
AMPK and Diseases: State of the Art Regulation by AMPK-Targeting Molecules




Abstract
:Simple Summary
Abstract
1. AMPK: An Important Player in Regulating Cellular Processes
2. AMPK Activators
2.1. Indirect AMPK Activators
2.2. Direct AMPK Activators
2.3. AMP Mimicking AMPK Activators
3. Gap of Knowledge for AMP Mimicking Drug Development
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheung, P.C.; Salt, I.P.; Davies, S.P.; Hardie, D.G.; Carling, D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem. J. 2000, 346 Pt 3, 659–669. [Google Scholar] [CrossRef]
- Thornton, C.; Snowden, M.A.; Carling, D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J. Biol. Chem. 1998, 273, 12443–12450. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, D.; Gao, G.; Michell, B.J.; Widmer, J.; Mitchelhill, K.; Teh, T.; House, C.M.; Witters, L.A.; Kemp, B.E. Mammalian 5’-AMP-activated protein kinase non-catalytic subunits are homologs of proteins that interact with yeast Snf1 protein kinase. J. Biol. Chem. 1994, 269, 29343–29346. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, A.; Ghilagaber, S.; Nikolaev, A.; Hardie, D.G. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009, 9, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, J.; Zhang, Y.Y.; Yan, S.F.; Neumann, D.; Schlattner, U.; Wang, Z.X.; Wu, J.W. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat. Struct. Mol. Biol. 2012, 19, 716–718. [Google Scholar] [CrossRef]
- Yan, Y.; Mukherjee, S.; Harikumar, K.G.; Strutzenberg, T.S.; Zhou, X.E.; Suino-Powell, K.; Xu, T.H.; Sheldon, R.D.; Lamp, J.; Brunzelle, J.S.; et al. Structure of an AMPK complex in an inactive, ATP-bound state. Science 2021, 373, 413–419. [Google Scholar] [CrossRef]
- Calabrese, M.F.; Rajamohan, F.; Harris, M.S.; Caspers, N.L.; Magyar, R.; Withka, J.M.; Wang, H.; Borzilleri, K.A.; Sahasrabudhe, P.V.; Hoth, L.R.; et al. Structural basis for AMPK activation: Natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 2014, 22, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Ngoei, K.R.W.; Langendorf, C.G.; Ling, N.X.Y.; Hoque, A.; Varghese, S.; Camerino, M.A.; Walker, S.R.; Bozikis, Y.E.; Dite, T.A.; Ovens, A.J.; et al. Structural Determinants for Small-Molecule Activation of Skeletal Muscle AMPK α2β2γ1 by the Glucose Importagog SC4. Cell Chem. Biol. 2018, 25, 728–737.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhou, X.E.; Novick, S.J.; Shaw, S.J.; Li, Y.; Brunzelle, J.S.; Hitoshi, Y.; Griffin, P.R.; Xu, H.E.; Melcher, K. Structures of AMP-activated protein kinase bound to novel pharmacological activators in phosphorylated, non-phosphorylated, and nucleotide-free states. J. Biol. Chem. 2019, 294, 953–967. [Google Scholar] [CrossRef] [Green Version]
- Langendorf, C.G.; Ngoei, K.R.W.; Scott, J.W.; Ling, N.X.Y.; Issa, S.M.A.; Gorman, M.A.; Parker, M.W.; Sakamoto, K.; Oakhill, J.S.; Kemp, B.E. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat. Commun. 2016, 7, 10912. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [Green Version]
