
Identification of Potentially Related Genes and Mechanisms Involved in Skeletal Muscle Atrophy Induced by Excessive Exercise in Zebrafish

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Swimming Exercises and Experimental Conditions
2.3. Hematoxylin and Eosin Staining
2.4. Swimming Performance and Oxygen Consumption
2.5. Identification of Differentially Expressed Genes
2.6. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Enrichment Analysis of DEGs
2.7. Clusters of Orthologous Groups Analysis of DEGs
2.8. Total RNA Extraction and qRT-PCR
2.9. Western Blot
2.10. Protein–Protein Interaction Network
2.11. Statistical Analysis
3. Results
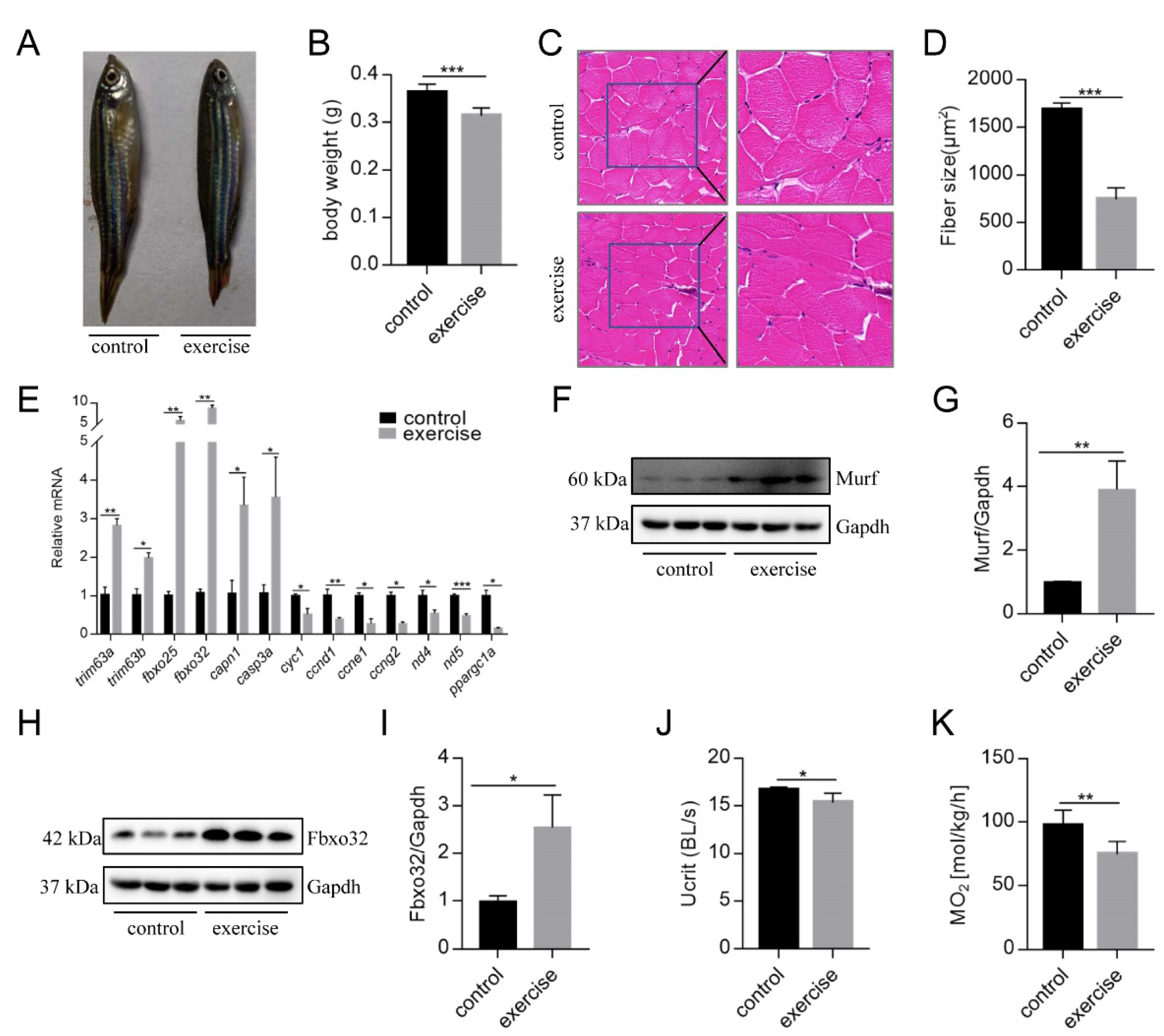
3.1. Excessive Exercise Caused Muscle Atrophy of Zebrafish Skeletal Muscle
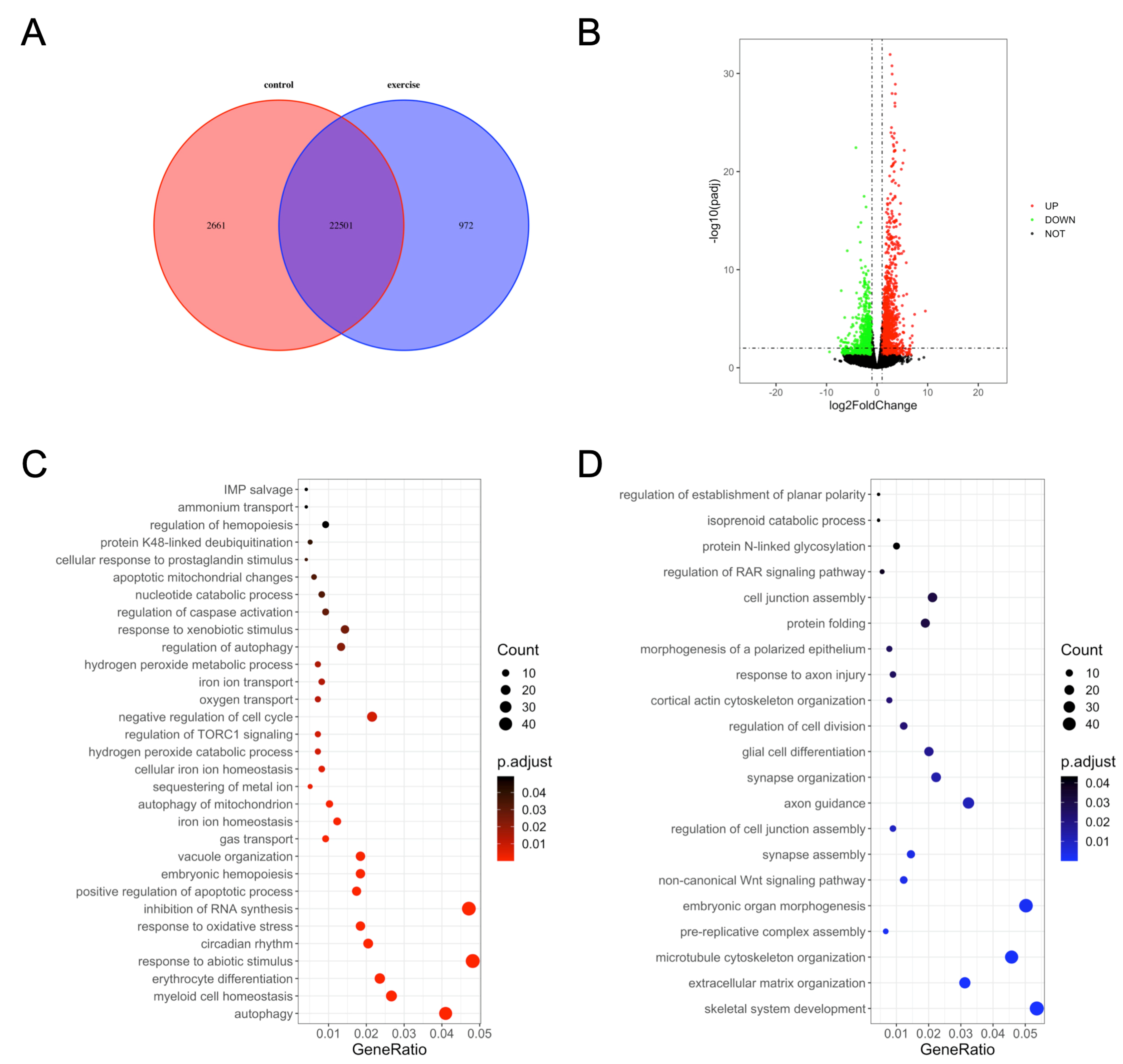
3.2. Functional Annotation of DEGs
3.3. KEGG Pathway and COG Analysis of DEGs
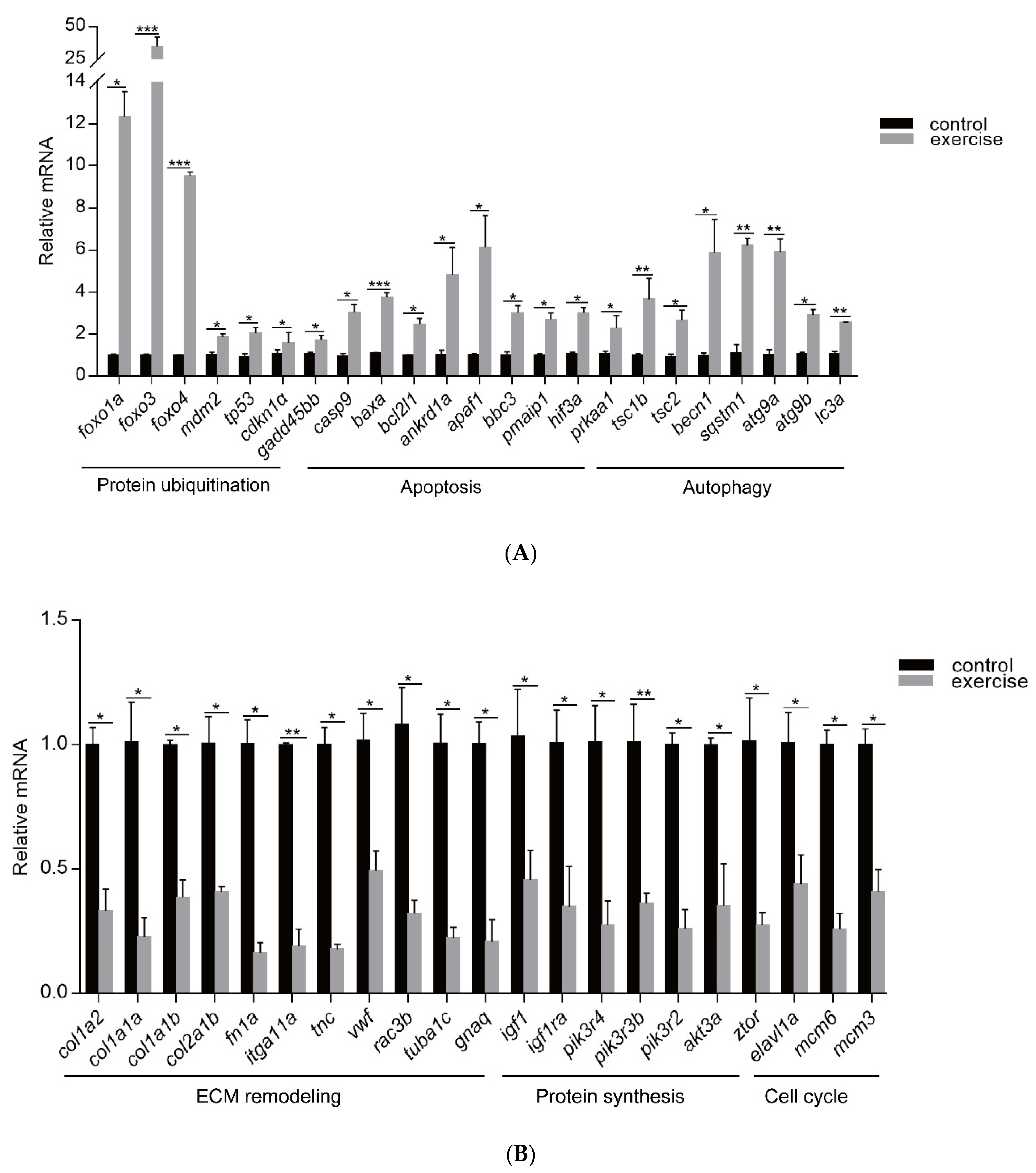
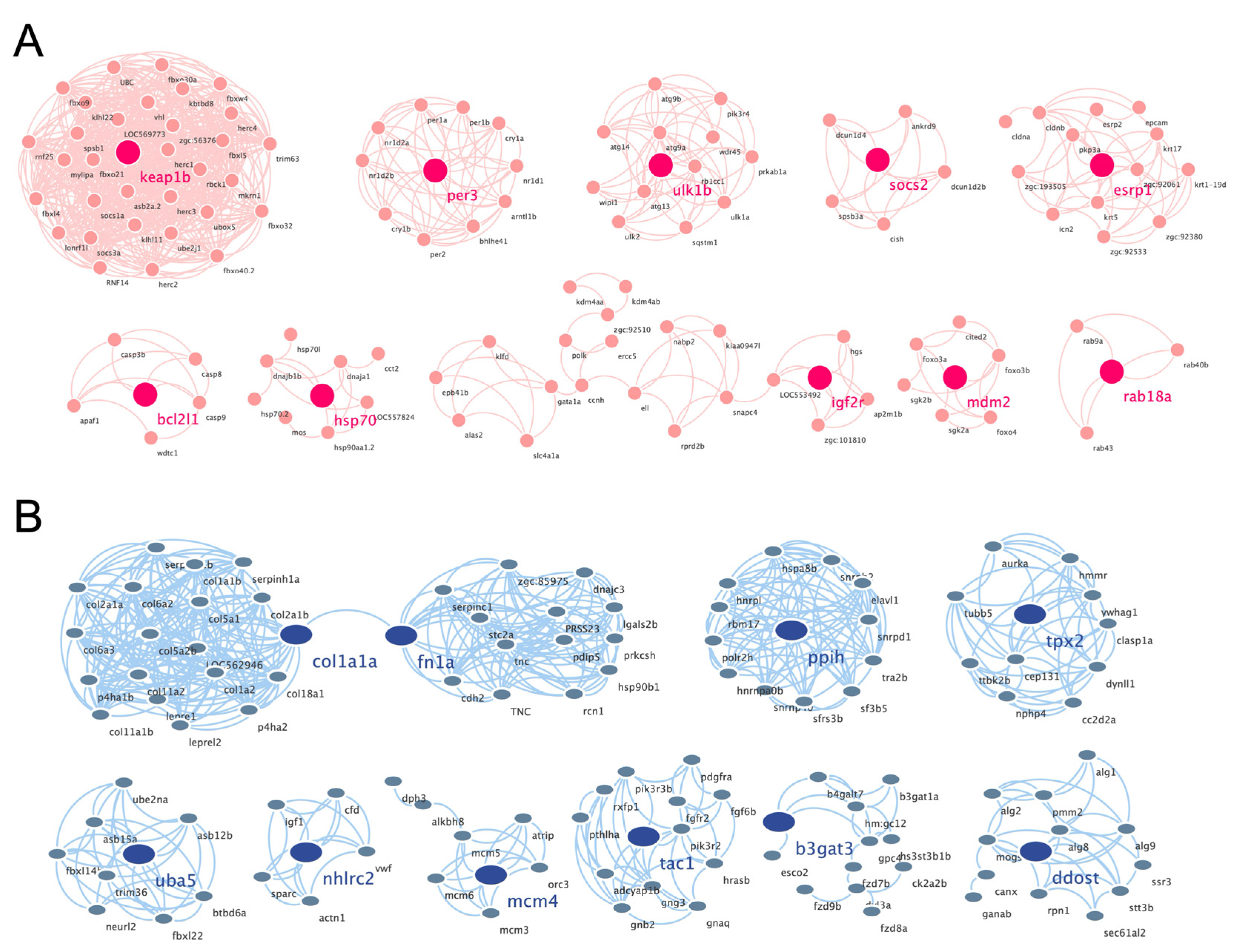
3.4. Construction of PPI Network and Cluster Identification
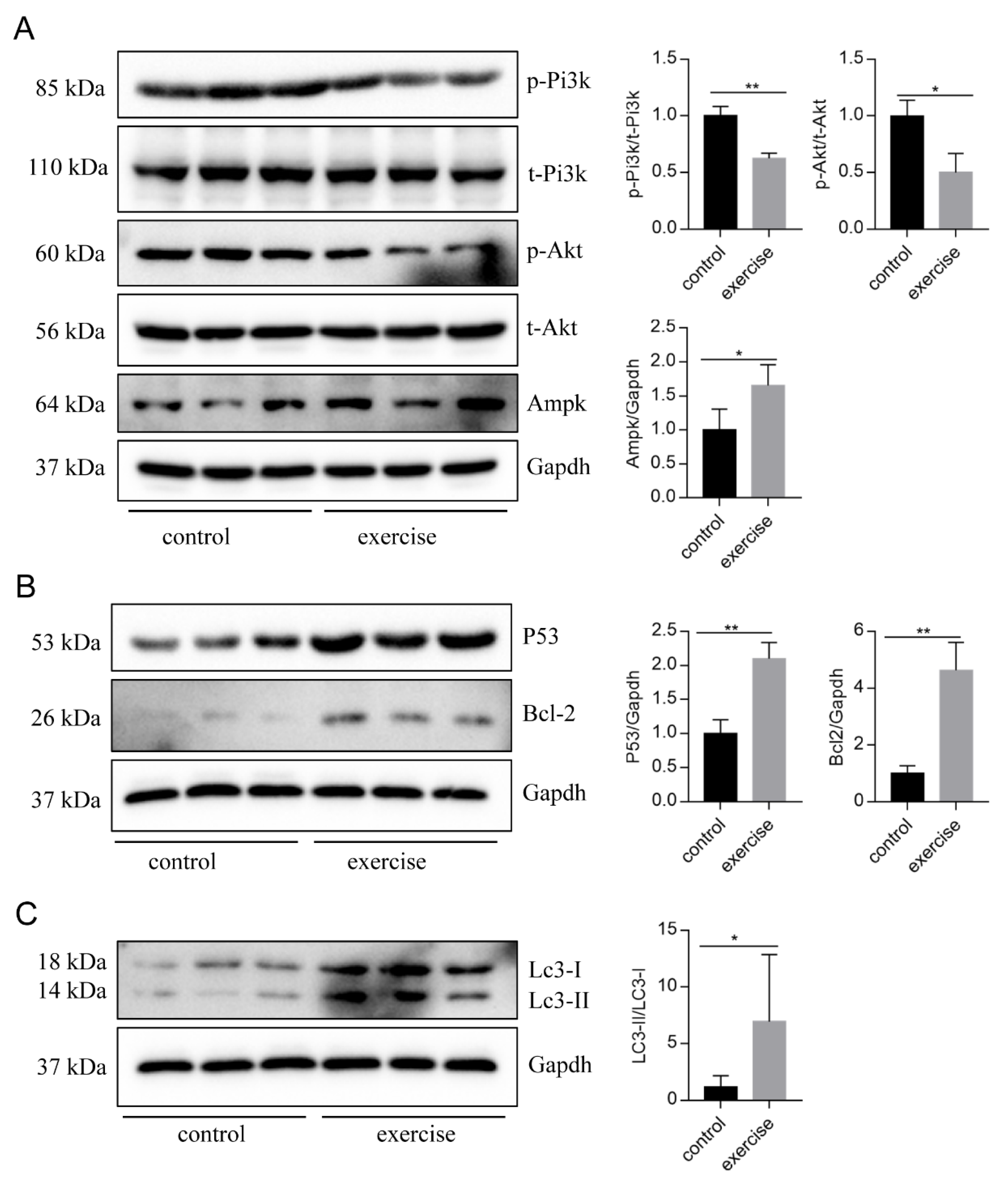
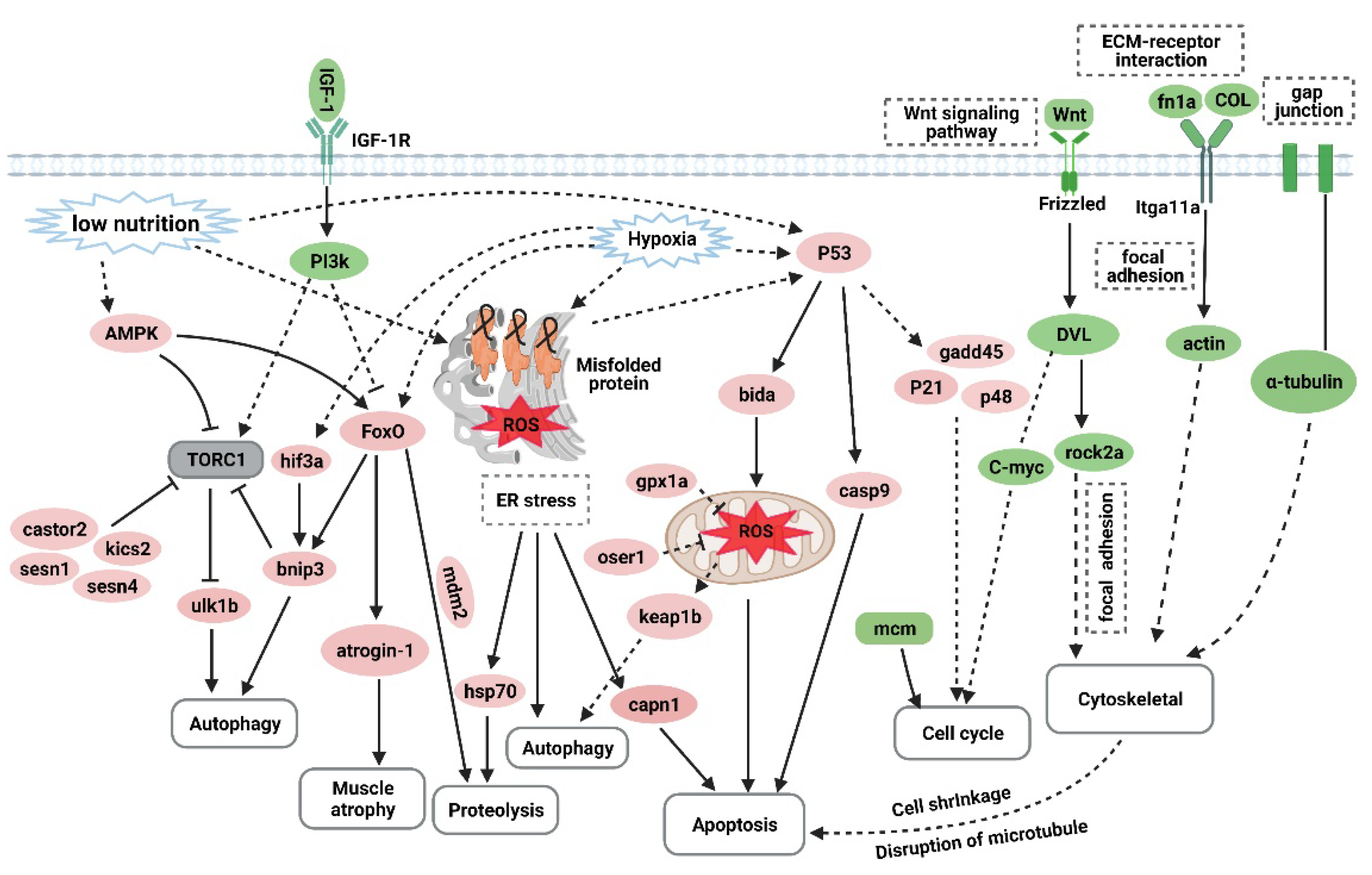
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deldicque, L. Protein intake and exercise-induced skeletal muscle hypertrophy: An Update. Nutrients 2020, 12, 2023. [Google Scholar] [CrossRef]
- Konopka, A.R.; Harber, M.P. Skeletal Muscle hypertrophy after aerobic exercise training. Exerc. Sport Sci. Rev. 2014, 42, 53–61. [Google Scholar] [CrossRef]
