinha, R.A. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. Landmark 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1. [Google Scholar]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2020, 21, 157. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Leibel, R.L. Role of leptin in energy homeostasis in humans. J. Endocrinol. 2014, 223, T83–T96. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Uña, M.; López-Mancheño, Y.; Diéguez, C.; Fernández-Rojo, M.A.; Novelle, M.G. Unraveling the role of leptin in liver function and its relationship with liver diseases. Int. J. Mol. Sci. 2020, 21, 9368. [Google Scholar] [CrossRef]
- Jiménez-Cortegana, C.; García-Galey, A.; Tami, M.; Del Pino, P.; Carmona, I.; López, S.; Alba, G.; Sánchez-Margalet, V. Role of leptin in non-alcoholic fatty liver disease. Biomedicines 2021, 9, 762. [Google Scholar] [CrossRef] [PubMed]
- Heydari, M.; Cornide-Petronio, M.E.; Jiménez-Castro, M.B.; Peralta, C. Data on adiponectin from 2010 to 2020: Therapeutic target and prognostic factor for liver diseases? Int. J. Mol. Sci. 2020, 21, 5242. [Google Scholar] [CrossRef]
- Nguyen, T.M.D. Adiponectin: Role in Physiology and Pathophysiology. Int. J. Prev. Med. 2020, 11, 136. [Google Scholar] [CrossRef]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef]
- Quiñones, M.; Fernø, J.; Al-Massadi, O. Ghrelin and liver disease. Rev. Endocr. Metab. Disord. 2020, 21, 45–56. [Google Scholar] [CrossRef]
- Acquarone, E.; Monacelli, F.; Borghi, R.; Nencioni, A.; Odetti, P. Resistin: A reappraisal. Mech. Ageing Dev. 2019, 178, 46–63. [Google Scholar] [CrossRef]
- Nono Nankam, P.A.; Blüher, M. Retinol-binding protein 4 in obesity and metabolic dysfunctions. Mol. Cell. Endocrinol. 2021, 531, 111312. [Google Scholar] [CrossRef]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef]
- Carbone, F.; Liberale, L.; Bonaventura, A.; Vecchié, A.; Casula, M.; Cea, M.; Monacelli, F.; Caffa, I.; Bruzzone, S.; Montecucco, F.; et al. Regulation and Function of Extracellular Nicotinamide Phosphoribosyltransferase/Visfatin. Compr. Physiol. 2017, 7, 603–621. [Google Scholar] [PubMed]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Wu, X.; Xu, A.; Hoo, R.L.C. A-FABP in Metabolic Diseases and the Therapeutic Implications: An Update. Int. J. Mol. Sci. 2021, 22, 9386. [Google Scholar] [CrossRef] [PubMed]
- Micioni Di Bonaventura, E.; Botticelli, L.; Del Bello, F.; Giorgioni, G.; Piergentili, A.; Quaglia, W.; Cifani, C.; Micioni Di Bonaventura, M.V. Assessing the role of ghrelin and the enzyme ghrelin O-acyltransferase (GOAT) system in food reward, food motivation, and binge eating behavior. Pharmacol. Res. 2021, 172, 105847. [Google Scholar] [CrossRef]
- Li, Y.; Hai, J.; Li, L.; Chen, X.; Peng, H.; Cao, M.; Zhang, Q. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine 2013, 43, 376–386. [Google Scholar] [CrossRef]
- Mao, Y.; Cheng, J.; Yu, F.; Li, H.; Guo, C.; Fan, X. Ghrelin Attenuated Lipotoxicity via Autophagy Induction and Nuclear Factor-κB Inhibition. Cell. Physiol. Biochem. 2015, 37, 563–576. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, Q.; Qi, M.; Zhang, C.; Li, Z.; Zhang, W. Ghrelin ameliorates nonalcoholic steatohepatitis induced by chronic low-grade inflammation via blockade of Kupffer cell M1 polarization. J. Cell. Physiol. 2021, 236, 5121–5133. [Google Scholar] [CrossRef]
- Guillory, B.; Jawanmardi, N.; Iakova, P.; Anderson, B.; Zang, P.; Timchenko, N.A.; Garcia, J.M. Ghrelin deletion protects against age-associated hepatic steatosis by downregulating the C/EBPα-p300/DGAT1 pathway. Aging Cell 2018, 17, e12688. [Google Scholar] [CrossRef]
- Ezquerro, S.; Becerril, S.; Tuero, C.; Méndez-Giménez, L.; Mocha, F.; Moncada, R.; Valentí, V.; Cienfuegos, J.A.; Catalán, V.; Gómez-Ambrosi, J.; et al. Role of ghrelin isoforms in the mitigation of hepatic inflammation, mitochondrial dysfunction, and endoplasmic reticulum stress after bariatric surgery in rats. Int. J. Obes. 2020, 44, 475–487. [Google Scholar] [CrossRef]
- Ezquerro, S.; Méndez-Giménez, L.; Becerril, S.; Moncada, R.; Valentí, V.; Catalán, V.; Gómez-Ambrosi, J.; Frühbeck, G.; Rodríguez, A. Acylated and desacyl ghrelin are associated with hepatic lipogenesis, β-oxidation and autophagy: Role in NAFLD amelioration after sleeve gastrectomy in obese rats. Sci. Rep. 2016, 6, 39942. [Google Scholar] [CrossRef]
- Dallak, M.A. Acylated ghrelin induces but deacylated ghrelin prevents hepatic steatosis and insulin resistance in lean rats: Effects on DAG/PKC/JNK pathway. Biomed. Pharmacother. 2018, 105, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.D.; Obici, S.; Bhanot, S.; Monia, B.P.; McKay, R.A.; Rajala, M.W.; Scherer, P.E.; Rossetti, L. Role of resistin in diet-induced hepatic insulin resistance. J. Clin. Investig. 2004, 114, 232–239. [Google Scholar] [CrossRef]
- Wen, F.; Shi, Z.; Liu, X.; Tan, Y.; Wei, L.; Zhu, X.; Zhang, H.; Zhu, X.; Meng, X.; Ji, W.; et al. Acute Elevated Resistin Exacerbates Mitochondrial Damage and Aggravates Liver Steatosis through AMPK/PGC-1α Signaling Pathway in Male NAFLD Mice. Horm. Metab. Res. 2021, 53, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Wang, J.; Whiteman, E.L.; Birnbaum, M.J.; Lazar, M.A. Activation of SOCS-3 by resistin. Mol. Cell. Biol. 2005, 25, 1569–1575. [Google Scholar] [CrossRef]