- Xin, F.J.; Wang, J.; Zhao, R.Q.; Wang, Z.X.; Wu, J.W. Coordinated regulation of AMPK activity by multiple elements in the α-subunit. Cell Res. 2013, 23, 1237–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Neumann, D. Is TAK1 a Direct Upstream Kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef] [Green Version]
- Joseph, B.K.; Liu, H.Y.; Francisco, J.; Pandya, D.; Donigan, M.; Gallo-Ebert, C.; Giordano, C.; Bata, A.; Nickels, J.T. Inhibition of AMP Kinase by the Protein Phosphatase 2A Heterotrimer, PP2APpp2r2d. J. Biol. Chem. 2015, 290, 10588–10598. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.B.; Liu, Y.Y.; Cheng, L.B.; Lu, J.W.; Zeng, P.; Lu, P.H. AMPKα phosphatase Ppm1E upregulation in human gastric cancer is required for cell proliferation. Oncotarget 2017, 8, 31288–31296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willows, R.; Sanders, M.J.; Xiao, B.; Patel, B.R.; Martin, S.R.; Read, J.; Wilson, J.R.; Hubbard, J.; Gamblin, S.J.; Carling, D. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem. J. 2017, 474, 3059–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fediuc, S.; Gaidhu, M.P.; Ceddia, R.B. Regulation of AMP-activated protein kinase and acetyl-CoA carboxylase phosphorylation by palmitate in skeletal muscle cells. J. Lipid Res. 2006, 47, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.A.; Roach, W.G.; Keller, S.R.; Lane, W.S.; Lienhard, G.E. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 9187–9195. [Google Scholar] [CrossRef] [Green Version]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, S.; Bierhoff, H.; Cado, I.; Weber, A.; Tiebe, M.; Grummt, I.; Voit, R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl. Acad. Sci. USA 2009, 106, 17781–17786. [Google Scholar] [CrossRef] [Green Version]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.R.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Mizrachy-Schwartz, S.; Cohen, N.; Klein, S.; Kravchenko-Balasha, N.; Levitzki, A. Up-regulation of AMP-activated protein kinase in cancer cell lines is mediated through c-Src activation. J. Biol. Chem. 2011, 286, 15268–15277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Assefa, B.T.; Tafere, G.G.; Wondafrash, D.Z.; Gidey, M.T. The Bewildering Effect of AMPK Activators in Alzheimer’s Disease: Review of the Current Evidence. BioMed Res. Int. 2020, 2020, 9895121. [Google Scholar] [CrossRef]