- Barnett, A. Using recovery modalities between training sessions in elite athletes. Sports Med. 2006, 36, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, V.C.; Roberts, L.; Markworth, J.F.; Barnett, M.P.G.; Coombes, J.S.; Raastad, T.; Peake, J.M.; Cameron-Smith, D. Impact of resistance exercise on ribosome biogenesis is acutely regulated by post-exercise recovery strategies. Physiol. Rep. 2016, 4, e12670. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Jude, B.; Lanner, J.T. Intramuscular mechanisms of overtraining. Redox Biol. 2020, 35, 101480. [Google Scholar] [CrossRef]
- Pereira, B.C.; da Rocha, A.L.; Pinto, A.P.; Pauli, J.R.; Moura, L.P.; Mekary, R.A.; De Freitas, E.C.; Da Silva, A.S.R. Excessive training impairs the insulin signal transduction in mice skeletal muscles. J. Endocrinol. 2016, 230, 93–104. [Google Scholar] [CrossRef] [Green Version]
- da Rocha, A.L.; Pereira, B.C.; Pauli, J.R.; de Souza, C.T.; Teixeira, G.; Lira, F.S.; Cintra, D.E.; Ropelle, E.; Júnior, C.R.; da Silva, A.S. Downhill running excessive training inhibits hypertrophy in mice skeletal muscles with different fiber type composition. J. Cell. Physiol. 2016, 231, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.C.; da Rocha, A.L.; Pinto, A.P.; Pauli, J.R.; de Souza, C.T.; Cintra, D.E.; Ropelle, E.; de Freitas, E.C.; Zagatto, A.; da Silva, A.S. Excessive eccentric exercise-induced overtraining model leads to endoplasmic reticulum stress in mice skeletal muscles. Life Sci. 2016, 145, 144–151. [Google Scholar] [CrossRef]