- Banerjee, R.R.; Rangwala, S.M.; Shapiro, J.S.; Rich, A.S.; Rhoades, B.; Qi, Y.; Wang, J.; Rajala, M.W.; Pocai, A.; Scherer, P.E.; et al. Regulation of fasted blood glucose by resistin. Science 2004, 303, 1195–1198. [Google Scholar] [CrossRef]
- Singhal, N.S.; Patel, R.T.; Qi, Y.; Lee, Y.S.; Ahima, R.S. Loss of resistin ameliorates hyperlipidemia and hepatic steatosis in leptin-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E331–E338. [Google Scholar] [CrossRef][Green Version]
- Song, R.; Wang, X.; Mao, Y.; Li, H.; Li, Z.; Xu, W.; Wang, R.; Guo, T.; Jin, L.; Zhang, X.; et al. Resistin disrupts glycogen synthesis under high insulin and high glucose levels by down-regulating the hepatic levels of GSK3β. Gene 2013, 529, 50–56. [Google Scholar] [CrossRef][Green Version]
- Garcia, C.C.; Piotrkowski, B.; Baz, P.; Poncino, D.; Benavides, J.; Colombato, L.; Toso, M.L.R.; Yantorno, S.; Descalzi, V.; Gondolesi, G.E.; et al. A Decreased Response to Resistin in Mononuclear Leukocytes Contributes to Oxidative Stress in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2021, 1, 3006–3016. [Google Scholar] [CrossRef]
- Beier, J.I.; Guo, L.; Von Montfort, C.; Kaiser, J.P.; Joshi-Barve, S.; Arteel, G.E. New role of resistin in lipopolysaccharide-induced liver damage in mice. J. Pharmacol. Exp. Ther. 2008, 325, 801–808. [Google Scholar] [CrossRef]
- Saxena, N.K.; Anania, F.A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.X.; Su, L.; Brymora, J.; Bird, C.; Xie, Q.; George, J.; Wang, J.H. Resistin mediates the hepatic stellate cell phenotype. World J. Gastroenterol. 2013, 19, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mu, D.; Chen, H.; Li, D.; Song, J.; Zhong, Y.; Xia, M. Retinol-Binding Protein 4 Induces Hepatic Mitochondrial Dysfunction and Promotes Hepatic Steatosis. J. Clin. Endocrinol. Metab. 2016, 101, 4338–4348. [Google Scholar] [CrossRef]
- Saeed, A.; Bartuzi, P.; Heegsma, J.; Dekker, D.; Kloosterhuis, N.; de Bruin, A.; Jonker, J.W.; van de Sluis, B.; Faber, K.N. Impaired Hepatic Vitamin A Metabolism in NAFLD Mice Leading to Vitamin A Accumulation in Hepatocytes. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 309–325.e3. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Yuen, J.J.; Jiang, H.; Kahn, B.B.; Blaner, W.S. Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice. Hepatology 2016, 64, 1534–1546. [Google Scholar] [CrossRef]
- Cioffi, C.L.; Racz, B.; Varadi, A.; Freeman, E.E.; Conlon, M.P.; Chen, P.; Zhu, L.; Kitchen, D.B.; Barnes, K.D.; Martin, W.H.; et al. Design, Synthesis, and Preclinical Efficacy of Novel Nonretinoid Antagonists of Retinol-Binding Protein 4 in the Mouse Model of Hepatic Steatosis. J. Med. Chem. 2019, 62, 5470–5500. [Google Scholar] [CrossRef]
- Heo, Y.J.; Choi, S.E.; Lee, N.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Visfatin exacerbates hepatic inflammation and fibrosis in a methionine-choline-deficient diet mouse model. J. Gastroenterol. Hepatol. 2021, 36, 2592–2600. [Google Scholar] [CrossRef]
- Heo, Y.J.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Visfatin Induces Inflammation and Insulin Resistance via the NF- κ B and STAT3 Signaling Pathways in Hepatocytes. J. Diabetes Res. 2019, 2019, 4021623. [Google Scholar] [CrossRef]
- Wang, L.F.; Wang, X.N.; Huang, C.C.; Hu, L.; Xiao, Y.F.; Guan, X.H.; Qian, Y.S.; Deng, K.Y.; Xin, H.B. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKα/SREBP1 signaling pathway. Lipids Health Dis. 2017, 16, 82. [Google Scholar] [CrossRef]
- Ilbeigi, D.; Nourbakhsh, M.; Pasalar, P.; Meshkani, R.; Afra, H.S.; Panahi, G.; Borji, M.; Sharifi, R. Nicotinamide phosphoribosyltransferase knockdown leads to lipid accumulation in HepG2 cells through the SIRT1-AMPK pathway. Cell J. 2020, 22, 125–132. [Google Scholar]
- Dall, M.; Hassing, A.S.; Niu, L.; Nielsen, T.S.; Ingerslev, L.R.; Sulek, K.; Trammell, S.A.J.; Gillum, M.P.; Barrès, R.; Larsen, S.; et al. Hepatocyte-specific perturbation of NAD+ biosynthetic pathways in mice induces reversible nonalcoholic steatohepatitis-like phenotypes. J. Biol. Chem. 2021, 297, 101388. [Google Scholar] [CrossRef] [PubMed]
- Bondue, B.; Wittamer, V.; Parmentier, M. Chemerin and its receptors in leukocyte trafficking, inflammation and metabolism. Cytokine Growth Factor Rev. 2011, 22, 331–338. [Google Scholar] [CrossRef]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Divoux, A.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Bedossa, P.; Tordjman, J.; Eckel, J.; Clément, K. Chemerin Correlates with Markers for Fatty Liver in Morbidly Obese Patients and Strongly Decreases after Weight Loss Induced by Bariatric Surgery. J. Clin. Endocrinol. Metab. 2010, 95, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Liu, J.; Li, Y.; Dou, Z.; Li, N.; Suo, Y.; Ma, Y.; Sun, M.; Tian, Z.; Xu, L. Chemerin/CMKLR1 ameliorates nonalcoholic steatohepatitis by promoting autophagy and alleviating oxidative stress through the JAK2-STAT3 pathway. Peptides 2021, 135, 170422. [Google Scholar] [CrossRef] [PubMed]