- Hardie, D.G.; Alessi, D.R. LKB1 and AMPK and the cancer-metabolism link—ten years after. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Park, S.Y.; Kim, Y.M.; Park, O.J. Regulatory effect of the AMPK-COX-2 signaling pathway in curcumin-induced apoptosis in HT-29 colon cancer cells. Ann. N Y Acad. Sci. 2009, 1171, 489–494. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Scheffler, T.L.; Gunawan, A.M.; Shi, H.; Zeng, C.; Hannon, K.M.; Grant, A.L.; Gerrard, D.E. Chronic elevated calcium blocks AMPK-induced GLUT-4 expression in skeletal muscle. Am. J. Physiol. Cell Physiol. 2009, 296, C106–C115. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. (Lausanne) 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrigal-Perez, L.A.; Ramos-Gomez, M. Resveratrol Inhibition of Cellular Respiration: New Paradigm for an Old Mechanism. Int. J. Mol. Sci. 2016, 17, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMP-activated protein kinase as a drug target. Annu. Rev. Pharm. Toxicol. 2007, 47, 185–210. [Google Scholar] [CrossRef]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- García Rubiño, M.E.; Carrillo, E.; Ruiz Alcalá, G.; Domínguez-Martín, A.; Marchal, J.A.; Boulaiz, H. Phenformin as an Anticancer Agent: Challenges and Prospects. Int. J. Mol. Sci. 2019, 20, 3316. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Bridges, M.D.; Yan, Y.; de Waal, P.W.; Zhou, X.E.; Suino-Powell, K.M.; Xu, H.E.; Hubbell, W.L.; Melcher, K. Conformational heterogeneity of the allosteric drug and metabolite (ADaM) site in AMP-activated protein kinase (AMPK). J. Biol. Chem. 2018, 293, 16994–17007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, M.J.; Ali, Z.S.; Hegarty, B.D.; Heath, R.; Snowden, M.A.; Carling, D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 2007, 282, 32539–32548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Dandapani, M.; Hardie, D.G. AMPK: Mediating the metabolic effects of salicylate-based drugs? Trends Endocrinol. Metab. 2013, 24, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, E.E.; Coelho, P.P.; Blagih, J.; Griss, T.; Viollet, B.; Jones, R.G. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 2015, 34, 3627–3639. [Google Scholar] [CrossRef] [Green Version]
- Moreno, D.; Knecht, E.; Viollet, B.; Sanz, P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008, 582, 2650–2654. [Google Scholar] [CrossRef] [Green Version]
- Benziane, B.; Björnholm, M.; Lantier, L.; Viollet, B.; Zierath, J.R.; Chibalin, A.V. AMP-activated protein kinase activator A-769662 is an inhibitor of the Na(+)-K(+)-ATPase. Am. J. Physiol. Cell Physiol. 2009, 297, C1554–C1566. [Google Scholar] [CrossRef] [Green Version]
- Pang, T.; Zhang, Z.S.; Gu, M.; Qiu, B.Y.; Yu, L.F.; Cao, P.R.; Shao, W.; Su, M.B.; Li, J.Y.; Nan, F.J.; et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J. Biol. Chem. 2008, 283, 16051–16060. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.E.; Ross, F.A.; Kleinert, M.; Sylow, L.; Knudsen, J.R.; Gowans, G.J.; Hardie, D.G.; Richter, E.A. PT-1 selectively activates AMPK-γ1 complexes in mouse skeletal muscle, but activates all three γ subunit complexes in cultured human cells by inhibiting the respiratory chain. Biochem. J. 2015, 467, 461–472. [Google Scholar] [CrossRef]