- da Rocha, A.L.; Pereira, B.C.; Teixeira, G.; Pinto, A.P.; Frantz, F.; Elias, L.; Lira, F.S.; Pauli, J.R.; Cintra, D.E.; Ropelle, E.R.; et al. Treadmill Slope Modulates Inflammation, Fiber Type Composition, Androgen, and Glucocorticoid Receptors in the Skeletal Muscle of Overtrained Mice. Front. Immunol. 2017, 8, 1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennison, E.; Sayer, A.A.; Cooper, C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Lei, S.; Jiang, T.; She, Y.; Shi, H. Regulation of Skeletal Muscle Atrophy in Cachexia by MicroRNAs and Long Non-coding RNAs. Front. Cell Dev. Biol. 2020, 8, 577010. [Google Scholar] [CrossRef]
- Prado, C.M.; Lieffers, J.R.; McCargar, L.J.; Reiman, T.; Sawyer, M.B.; Martin, L.; Baracos, V.E. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: A population-based study. Lancet Oncol. 2008, 9, 629–635. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Yang, X.; Xue, P.; Chen, H.; Yuan, M.; Kang, Y.; Duscher, D.; Machens, H.-G.; Chen, Z. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR-142a-5p/MFN1 axis. Theranostics 2020, 10, 1415–1432. [Google Scholar] [CrossRef] [PubMed]
- Perry, B.D.; Caldow, M.; Brennan-Speranza, T.C.; Sbaraglia, M.; Jerums, G.; Garnham, A.; Wong, C.; Levinger, P.; Haq, M.A.U.; Hare, D.L.; et al. Muscle atrophy in patients with Type 2 Diabetes Mellitus: Roles of inflammatory pathways, physical activity and exercise. Exerc. Immunol. Rev. 2016, 22, 94–109. [Google Scholar] [PubMed]