- Pohl, R.; Rein-Fischboeck, L.; Meier, E.M.; Eisinger, K.; Krautbauer, S.; Buechler, C. Resolvin E1 and chemerin C15 peptide do not improve rodent non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2015, 98, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Gruben, N.; Vergara, M.A.; Kloosterhuis, N.J.; Van Der Molen, H.; Stoelwinder, S.; Youssef, S.; De Bruin, A.; Delsing, D.J.; Kuivenhoven, J.A.; Van De Sluis, B.; et al. Chemokine-like receptor 1 deficiency does not affect the development of insulin resistance and nonalcoholic fatty liver disease in mice. PLoS ONE 2014, 9, e96345. [Google Scholar] [CrossRef]
- Krautbauer, S.; Wanninger, J.; Eisinger, K.; Hader, Y.; Beck, M.; Kopp, A.; Schmid, A.; Weiss, T.S.; Dorn, C.; Buechler, C. Chemerin is highly expressed in hepatocytes and is induced in non-alcoholic steatohepatitis liver. Exp. Mol. Pathol. 2013, 95, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Hao, J.; Yi, Y.; Sauter, E.; Li, B. Regulation of macrophage functions by FABP-mediated inflammatory and metabolic pathways. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158964. [Google Scholar] [CrossRef]
- Hoo, R.L.C.; Lee, I.P.C.; Zhou, M.; Wong, J.Y.L.; Hui, X.; Xu, A.; Lam, K.S.L. Pharmacological inhibition of adipocyte fatty acid binding protein alleviates both acute liver injury and non-alcoholic steatohepatitis in mice. J. Hepatol. 2013, 58, 358–364. [Google Scholar] [CrossRef]
- Wu, X.; Shu, L.; Zhang, Z.; Li, J.; Zong, J.; Cheong, L.Y.; Ye, D.; Lam, K.S.L.; Song, E.; Wang, C.; et al. Adipocyte Fatty Acid Binding Protein Promotes the Onset and Progression of Liver Fibrosis via Mediating the Crosstalk between Liver Sinusoidal Endothelial Cells and Hepatic Stellate Cells. Adv. Sci. 2021, 8, 2003721. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, D.H.; Karelis, A.D.; Coderre, L.; Malita, F.; Fontaine, J.; Mignault, D.; Brochu, M.; Bastard, J.P.; Cianflone, K.; Doucet, E.; et al. Association of acylated and nonacylated ghrelin with insulin sensitivity in overweight and obese postmenopausal women. J. Clin. Endocrinol. Metab. 2007, 92, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Barazzoni, R.; Zanetti, M.; Ferreira, C.; Vinci, P.; Pirulli, A.; Mucci, M.P.; Dore, F.; Fonda, M.; Ciocchi, B.; Cattin, L.; et al. Relationships between desacylated and acylated ghrelin and insulin sensitivity in the metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3935–3940. [Google Scholar] [CrossRef]
- Pacifico, L.; Poggiogalle, E.; Costantino, F.; Anania, C.; Ferraro, F.; Chiarelli, F.; Chiesa, C. Acylated and nonacylated ghrelin levels and their associations with insulin resistance in obese and normal weight children with metabolic syndrome. Eur. J. Endocrinol. 2009, 161, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Arslan, N.; Sayin, O.; Tokgoz, Y. Evaluation of serum xenin and ghrelin levels and their relationship with nonalcoholic fatty liver disease and insulin resistance in obese adolescents. Acad. Psychiatry 2014, 37, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- MacHado, M.V.; Coutinho, J.; Carepa, F.; Costa, A.; Proença, H.; Cortez-Pinto, H. How adiponectin, leptin, and ghrelin orchestrate together and correlate with the severity of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1166–1172. [Google Scholar] [CrossRef]
- Marchesini, G.; Pagotto, U.; Bugianesi, E.; De Iasio, R.; Manini, R.; Vanni, E.; Pasquali, R.; Melchionda, N.; Rizzetto, M. Low Ghrelin Concentrations in Nonalcoholic Fatty Liver Disease Are Related to Insulin Resistance. J. Clin. Endocrinol. Metab. 2003, 88, 5674–5679. [Google Scholar] [CrossRef]
- Estep, M.; Abawi, M.; Jarrar, M.; Wang, L.; Stepanova, M.; Elariny, H.; Moazez, A.; Goodman, Z.; Chandhoke, V.; Baranova, A.; et al. Association of obestatin, ghrelin, and inflammatory cytokines in obese patients with non-alcoholic fatty liver disease. Obes. Surg. 2011, 21, 1750–1757. [Google Scholar] [CrossRef]
- Tabaeian, S.P.; Mahmoudi, T.; Sabzikarian, M.; Rezamand, G.; Dabiri, R.; Nobakht, H.; Asadi, A.; Farahani, H.; Mansour-Ghanaei, F.; Zali, M.R. The leu72met (Rs696217 g>t) polymorphism of the ghrelin gene might be a protective factor for nonalcoholic fatty liver disease. J. Gastrointest. Liver Dis. 2021, 30, 233–239. [Google Scholar] [CrossRef]
- Rezamand, G.; Mahmoudi, T.; Tabaeian, S.P.; Farahani, H.; Shahinmehr, F.; Nobakht, H.; Dabiri, R.; Asadi, A.; Mansour-Ghanaei, F.; Zali, M.R. The “GG” genotype of rs26802 variant in the ghrelin gene is a potential protective factor against nonalcoholic fatty liver disease. Physiol. Int. 2021, 108, 342–352. [Google Scholar] [CrossRef]