- Feng, D.; Biftu, T.; Romero, F.A.; Kekec, A.; Dropinski, J.; Kassick, A.; Xu, S.; Kurtz, M.M.; Gollapudi, A.; Shao, Q.; et al. Discovery of MK-8722: A Systemic, Direct Pan-Activator of AMP-Activated Protein Kinase. ACS Med. Chem. Lett. 2018, 9, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
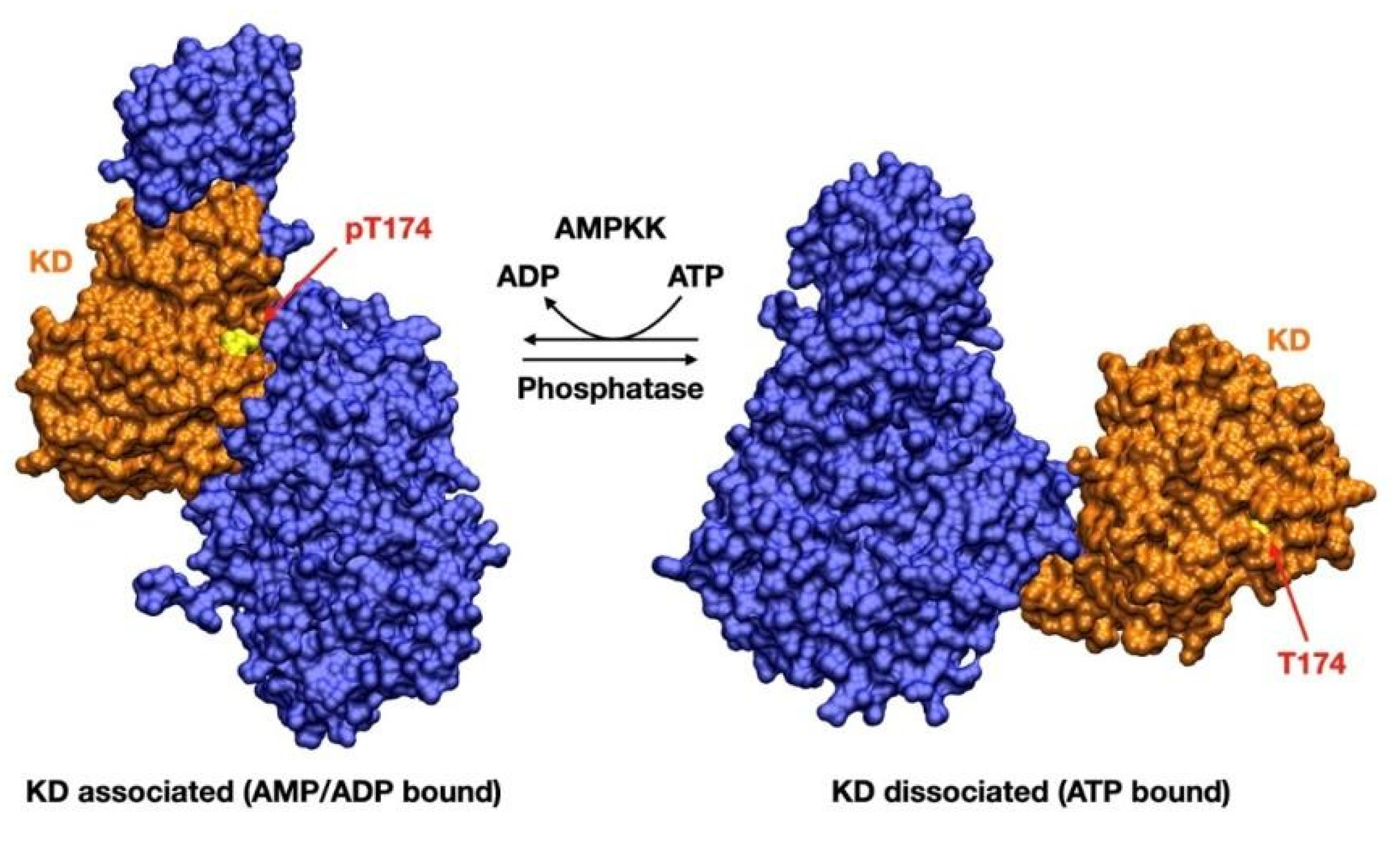
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.C.; Woods, A.; Jones, N.A.; Davison, M.D.; Carling, D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 2000, 345 Pt 3, 437–443. [Google Scholar] [CrossRef]
- Grahame Hardie, D. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Buhl, E.S.; Jessen, N.; Pold, R.; Ledet, T.; Flyvbjerg, A.; Pedersen, S.B.; Pedersen, O.; Schmitz, O.; Lund, S. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes 2002, 51, 2199–2206. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Ross, F.A.; Russell, F.M.; Atrih, A.; Lamont, D.J.; Hardie, D.G. Mechanism of Activation of AMPK by Cordycepin. Cell Chem. Biol. 2020, 27, 214–222.e4. [Google Scholar] [CrossRef] [Green Version]
- Steneberg, P.; Lindahl, E.; Dahl, U.; Lidh, E.; Straseviciene, J.; Backlund, F.; Kjellkvist, E.; Berggren, E.; Lundberg, I.; Bergqvist, I.; et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 2018, 3, e99114. [Google Scholar] [CrossRef]
- Bung, N.; Surepalli, S.; Seshadri, S.; Patel, S.; Peddasomayajula, S.; Kummari, L.K.; Kumar, S.T.; Babu, P.P.; Parsa, K.V.L.; Poondra, R.R.; et al. 2-[2-(4-(trifluoromethyl)phenylamino)thiazol-4-yl]acetic acid (Activator-3) is a potent activator of AMPK. Sci. Rep. 2018, 8, 9599. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Galeno, J.E.; Dang, Q.; Nguyen, T.H.; Boyer, S.H.; Grote, M.P.; Sun, Z.; Chen, M.; Craigo, W.A.; van Poelje, P.D.; MacKenna, D.A.; et al. A Potent and Selective AMPK Activator That Inhibits de Novo Lipogenesis. ACS Med. Chem. Lett. 2010, 1, 478–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.W.; Foretz, M.; Bultot, L.; Fullerton, M.D.; Deak, M.; Ross, F.A.; Hawley, S.A.; Shpiro, N.; Viollet, B.; Barron, D.; et al. Mechanism of action of compound-13: An α1-selective small molecule activator of AMPK. Chem. Biol. 2014, 21, 866–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.W.; Ling, N.; Issa, S.M.; Dite, T.A.; O’Brien, M.T.; Chen, Z.P.; Galic, S.; Langendorf, C.G.; Steinberg, G.R.; Kemp, B.E.; et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem. Biol. 2014, 21, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducommun, S.; Ford, R.J.; Bultot, L.; Deak, M.; Bertrand, L.; Kemp, B.E.; Steinberg, G.R.; Sakamoto, K. Enhanced activation of cellular AMPK by dual-small molecule treatment: AICAR and A769662. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E688–E696. [Google Scholar] [CrossRef] [Green Version]