- Chen, H.; Qian, Z.; Zhang, S.; Tang, J.; Fang, L.; Jiang, F.; Ge, D.; Chang, J.; Cao, J.; Yang, L.; et al. Silencing COX-2 blocks PDK1/TRAF4-induced AKT activation to inhibit fibrogenesis during skeletal muscle atrophy. Redox Biol. 2021, 38, 101774. [Google Scholar] [CrossRef]
- Goldbraikh, D.; Neufeld, D.; Eid-Mutlak, Y.; Lasry, I.; Gilda, J.E.; Parnis, A.; Cohen, S. USP 1 deubiquitinates Akt to inhibit PI 3K-Akt-FoxO signaling in muscle during prolonged starvation. EMBO Rep. 2020, 21, e48791. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Bohnert, K.R.; McMillan, J.D.; Kumar, A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J. Cell. Physiol. 2018, 233, 67–78. [Google Scholar] [CrossRef]
- Nath, S.R.; Lieberman, M.L.; Yu, Z.; Marchioretti, C.; Jones, S.T.; Danby, E.C.E.; Van Pelt, K.M.; Sorarù, G.; Robins, D.M.; Bates, G.P.; et al. MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease. Acta Neuropathol. 2020, 140, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Hao, M.; Li, B.; Chen, M.; Chen, J.; Tang, J.; Hong, S.; Min, J.; Hu, M.; Hong, L. CACNA1H downregulation induces skeletal muscle atrophy involving endoplasmic reticulum stress activation and autophagy flux blockade. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Daya, A.; Donaka, R.; Karasik, D. Zebrafish models of sarcopenia. Dis. Model. Mech. 2020, 13, dmm042689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.; Kawahara, G.; Gundry, S.R.; Chen, A.T.; Lencer, W.I.; Zhou, Y.; Zon, L.I.; Kunkel, L.M.; Beggs, A.H. The zebrafish dag1 mutant: A novel genetic model for dystroglycanopathies. Hum. Mol. Genet. 2011, 20, 1712–1725. [Google Scholar] [CrossRef] [Green Version]
- Bassett, D.; Currie, P.D. Identification of a zebrafish model of muscular dystrophy. Clin. Exp. Pharmacol. Physiol. 2004, 31, 537–540. [Google Scholar] [CrossRef]
- Bassett, D.I.; Bryson-Richardson, R.; Daggett, D.F.; Gautier, P.; Keenan, D.G.; Currie, P. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003, 130, 5851–5860. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zheng, L.; Tang, C.; Chen, Z.; Zhu, R.; Peng, X.; Wu, X.; Zhu, P. Identification of Potentially Relevant Genes for Excessive Exercise-Induced Pathological Cardiac Hypertrophy in Zebrafish. Front. Physiol. 2020, 11, 565307. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Sun, C.-C.; Duan, J.-X.; Yang, H.-H.; Zhang, C.-Y.; Xiong, J.-B.; Zhong, W.-J.; Zu, C.; Guan, X.-X.; Jiang, H.-L.; et al. A COX-2/sEH dual inhibitor PTUPB ameliorates cecal ligation and puncture-induced sepsis in mice via anti-inflammation and anti-oxidative stress. Biomed. Pharmacother. 2020, 126, 109907. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, M.; Alfò, M.; Martella, F.; Pappalardo, E.; Musarò, A. Finite mixture clustering of human tissues with different levels of IGF-1 splice variants mRNA transcripts. BMC Bioinform. 