- Bekaert, M.; Verhelst, X.; Geerts, A.; Lapauw, B.; Calders, P. Association of recently described adipokines with liver histology in biopsy-proven non-alcoholic fatty liver disease: A systematic review. Obes. Rev. 2016, 17, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, C.; Ardicoglu, Y.; Bulut, S.; Boyacioglu, S. Serum retinol-binding protein 4 in patients with nonalcoholic fatty liver disease: Does it have a significant impact on pathogenesis? Eur. J. Gastroenterol. Hepatol. 2010, 22, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; De Luis, D.A.; Fernandez, L.; Calle, F.; Velayos, B.; Olcoz, J.L.; Izaola, O.; Sagrado, M.G.; Conde, R.; Gonzalez, J.M. Influence of insulin resistance and adipokines in the grade of steatosis of nonalcoholic fatty liver disease. Dig. Dis. Sci. 2008, 53, 1088–1092. [Google Scholar] [CrossRef]
- Pagano, C.; Soardo, G.; Pilon, C.; Milocco, C.; Basan, L.; Milan, G.; Donnini, D.; Faggian, D.; Mussap, M.; Plebani, M.; et al. Increased serum resistin in nonalcoholic fatty liver disease is related to liver disease severity and not to insulin resistance. J. Clin. Endocrinol. Metab. 2006, 91, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Şenateş, E.; Çolak, Y.; Yeşil, A.; Coşkunpinar, E.; Şahin, Ö.; Kahraman, Ö.T.; Erkalma Şenateş, B.; Tuncer, I. Circulating resistin is elevated in patients with non-alcoholic fatty liver disease and is associated with steatosis, portal inflammation, insulin resistance and nonalcoholic steatohepatitis scores. Minerva Med. 2012, 103, 369–376. [Google Scholar] [PubMed]
- Younossi, Z.M.; Jarrar, M.; Nugent, C.; Randhawa, M.; Afendy, M.; Stepanova, M.; Rafiq, N.; Goodman, Z.; Chandhoke, V.; Baranova, A. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH). Obes. Surg. 2008, 18, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Page, S.; Rafiq, N.; Birerdinc, A.; Stepanova, M.; Hossain, N.; Afendy, A.; Younoszai, Z.; Goodman, Z.; Baranova, A. A biomarker panel for non-alcoholic steatohepatitis (NASH) and NASH-related fibrosis. Obes. Surg. 2011, 21, 431–439. [Google Scholar] [CrossRef]
- Argentou, M.; Tiniakos, D.G.; Karanikolas, M.; Melachrinou, M.; Makri, M.G.; Kittas, C.; Kalfarentzos, F. Adipokine serum levels are related to liver histology in severely obese patients undergoing bariatric surgery. Obes. Surg. 2009, 19, 1313–1323. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Durazzo, M.; Biroli, G.; Carello, M.; Fagà, E.; Pacini, G.; De Michieli, F.; Rabbione, L.; Premoli, A.; et al. Adipokines in NASH: Postprandial lipid metabolism as a link between adiponectin and liver disease. Hepatology 2005, 42, 1175–1183. [Google Scholar] [CrossRef]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef]
- Wong, V.W.S.; Hui, A.Y.; Tsang, S.W.C.; Chan, J.L.Y.; Tse, A.M.L.; Chan, K.F.; So, W.Y.; Cheng, A.Y.S.; Ng, W.F.; Wong, G.L.H.; et al. Metabolic and adipokine profile of Chinese patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2006, 4, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Koehler, E.; Swain, J.; Sanderson, S.; Krishnan, A.; Watt, K.; Charlton, M. Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: An endocrine signature for advanced fibrosis in obese patients. Liver Int. 2012, 32, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Baltieri, L.; Chaim, E.A.; Chaim, F.D.M.; Utrini, M.P.; Gestic, M.A.; Cazzo, E. Correlation between nonalcoholic fatty liver disease features and levels of adipokines and inflammatory cytokines among morbidly obese individuals. Arq. Gastroenterol. 2018, 55, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Ajmera, V.; Perito, E.R.; Bass, N.M.; Terrault, N.A.; Yates, K.P.; Gill, R.; Loomba, R.; Diehl, A.M.; Aouizerat, B.E. Novel plasma biomarkers associated with liver disease severity in adults with nonalcoholic fatty liver disease. Hepatology 2017, 65, 65–77. [Google Scholar] [CrossRef]
- Shen, C.; Zhao, C.Y.; Wang, W.; Wang, Y.D.; Sun, H.; Cao, W.; Yu, W.Y.; Zhang, L.; Ji, R.; Li, M.; et al. The relationship between hepatic resistin overexpression and inflammation in patients with nonalcoholic steatohepatitis. BMC Gastroenterol. 2014, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; De Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar] [CrossRef]
- Gierej, P.; Gierej, B.; Kalinowski, P.; Wróblewski, T.; Paluszkiewicz, R.; Kobryń, K.; Ziarkiewicz-Wróblewska, B. Expression of resistin in the liver of patients with Non-Alcoholic fatty liver disease. Pol. J. Pathol. 2017, 68, 225–233. [Google Scholar] [CrossRef]
- Kohan, L.; Safarpur, M.; Abdollahi, H. Omentin-1 rs2274907 and resistin rs1862513 polymorphisms influence genetic susceptibility to nonalcoholic fatty liver disease. Mol. Biol. Res. Commun. 2016, 5, 11. [Google Scholar]
- Balagopal, P.; Graham, T.E.; Kahn, B.B.; Altomare, A.; Funanage, V.; George, D. Reduction of elevated serum retinol binding protein in obese children by lifestyle intervention: Association with subclinical inflammation. J. Clin. Endocrinol. Metab. 2007, 92, 1971–1974. [Google Scholar] [CrossRef]
- Haider, D.G.; Schindler, K.; Prager, G.; Bohdjalian, A.; Luger, A.; Wolzt, M.; Ludvik, B. Serum retinol-binding protein 4 is reduced after weight loss in morbidly obese subjects. J. Clin. Endocrinol. Metab. 2007, 92, 1168–1171. [Google Scholar] [CrossRef]