- Dunn, D.M.; Munger, J. Interplay Between Calcium and AMPK Signaling in Human Cytomegalovirus Infection. Front. Cell Infect. Microbiol. 2020, 10, 384. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Park, W.H.; You, B.R. Antimycin A induces death of the human pulmonary fibroblast cells via ROS increase and GSH depletion. Int. J. Oncol. 2016, 48, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Giaccari, A.; Solini, A.; Frontoni, S.; Del Prato, S. Metformin Benefits: Another Example for Alternative Energy Substrate Mechanism? Diabetes Care 2021, 44, 647–654. [Google Scholar] [CrossRef]
- Smith, R.M.; Peterson, W.H.; Mccoy, E. Oligomycin, a new antifungal antibiotic. Antibiot. Chemother. 1954, 4, 962–970. [Google Scholar]
- Kobayashi, K.; Nishino, C.; Ohya, J.; Sato, S.; Mikawa, T.; Shiobara, Y.; Kodama, M.; Nishimoto, N. Oligomycin E, a new antitumor antibiotic produced by Streptomyces sp. MCI-2225. J. Antibiot. 1987, 40, 1053–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermans, A.D.; Balteau, M.; Gélinas, R.; Renguet, E.; Ginion, A.; de Meester, C.; Sakamoto, K.; Balligand, J.L.; Bontemps, F.; Vanoverschelde, J.L.; et al. A-769662 potentiates the effect of other AMP-activated protein kinase activators on cardiac glucose uptake. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1619–H1630. [Google Scholar] [CrossRef] [Green Version]
- Bultot, L.; Jensen, T.E.; Lai, Y.C.; Madsen, A.L.; Collodet, C.; Kviklyte, S.; Deak, M.; Yavari, A.; Foretz, M.; Ghaffari, S.; et al. Benzimidazole derivative small-molecule 991 enhances AMPK activity and glucose uptake induced by AICAR or contraction in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E706–E719. [Google Scholar] [CrossRef] [Green Version]
- Yeasmin, F.; Choi, H.W. Natural Salicylates and Their Roles in Human Health. Int. J. Mol. Sci. 2020, 21, 9049. [Google Scholar] [CrossRef] [PubMed]
- Višnjić, D.; Lalić, H.; Dembitz, V.; Tomić, B.; Smoljo, T. AICAr, a Widely Used AMPK Activator with Important AMPK-Independent Effects: A Systematic Review. Cells 2021, 10, 1095. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.A.; Elkhalifa, A.E.O.; Siddiqui, A.J.; Patel, M.; Awadelkareem, A.M.; Snoussi, M.; Ashraf, M.S.; Adnan, M.; Hadi, S. Cordycepin for Health and Wellbeing: A Potent Bioactive Metabolite of an Entomopathogenic. Molecules 2020, 25, 2735. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, M.; Steneberg, P.; Nyrén, R.; Edlund, H. AMPK activator O304 improves metabolic and cardiac function, and exercise capacity in aged mice. Commun. Biol. 2021, 4, 1306. [Google Scholar] [CrossRef]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Coccimiglio, I.F.; Clarke, D.C. ADP is the dominant controller of AMP-activated protein kinase activity dynamics in skeletal muscle during exercise. PLoS Comput. Biol. 2020, 16, e1008079. [Google Scholar] [CrossRef]

{kind=link}
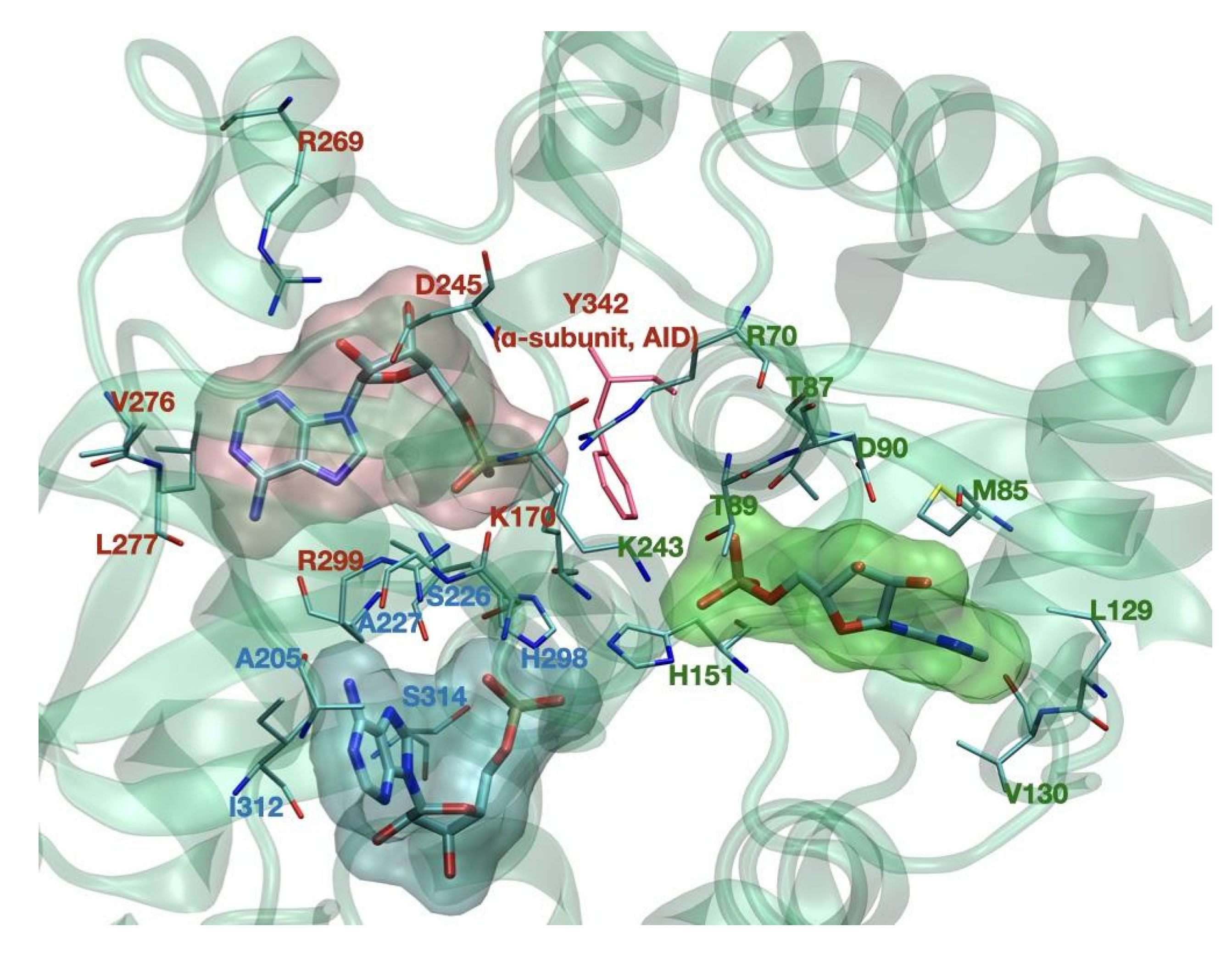{kind=link}
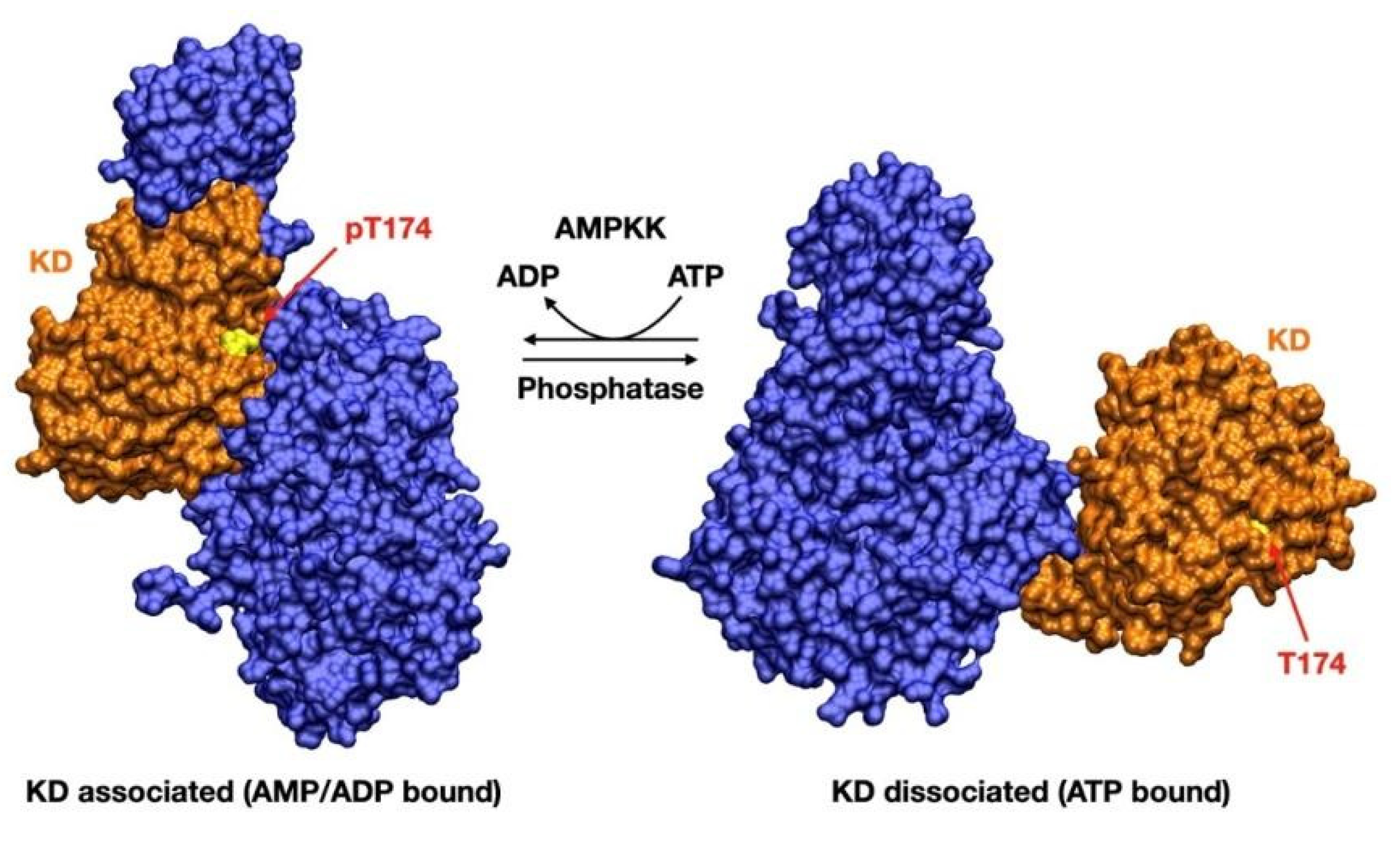{kind=link}