- Stefan, N.; Hennige, A.M.; Staiger, H.; Machann, J.; Schick, F.; Schleicher, E.; Fritsche, A.; Häring, H.U. High circulating retinol-binding protein 4 is associated with elevated liver fat but not with total, subcutaneous, visceral, or intramyocellular fat in humans. Diabetes Care 2007, 30, 1173–1178. [Google Scholar] [CrossRef][Green Version]
- Seo, J.A.; Kim, N.H.; Park, S.Y.; Kim, H.Y.; Ryu, O.H.; Lee, K.W.; Lee, J.; Kim, D.L.; Choi, K.M.; Baik, S.H.; et al. Serum retinol-binding protein 4 levels are elevated in non-alcoholic fatty liver disease. Clin. Endocrinol. 2008, 68, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Yang, Y.J. Serum retinol-binding protein 4 is independently associated with pediatric NAFLD and fasting triglyceride level. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, H.; Ju, H.; Sun, M. Circulating retinol binding protein 4 levels in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Lipids Health Dis. 2017, 16, 180. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Zhang, H.; Pang, J.; Lin, J.; Xu, X.; Yang, L.; Ma, J.; Ling, W.; Chen, Y. Circulating retinol-binding protein 4 is associated with the development and regression of non-alcoholic fatty liver disease. Diabetes Metab. 2020, 46, 119–128. [Google Scholar] [CrossRef]
- Karamfilova, V.; Gateva, A.; Alexiev, A.; Zheleva, N.; Velikova, T.; Ivanova-Boyanova, R.; Ivanova, R.; Cherkezov, N.; Kamenov, Z.; Mateva, L. The association between retinol-binding protein 4 and prediabetes in obese patients with nonalcoholic fatty liver disease. Arch. Physiol. Biochem. 2022, 128, 217–222. [Google Scholar] [CrossRef]
- Ismaiel, A.; Leucuta, D.C.; Popa, S.L.; Dumitrascu, D.L. Serum visfatin levels in nonalcoholic fatty liver disease and liver fibrosis: Systematic review and meta-analysis. J. Clin. Med. 2021, 10, 3029. [Google Scholar] [CrossRef]
- Genc, H.; Dogru, T.; Kara, M.; Tapan, S.; Ercin, C.N.; Acikel, C.; Karslioglu, Y.; Bagci, S. Association of plasma visfatin with hepatic and systemic inflammation in nonalcoholic fatty liver disease. Ann. Hepatol. 2013, 12, 380–387. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Papatheodorou, A.; Katsiki, E.; Patsiaoura, K.; Zafeiriadou, E.; Zavos, C.; Papadopoulou, E.; Terpos, E. Adipocytokines and cytokeratin-18 in patients with nonalcoholic fatty liver disease: Introduction of CHA index. Ann. Hepatol. 2013, 12, 749–757. [Google Scholar] [CrossRef]
- Yoon, M.Y.; Sung, J.M.; Song, C.S.; Lee, W.Y.; Rhee, E.J.; Shin, J.H.; Yoo, C.H.; Chae, S.W.; Kim, J.Y.; Jin, W.; et al. Enhanced A-FABP expression in visceral fat: Potential contributor to the progression of NASH. Clin. Mol. Hepatol. 2012, 18, 279–286. [Google Scholar] [CrossRef]
- Dahl, T.B.; Haukeland, J.W.; Yndestad, A.; Ranheim, T.; Gladhaug, I.P.; Damås, J.K.; Haaland, T.; Løberg, E.M.; Arntsen, B.; Birkeland, K.; et al. Intracellular Nicotinamide Phosphoribosyltransferase Protects against Hepatocyte Apoptosis and Is Down-Regulated in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2010, 95, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Kukla, M.; Ciupińska-Kajor, M.; Kajor, M.; Wylezoł, M.; Zwirska-Korczala, K.; Hartleb, M.; Berdowska, A.; Mazur, W. Liver visfatin expression in morbidly obese patients with nonalcoholic fatty liver disease undergoing bariatric surgery. Polish J. Pathol. 2010, 61, 147–153. [Google Scholar]
- Auguet, T.; Terra, X.; Porras, J.A.; Orellana-Gavaldà, J.M.; Martinez, S.; Aguilar, C.; Lucas, A.; Pellitero, S.; Hernández, M.; Del Castillo, D.; et al. Plasma visfatin levels and gene expression in morbidly obese women with associated fatty liver disease. Clin. Biochem. 2013, 46, 202–208. [Google Scholar] [CrossRef]
- Gaddipati, R.; Mitnala, S.; Padaki, N.; Mukherjee, R.M.; Sekaran, A.; Jayaraj-Mansard, M.; Rabella, P.; Rao-Guduru, V.; Reddy-Duwuru, N. Visceral adipose tissue visfatin in nonalcoholic fatty liver disease. Ann. Hepatol. 2010, 9, 266–270. [Google Scholar] [CrossRef]
- Amirkalali, B.; Sohrabi, M.R.; Esrafily, A.; Jalali, M.; Gholami, A.; Hosseinzadeh, P.; Keyvani, H.; Shidfar, F.; Zamani, F. Association between Nicotinamide Phosphoribosyltransferase and de novo Lipogenesis in Nonalcoholic Fatty Liver Disease. Med. Princ. Pract. 2017, 26, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; De Luis, D.A.; Izaola, O.; Sagrado, M.G.; Conde, R.; Velasco, M.C.; Alvarez, T.; Pacheco, D.; González, J.M. Influence of visfatin on histopathological changes of non-alcoholic fatty liver disease. Dig. Dis. Sci. 2009, 54, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Elkabany, Z.A.; Hamza, R.T.; Ismail, E.A.R.; Elsharkawy, A.; Yosry, A.; Musa, S.; Khalaf, M.A.; Elgawesh, R.M.; Esmat, G. Serum visfatin level as a noninvasive marker for nonalcoholic fatty liver disease in children and adolescents with obesity: Relation to transient elastography with controlled attenuation parameter. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1008–1016. [Google Scholar] [CrossRef]
- Johannsen, K.; Flechtner-Mors, M.; Kratzer, W.; Koenig, W.; Boehm, B.O.; Schmidberger, J. Association Between Visfatin and Hepatic Steatosis in the General Population During Long-Term Follow-Up. Horm. Metab. Res. 2019, 51, 602–607. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Wang, H. Correlation of blood glucose, serum chemerin and insulin resistance with NAFLD in patients with type 2 diabetes mellitus. Exp. Ther. Med. 2018, 15, 2936. [Google Scholar] [CrossRef]
- Kłusek-Oksiuta, M.; Bialokoz-Kalinowska, I.; Tarasów, E.; Wojtkowska, M.; Werpachowska, I.; Lebensztejn, D.M. Chemerin as a novel non-invasive serum marker of intrahepatic lipid content in obese children. Ital. J. Pediatr. 2014, 40, 84. [Google Scholar] [CrossRef]
- Ren, Q.; Wang, H.; Zeng, Y.; Fang, X.; Wang, M.; Li, D.; Huang, W.; Xu, Y. Circulating chemerin levels in metabolic-associated fatty liver disease: A systematic review and meta-analysis. Lipids Health Dis. 2022, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, M.; Ouwens, D.M.; Hörbelt, T.; Van de Velde, F.; Fahlbusch, P.; Herzfeld de Wiza, D.; Van Nieuwenhove, Y.; Calders, P.; Praet, M.; Hoorens, A.; et al. Reduced expression of chemerin in visceral adipose tissue associates with hepatic steatosis in patients with obesity. Obesity 2016, 24, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Pohl, R.; Haberl, E.M.; Rein-Fischboeck, L.; Zimny, S.; Neumann, M.; Aslanidis, C.; Schacherer, D.; Krautbauer, S.; Eisinger, K.; Weiss, T.S.; et al. Hepatic chemerin mRNA expression is reduced in human nonalcoholic steatohepatitis. Eur. J. Clin. Investig. 2017, 47, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kajor, M.; Kukla, M.; Waluga, M.; Liszka, Ł.; Dyaczyński, M.; Kowalski, G.; Żądło, D.; Berdowska, A.; Chapuła, M.; Kostrząb-Zdebel, A.; et al. Hepatic chemerin mRNA in morbidly obese patients with nonalcoholic fatty liver disease. Pol. J. Pathol. 2017, 68, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Döcke, S.; Lock, J.F.; Birkenfeld, A.L.; Hoppe, S.; Lieske, S.; Rieger, A.; Raschzok, N.; Sauer, I.M.; Florian, S.; Osterhoff, M.A.; et al. Elevated hepatic chemerin mRNA expression in human non-alcoholic fatty liver disease. Eur. J. Endocrinol. 2013, 169, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.B.; Kim, S.M.; Cho, G.J.; Choi, K.M. Serum AFBP levels are elevated in patients with nonalcoholic fatty liver disease. Scand. J. Gastroenterol. 2014, 49, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, X.; Pan, X.; He, X.; Wang, Y.; Bao, Y. Serum adipocyte fatty acid-binding protein levels: An indicator of non-alcoholic fatty liver disease in Chinese individuals. Liver Int. 2019, 39, 568–574. [Google Scholar] [CrossRef]
- Milner, K.L.; van der Poorten, D.; Xu, A.; Bugianesi, E.; Kench, J.G.; Lam, K.S.L.; Chisholm, D.J.; George, J. Adipocyte fatty acid binding protein levels relate to inflammation and fibrosis in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1926–1934. [Google Scholar] [CrossRef]
- Shen, J.; Chan, H.L.Y.; Wong, G.L.H.; Choi, P.C.L.; Chan, A.W.H.; Chan, H.Y.; Chim, A.M.L.; Yeung, D.K.W.; Chan, F.K.L.; Woo, J.; et al. Non-invasive diagnosis of non-alcoholic steatohepatitis by combined serum biomarkers. J. Hepatol. 2012, 56, 1363–1370. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef]
- Salman, A.A.; Sultan, A.A.E.A.; Abdallah, A.; Abdelsalam, A.; Mikhail, H.M.S.; Tourky, M.; Omar, M.G.; Youssef, A.; Ahmed, R.A.; Elkassar, H.; et al. Effect of weight loss induced by laparoscopic sleeve gastrectomy on liver histology and serum adipokine levels. J. Gastroenterol. Hepatol. 2020, 35, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, V.; Wills, R.; DeLany, J.P.; Kershaw, E.E.; Behari, J. Differential Impact of Weight Loss on Nonalcoholic Fatty Liver Resolution in a North American Cohort with Obesity. Obesity 2017, 25, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Kader, S.M.; Al-Shreef, F.M.; Al-Jiffri, O.H. Biochemical parameters response to weight loss in patients with non-alcoholic steatohepatitis. Afr. Health Sci. 2016, 16, 242. [Google Scholar] [CrossRef]
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol. Metab. 2018, 33, 33–43. [Google Scholar] [CrossRef]
- Meir, A.Y.; Rinott, E.; Tsaban, G.; Zelicha, H.; Kaplan, A.; Rosen, P.; Shelef, I.; Youngster, I.; Shalev, A.; Blüher, M.; et al. Effect of green-Mediterranean diet on intrahepatic fat: The DIRECT PLUS randomised controlled trial. Gut 2021, 70, 2085–2095. [Google Scholar] [CrossRef]
- Deibert, P.; Lazaro, A.; Schaffner, D.; Berg, A.; Koenig, D.; Kreisel, W.; Baumstark, M.W.; Steinmann, D.; Buechert, M.; Lange, T. Comprehensive lifestyle intervention vs soy protein-based meal regimen in non-alcoholic steatohepatitis. World J. Gastroenterol. 2019, 25, 1116. [Google Scholar] [CrossRef]