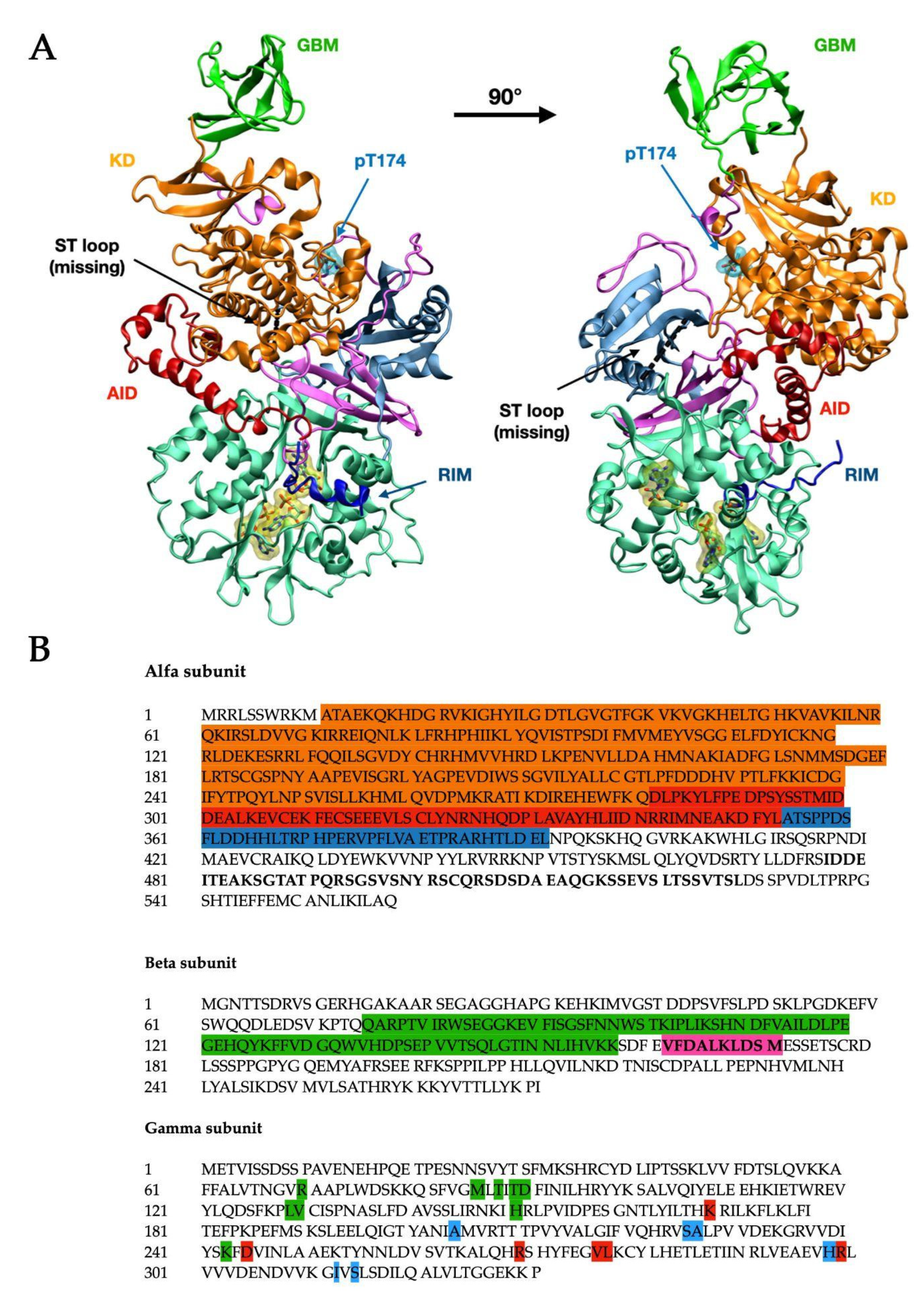
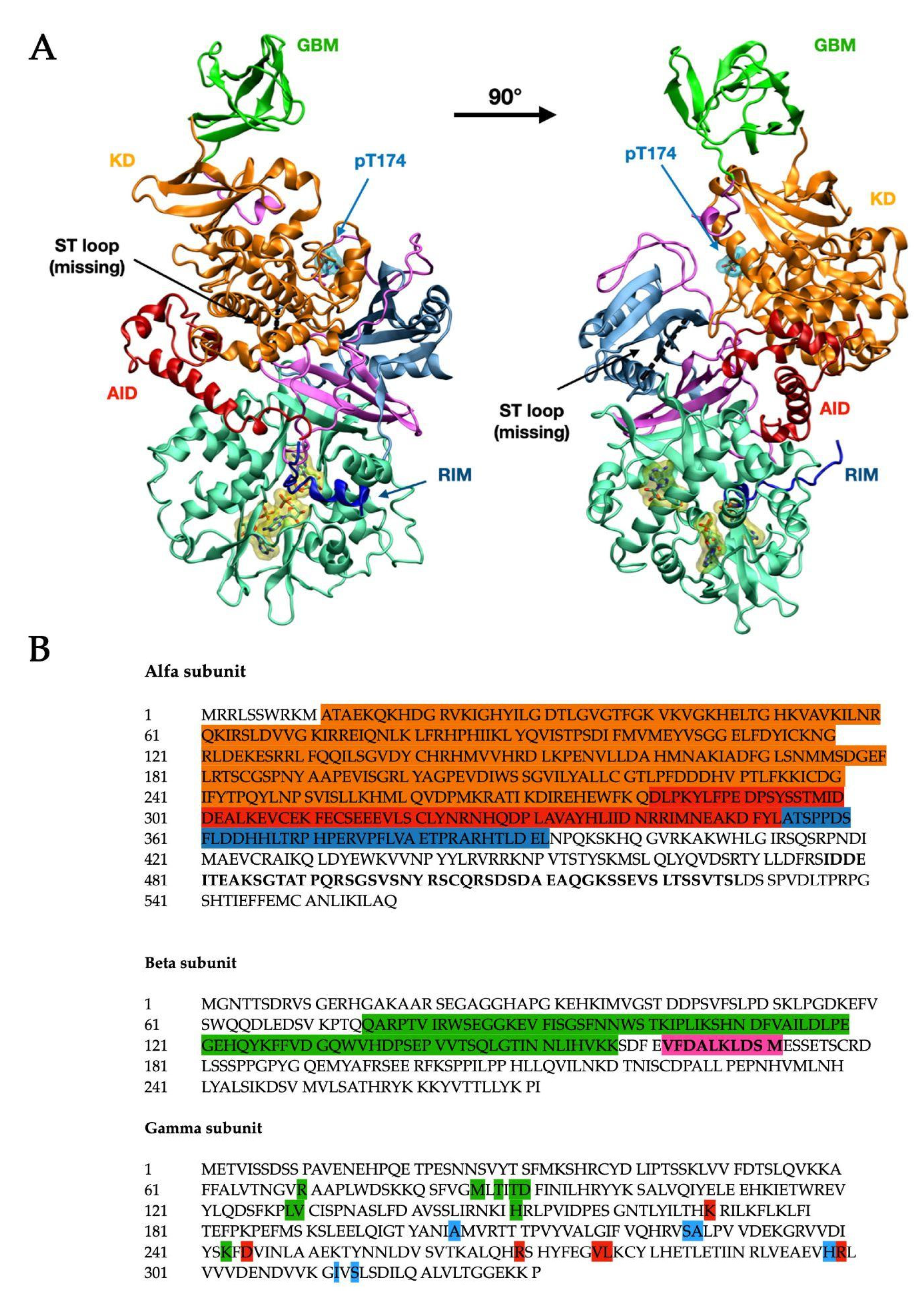
| Subunit | Domain | Residues (aa) | Color Code (Figure 1) |
|---|---|---|---|
| Alfa | Kinase Domain | 11–281 [4] | Orange |
| AID | 282–353 [4] | Red | |
| RIMST loop (black dashed line) | 354–392 [4] 477–528 [9] | Blue | |
| Beta | GBM | 75–157 [4] | Bright green |
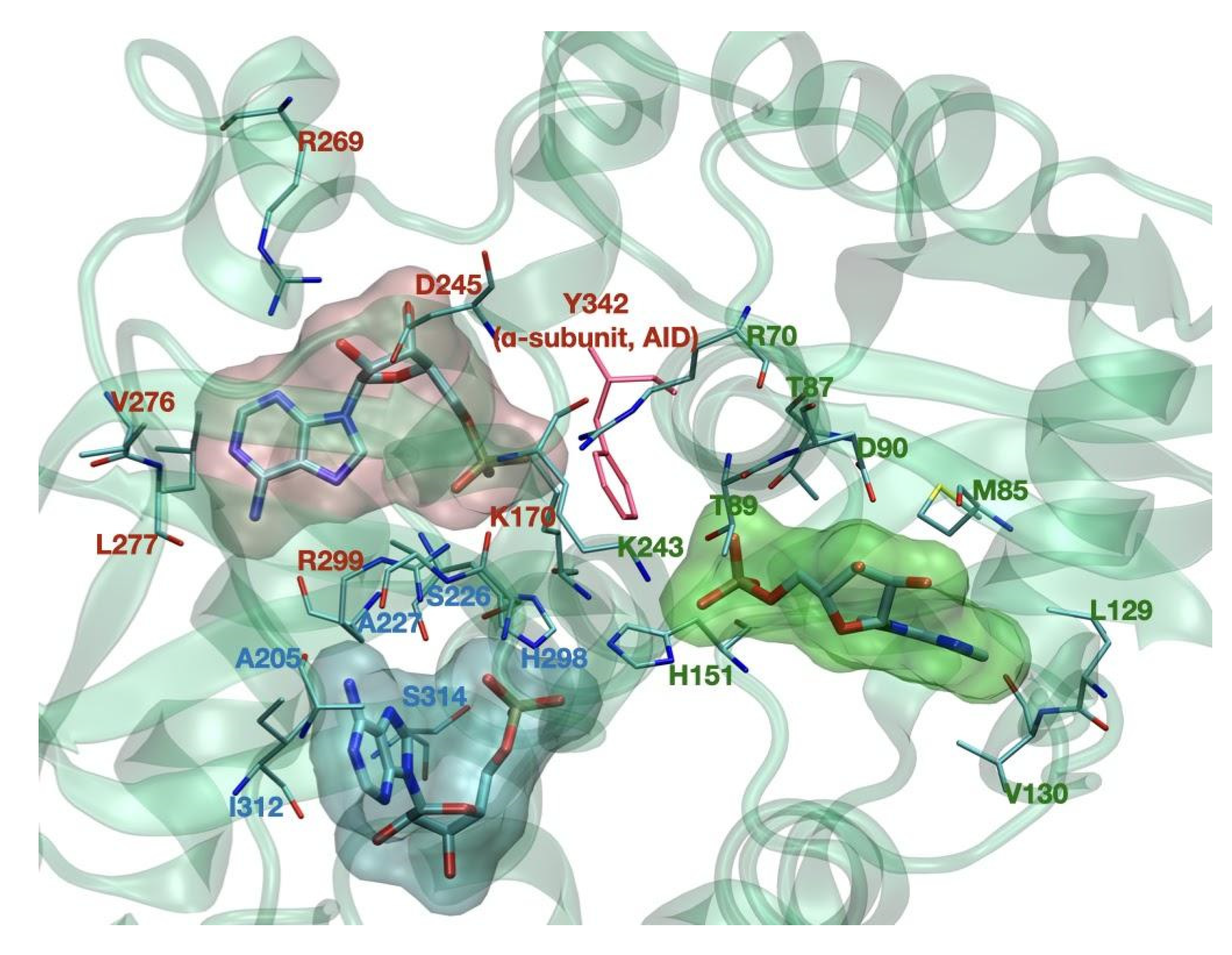
| Allosteric drug and metabolite binding site—ADaM site (see below in the text—not completely resolved) | 162–171 [10] | n/a |
| PDB ID | Title | Subunits | Method | Release Date | Reference |
|---|---|---|---|---|---|
| 7JHG | Cryo-EM structure of ATP-bound fully inactive AMPK in complex with Dorsomorphin (Compound C) and Fab-nanobody | α1; β2; γ1 | Cryo-EM | 2021 | [9] |
| 7JHH | Cryo-EM structure of ATP-bound fully inactive AMPK in complex with Fab and nanobody | α1; β2; γ1 | Cryo-EM | 2021 | [9] |
| 7JIJ | ATP-bound AMP-activated protein kinase | α1; β2; γ1 | X-ray diffraction | 2021 | [9] |
| 7M74 | ATP-bound AMP-activated protein kinase | α1; β2; γ1 | Cryo-EM | 2021 | [9] |
| 6B2E | Structure of full-length human AMPK (a2b2g1) in complex with a small molecule activator SC4 | α2; β2; γ1 | X-ray diffraction | 2018 | [11] |
| 6B1U | Structure of full-length human AMPK (a2b2g1) in complex with a small molecule activator SC4 | α2; β1; γ1 | X-ray diffraction | 2018 | [11] |
| 6C9F | AMP-activated protein kinase bound to pharmacological activator R734 | α1; β1; γ1 | X-ray diffraction | 2018 | [12] |
| 6C9G | AMP-activated protein kinase bound to pharmacological activator R739 | α1; β1; γ1 | X-ray diffraction | 2018 | [12] |
| 6C9H | Non-phosphorylated AMP-activated protein kinase bound to pharmacological activator R734 | α1; β1; γ1 | X-ray diffraction | 2018 | [12] |
| 6C9J | AMP-activated protein kinase bound to pharmacological activator R734 | α1; β1; γ1 | X-ray diffraction | 2018 | [12] |
| 5ISO | Structure of full-length human AMPK (non-phosphorylated at T-loop) in complex with a small molecule activator, a benzimidazole derivative (991) | α2; β1; γ1 | X-ray diffraction | 2017 | to be published |
| 5EZV | X-ray crystal structure of AMP-activated protein kinase alpha-2/alpha-1 RIM chimaera (alpha-2(1–347)/alpha-1(349–401)/alpha-2(397-end) beta-1 gamma-1) co-crystallized with C2 (5-(5-hydroxyl-isoxazol-3-yl)-furan-2-phosphonic acid) | α2/α1; β1; γ1 | X-ray diffraction | 2016 | [13] |
| 4ZHX | Novel binding site for allosteric activation of AMPK | α2; β1; γ1 | X-ray diffraction | 2016 | [13] |
| 4RER | Crystal structure of the phosphorylated human alpha1 beta2 gamma1 holo-AMPK complex bound to AMP and cyclodextrin | α1; β2; γ1 | X-ray diffraction | 2014 | [4] |
| 4REW | Crystal structure of the non-phosphorylated human alpha1 beta2 gamma1 holo-AMPK complex | α1; β2; γ1 | X-ray diffraction | 2014 | [4] |