- Nanjan, M.J.; Mohammed, M.; Prashantha Kumar, B.R.; Chandrasekar, M.J.N. Thiazolidinediones as antidiabetic agents: A critical review. Bioorg. Chem. 2018, 77, 548–567. [Google Scholar] [CrossRef]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients with Nonalcoholic Steatohepatitis with vs Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566.e2. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Mantzoros, C.S. Adiponectin as a target for the treatment of nonalcoholic steatohepatitis with thiazolidinediones: A systematic review. Metabolism 2016, 65, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Sabatini, S.; Carli, F.; Gaggini, M.; Bril, F.; Belfort-DeAguiar, R.; Positano, V.; Barb, D.; Kadiyala, S.; Harrison, S.; et al. PPAR-γ-induced changes in visceral fat and adiponectin levels are associated with improvement of steatohepatitis in patients with NASH. Liver Int. 2021, 41, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Lutchman, G.; Modi, A.; Kleiner, D.E.; Promrat, K.; Heller, T.; Ghany, M.; Borg, B.; Loomba, R.; Liang, T.J.; Premkumar, A.; et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology 2007, 46, 424–429. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med. 2010, 170, 1191–1201. [Google Scholar] [CrossRef]
- Lewis, J.D.; Ferrara, A.; Peng, T.; Hedderson, M.; Bilker, W.B.; Quesenberry, C.P.; Vaughn, D.J.; Nessel, L.; Selby, J.; Strom, B.L. Risk of Bladder Cancer Among Diabetic Patients Treated With PioglitazoneInterim report of a longitudinal cohort study. Diabetes Care 2011, 34, 916–922. [Google Scholar] [CrossRef]
- Athyros, V.G.; Polyzos, S.A.; Kountouras, J.; Katsiki, N.; Anagnostis, P.; Doumas, M.; Mantzoros, C.S. Non-Alcoholic Fatty Liver Disease Treatment in Patients with Type 2 Diabetes Mellitus; New Kids on the Block. Curr. Vasc. Pharmacol. 2020, 18, 172–181. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An Old Drug with New Applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef]
- Said, A.; Akhter, A. Meta-analysis of randomized controlled trials of pharmacologic agents in non-alcoholic steatohepatitis. Ann. Hepatol. 2017, 16, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Sun, F.; Li, L.; Jiang, D.; Li, X.; Sun, A.; Pan, Z.; Lou, N.; Zhang, L.; Lou, F. Therapeutic Effect of Metformin on Chemerin in Non-Obese Patients with Non-Alcoholic Fatty Liver Disease (NAFLD). Clin. Lab. 2015, 61, 1409–1414. [Google Scholar] [PubMed]
- Yang, L.; Song, M.Q.; Zhang, Q.L.; Shou, L.; Zang, S.F.; Yang, Y.L. Effect of piglitazone and metformin on retinol-binding protein-4 and adiponectin in patients with type 2 diabetes mellitus complicated with non-alcohol fatty acid liver diseases. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2014, 36, 309–312. [Google Scholar] [PubMed]
- Fan, H.; Pan, Q.; Xu, Y.; Yang, X. Exenatide improves type 2 diabetes concomitant with non-alcoholic fatty liver disease. Arq. Bras. Endocrinol. Metabol. 2013, 57, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Shargorodsky, M.; Omelchenko, E.; Matas, Z.; Boaz, M.; Gavish, D. Relation between augmentation index and adiponectin during one-year metformin treatment for nonalcoholic steatohepatosis: Effects beyond glucose lowering? Cardiovasc. Diabetol. 2012, 11, 61. [Google Scholar] [CrossRef]
- Wei, Q.; Xu, X.; Guo, L.; Li, J.; Li, L. Effect of SGLT2 Inhibitors on Type 2 Diabetes Mellitus With Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Front. Endocrinol. 2021, 12, 635556. [Google Scholar] [CrossRef]
- Mo, M.; Huang, Z.; Liang, Y.; Liao, Y.; Xia, N. The safety and efficacy evaluation of sodium-glucose co-transporter 2 inhibitors for patients with non-alcoholic fatty liver disease: An updated meta-analysis. Dig. Liver Dis. 2022, 54, 461–468. [Google Scholar] [CrossRef]
- Phrueksotsai, S.; Pinyopornpanish, K.; Euathrongchit, J.; Leerapun, A.; Phrommintikul, A.; Buranapin, S.; Chattipakorn, N.; Thongsawat, S. The effects of dapagliflozin on hepatic and visceral fat in type 2 diabetes patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2021, 36, 2952–2959. [Google Scholar] [CrossRef]
- Liu, M.; Liu, F. Regulation of adiponectin multimerization, signaling and function. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 25–31. [Google Scholar] [CrossRef]
- Narasimhan, M.L.; Coca, M.A.; Jin, J.; Yamauchi, T.; Ito, Y.; Kadowaki, T.; Kim, K.K.; Pardo, J.M.; Damsz, B.; Hasegawa, P.M.; et al. Osmotin Is a Homolog of Mammalian Adiponectin and Controls Apoptosis in Yeast through a Homolog of Mammalian Adiponectin Receptor. Mol. Cell 2005, 17, 171–180. [Google Scholar] [CrossRef]
- Ahmad, A.; Ali, T.; Kim, M.W.; Khan, A.; Jo, M.H.; Rehman, S.U.; Khan, M.S.; Abid, N.B.; Khan, M.; Ullah, R.; et al. Adiponectin homolog novel osmotin protects obesity/diabetes-induced NAFLD by upregulating AdipoRs/PPARα signaling in ob/ob and db/db transgenic mouse models. Metabolism 2019, 90, 31–43. [Google Scholar] [CrossRef]
- Meehan, C.A.; Cochran, E.; Kassai, A.; Brown, R.J.; Gorden, P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev. Clin. Pharmacol. 2016, 9, 59–68. [Google Scholar] [CrossRef]
- Akinci, B.; Subauste, A.; Ajluni, N.; Esfandiari, N.H.; Meral, R.; Neidert, A.H.; Eraslan, A.; Hench, R.; Rus, D.; Mckenna, B.; et al. Metreleptin therapy for nonalcoholic steatohepatitis: Open-label therapy interventions in two different clinical settings. Med 2021, 2, 814–835.e6. [Google Scholar] [CrossRef] [PubMed]