| 4CFE | Structure of full-length human AMPK in complex with a small molecule activator, a benzimidazole derivative (991) | α2; β1; γ1 | X-ray diffraction | 2013 | [14] |
| 4CFF | Structure of full-length human AMPK in complex with a small molecule activator, a thienopyridone derivative (A-769662) | α2; β1; γ1 | X-ray diffraction | 2013 | [14] |
| Mechanism of action | Drug name | Treatment | Disadvantage | Advantage | Ref. | |
|---|---|---|---|---|---|---|
| Indirect AMPK activation | intracellular accumulation of Ca2+ | upstream regulation of (CaMKK2) | calcium-AMPK signaling regulates Human Cytomegalovirus (HCMV) infection | disruption of Ca2+ balance can lead to various side effects. | Ca2+ plays an essential role in regulating many signaling pathways and cellular processes, such as cell growth and differentiation | [76,77] |
| inhibit mitochondrial ATP synthesis by inhibiting the respiratory chain | antimycin A | anti-tumoral | lack of specificity, quite toxic for normal cells | therapeutic advantage for the treatment of tumors | [78] | |
| metformin | type II diabetes | gastrointestinal side effects (diarrhea, nausea, abdominal discomfort) | glucose-lowering efficacy, modest body weight reduction, easy combination with almost any other glucose-lowering agent | [79] | ||
| phenformin | type II diabetes | develops life-threatening cases of lactic acidosis | potential effect on cancer treatment | [49] | ||
| oligomycin | anti-fungal, anti-tumoral | high lactate accumulating in the blood, urine | therapeutic advantage for the treatment of tumors | [80,81] | ||
| resveratrol | anti-inflammatory, anti-oxidative, antitumoral, neurological, cardiovascular diseases, diabetes, NAFLD, obesity | nausea, vomiting, diarrhea, and liver dysfunction in patients with NAFLD | high range of treatment applications, may promote heart health, lower cholesterol, promote brain health, slow cancer growth | [50] | ||
| Direct AMPK activation | ADaM site | A-769662 | cardiovascular disorders | can have few off-target effects, ineffective activation of β2-subunit | reduction of lactic acidosis, reduces infarct size, allows a better recovery of contractile function during reperfusion | [82] |
| ADaM site | compound 991 | skeletal muscle glucose uptake, type II diabetes, obesity | activate β1-isoform 10 times stronger than β2 | 5-10-fold more potent than A-769662 in activating AMPK, minimal side effects | [83] | |
| ADaM site | MT 63–78 | anti-tumoral | low-affinity binding to β2 subunit | effective at low concentration | [54] | |
| ADaM site | salicylate | relieve pain and inflammation, reduce fever, prevent excessive blood clotting | difficult breathing, diarrhea, nausea, vomiting | lower risks of cancer, heart disease, and diabetes | [84] | |
| contradictive information | PT-1 | lower hepatic lipid content, type II diabetes, obesity | selective for γ1 and not γ3 isoform | promising AMPK activator, minimal side effects | [59,60] | |