- Javor, E.D.; Ghany, M.G.; Cochran, E.K.; Oral, E.A.; DePaoli, A.M.; Premkumar, A.; Kleiner, D.E.; Gorden, P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology 2005, 41, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Safar Zadeh, E.; Lungu, A.O.; Cochran, E.K.; Brown, R.J.; Ghany, M.G.; Heller, T.; Kleiner, D.E.; Gorden, P. The liver diseases of lipodystrophy: The long-term effect of leptin treatment. J. Hepatol. 2013, 59, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Pei, H.; Ma, L.; Ran, Y.; Chen, J.; Wang, G.; Chen, L. Synthesis and biological evaluation of novel urea-and guanidine-based derivatives for the treatment of obesity-related hepatic steatosis. Molecules 2014, 19, 6163–6183. [Google Scholar] [PubMed]
- Marchesini, G.; Day, C.P.; Dufour, J.F.; Canbay, A.; Nobili, V.; Ratziu, V.; Tilg, H.; Roden, M.; Gastaldelli, A.; Yki-Jarvinen, H.; et al. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Hardy, T.; Wonders, K.; Younes, R.; Aithal, G.P.; Aller, R.; Allison, M.; Bedossa, P.; Betsou, F.; Boursier, J.; Brosnan, M.J.; et al. The European NAFLD Registry: A real-world longitudinal cohort study of nonalcoholic fatty liver disease. Contemp. Clin. Trials 2020, 98, 106175. [Google Scholar] [CrossRef]
- Krieg, L.; Schaffert, A.; Kern, M.; Landgraf, K.; Wabitsch, M.; Beck-Sickinger, A.G.; Koerner, A.; Blüher, M.; von Bergen, M.; Schubert, K. An MRM-Based Multiplexed Quantification Assay for Human Adipokines and Apolipoproteins. Molecules 2020, 25, 775. [Google Scholar] [CrossRef]
- Magni, P.; Liuzzi, A.; Ruscica, M.; Dozio, E.; Ferrario, S.; Bussi, I.; Minocci, A.; Castagna, A.; Motta, M.; Savia, G. Free and bound plasma leptin in normal weight and obese men and women: Relationship with body composition, resting energy expenditure, insulin-sensitivity, lipid profile and macronutrient preference. Clin. Endocrinol. 2005, 62, 189–196. [Google Scholar] [CrossRef]
- Francisco, V.; Tovar, S.; Conde, J.; Pino, J.; Mera, A.; Lago, F.; González-Gay, M.A.; Dieguez, C.; Gualillo, O. Levels of the novel endogenous antagonist of ghrelin receptor, liver-enriched antimicrobial peptide-2, in patients with rheumatoid arthritis. Nutrients 2020, 12, 1006. [Google Scholar] [CrossRef]
- Schultz, A.; Saville, B.R.; Marsh, J.A.; Snelling, T.L. An introduction to clinical trial design. Paediatr. Respir. Rev. 2019, 32, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Hollis, S.; Fletcher, C.; Lynn, F.; Urban, H.J.; Branson, J.; Burger, H.U.; Tudur Smith, C.; Sydes, M.R.; Gerlinger, C. Best practice for analysis of shared clinical trial data. BMC Med. Res. Methodol. 2016, 16, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Reimer, K.C.; Wree, A.; Roderburg, C.; Tacke, F. New drugs for NAFLD: Lessons from basic models to the clinic. Hepatol. Int. 2020, 14, 8–23. [Google Scholar] [CrossRef]
- Mazza, G.; Al-Akkad, W.; Rombouts, K. Engineering in vitro models of hepatofibrogenesis. Adv. Drug Deliv. Rev. 2017, 121, 147–157. [Google Scholar] [CrossRef]
- Abdolahi, A.; Vahabzadeh, Z.; Izadpanah, E.; Moloudi, M.R. Vaspin attenuates steatosis-induced fibrosis via GRP78 receptor by targeting AMPK signaling pathway. J. Physiol. Biochem. 2022, 78, 185–197. [Google Scholar] [CrossRef]
- Waluga, M.; Kukla, M.; Kotulski, R.; Zorniak, M.; Boryczka, G.; Kajor, M.; Ciupinska-Kajor, M.; Lekstan, A.; Olczyk, P.; Waluga, E. Omentin, vaspin and irisin in chronic liver diseases. J. Physiol. Pharmacol. 2019, 70, 277–285. [Google Scholar]
- Aliasghari, F.; Izadi, A.; Jabbari, M.; Imani, B.; Gargari, B.P.; Asjodi, F.; Ebrahimi, S. Are Vaspin and Omentin-1 Related to Insulin Resistance, Blood Pressure and Inflammation in NAFLD Patients? J. Med. Biochem. 2018, 37, 470–475. [Google Scholar] [CrossRef]
- Gu, Y.; Luo, J.; Chen, Q.; Qiu, Y.; Zhou, Y.; Wang, X.; Qian, X.; Liu, Y.; Xie, J.; Xu, Z.; et al. Inverse Association of Serum Adipsin with the Remission of Nonalcoholic Fatty-Liver Disease: A 3-Year Community-Based Cohort Study. Ann. Nutr. Metab. 2022, 78, 21–32. [Google Scholar] [CrossRef]
- Wang, Y.; Song, J.; Bian, H.; Bo, J.; Lv, S.; Pan, W.; Lv, X. Apelin promotes hepatic fibrosis through ERK signaling in LX-2 cells. Mol. Cell. Biochem. 2019, 460, 205–215. [Google Scholar] [CrossRef]
- Montazerifar, F.; Bakhshipour, A.R.; Karajibani, M.; Torki, Z.; Dashipour, A.R. Serum omentin-1, vaspin, and apelin levels and central obesity in patients with nonalcoholic fatty liver disease. J. Res. Med. Sci. 2017, 22, 70. [Google Scholar]
- Khaleel, E.F.; Abdel-Aleem, G.A. Obestatin protects and reverses nonalcoholic fatty liver disease and its associated insulin resistance in rats via inhibition of food intake, enhancing hepatic adiponectin signaling, and blocking ghrelin acylation. Arch. Physiol. Biochem. 2019, 125, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L.; Shao, W.-H.; Vanniasinghe, A.S.; Amon, M.A.; Holub, M.C.; Kovalszky, I.; Wade, J.D.; Doll, M.; Cohen, P.L.; Manolios, N.; et al. Toward understanding the role of leptin and leptin receptor antagonism in preclinical models of rheumatoid arthritis. Peptides 2011, 32, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- DePaoli, A.M. Leptin in common obesity and associated disorders of metabolism. J. Endocrinol. 2014, 223, T71–T81. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).