| ADAM site | MK-8722 | increase glucose uptake into skeletal muscle, type II diabetes | induce reversible cardiac hypertrophy and increase cardiac glycogen | activate 12 AMPK complexes, induce robust, durable, insulin-independent glucose uptake and glycogen synthesis | [62] | |
| AMPK mimicking compounds | phosphorylated by adenosine kinase to ZMP, binds the same CBS domains as AMP | AICAR | anti-inflammatory, skeletal muscle glucose uptake, cardiovascular diseases | poor oral bioavailability, may have AMPK-independent effects | long-term treatment without side effects, reduce myocardial infarction | [85] |
| AMP analog CoMP binds γ1 subunit | Cordycepin | anti-tumoral, type II diabetes, obesity, anti-fungal, anti-inflammatory, antioxidant, anti-aging, antiviral, hepato-protective | mild gastrointestinal side effects | structure similarity with adenosine makes it an important bioactive component, a wide variety of positive effect | [68,86] | |
| mimics ADP, suppresses pAMPK. dephosphorylation | O304 | type II diabetes, obesity, cardiovascular diseases, peripheral microvascular perfusion | no particular side effects in clinical trials | reduces hyperglycemia, and hyperinsulinemia without inducing cardiac hypertrophy, mimics the beneficial effects of exercise, shows good safety | [87] | |
| interacts with R70 and R152 of the CBS1 domain on γ-subunit near AMP binding site. | Activator-3 | type II diabetes, obesity | mode of activation of Activator-3 is not completely understood | significantly enhance glucose consumption, increase lipid profiles, good pharmacokinetic profile in blood plasma, low brain penetration | [70] | |
| bind γ -subunit near AMP binding site | Compound-2 | metabolic disorders, obesity, and type II diabetes | selectivity for α1 rather than α2 subunit | >20-fold more potent than A-769662 and more than two orders of magnitude more potent than AMP, does not affect any of other AMP activating enzymes | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarasiuk, O.; Miceli, M.; Di Domizio, A.; Nicolini, G. AMPK and Diseases: State of the Art Regulation by AMPK-Targeting Molecules. Biology 2022, 11, 1041. https://doi.org/10.3390/biology11071041
Tarasiuk O, Miceli M, Di Domizio A, Nicolini G. AMPK and Diseases: State of the Art Regulation by AMPK-Targeting Molecules. Biology. 2022; 11(7):1041. https://doi.org/10.3390/biology11071041
Chicago/Turabian StyleTarasiuk, Olga, Matteo Miceli, Alessandro Di Domizio, and Gabriella Nicolini. 2022. "AMPK and Diseases: State of the Art Regulation by AMPK-Targeting Molecules" Biology 11, no. 7: 1041. https://doi.org/10.3390/biology11071041
APA StyleTarasiuk, O., Miceli, M., Di Domizio, A., & Nicolini, G. (2022). AMPK and Diseases: State of the Art Regulation by AMPK-Targeting Molecules. Biology, 11(7), 1041. https://doi.org/10.3390/biology11071041