2015, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.; Lee, K.Y.; Klaus, K.; Softic, S.; Krumpoch, M.T.; Fentz, J.; Stanford, K.I.; Robinson, M.M.; Cai, W.; Kleinridders, A.; et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J. Clin. Investig. 2016, 126, 3433–3446. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.S.; Ahmad, K.; Lee, E.J.; Lee, Y.-H.; Choi, I. Implications of Insulin-Like Growth Factor-1 in Skeletal Muscle and Various Diseases. Cells 2020, 9, 1773. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Moulin, M.; Ferreiro, A. Muscle redox disturbances and oxidative stress as pathomechanisms and therapeutic targets in early-onset myopathies. Semin. Cell Dev. Biol. 2017, 64, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Kourakis, S.; Timpani, C.A.; de Haan, J.B.; Gueven, N.; Fischer, D.; Rybalka, E. Targeting Nrf2 for the treatment of Duchenne Muscular Dystrophy. Redox Biol. 2021, 38, 101803. [Google Scholar] [CrossRef]
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Afroze, D.; Kumar, A. ER stress in skeletal muscle remodeling and myopathies. FEBS J. 2019, 286, 379–398. [Google Scholar] [CrossRef] [Green Version]
- Gallot, Y.; Bohnert, K. Confounding Roles of ER Stress and the Unfolded Protein Response in Skeletal Muscle Atrophy. Int. J. Mol. Sci. 2021, 22, 2567. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Khair, M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Ebert, S.M.; Dierdorff, J.M.; Meyerholz, D.K.; Bullard, S.A.; Al-Zougbi, A.; Delau, A.D.; Tomcheck, K.C.; Skopec, Z.P.; Marcotte, G.R.; Bodine, S.C.; et al. An investigation of p53 in skeletal muscle aging. J. Appl. Physiol. 2019, 127, 1075–1084. [Google Scholar] [CrossRef]
- Dai, J.; Xiang, Y.; Fu, D.; Xu, L.; Jiang, J.; Xu, J. Ficus carica L. Attenuates denervated skeletal muscle atrophy via PPARα/NF-κB Pathway. Front. Physiol. 2020, 11, 580223. [Google Scholar] [CrossRef]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging Mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zhang, Y.; Gu, L.; Lan, M.; Liu, C.; Wang, M.; Su, Y.; Ge, M.; Wang, T.; Yu, Y.; et al. Islr regulates canonical Wnt signaling-mediated skeletal muscle regeneration by stabilizing Dishevelled-2 and preventing autophagy. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ratnayake, D.; Nguyen, P.D.; Rossello, F.J.; Wimmer, V.C.; Tan, J.L.; Galvis, L.A.; Julier, Z.; Wood, A.J.; Boudier, T.; Isiaku, A.I.; et al. Macrophages provide a transient muscle stem cell niche via NAMPT secretion. Nat. Cell Biol. 2021, 591, 281–287. [Google Scholar] [CrossRef]
- Purslow, P.P. The Structure and role of intramuscular connective tissue in muscle function. Front. Physiol. 2020, 11, 495. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Reider, K.M.; D’Amore, A.; Beezhold, K.; Rothrauff, B.; Cavalli, L.; Wagner, W.; Vorp, D.A.; Tsamis, A.; Shinde, S.; Zhang, C.; et al. Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell 2017, 16, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lephart, E.D.; Naftolin, F. Menopause and the Skin: Old favorites and new innovations in cosmeceuticals for estrogen-deficient skin. Dermatol. Ther. 2021, 11, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, J.T.; Höke, A. Cellular and molecular features of neurogenic skeletal muscle atrophy. Exp. Neurol. 2020, 331, 113379. [Google Scholar] [CrossRef] [PubMed]
- Chédotal, A. Roles of axon guidance molecules in neuronal wiring in the developing spinal cord. Nat. Rev. Neurosci. 2019, 20, 380–396. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hub Gene | Description | |
|---|---|---|
| UP | keap1b | kelch-like ECH-associated protein 1b |
| per3 | period circadian clock 3 | |
| ulk1b | unc-51-like kinase 1b | |
| socs2 | suppressor of cytokine signaling 2 | |
| esrp1 | epithelial splicing regulatory protein 1 | |
| bcl2l1 | bcl2-like 1 | |
| hsp70 | heat shock cognate 70-kd protein | |
| igf2r | insulin-like growth factor 2 receptor | |
| mdm2 | MDM2 oncogene, E3 ubiquitin protein ligase | |
| rab18a | RAB18A, member RAS oncogene family | |
| DOWN | col1a1a | collagen, typeI, alpha1a |
| fn1a | fibronectin 1a | |
| ppih | peptidylprolyl isomerase h | |
| tpx2 | tpx2, microtubule-associated, homolog | |
| uba5 | ubiquitin-like modifier activating enzyme 5 | |
| nhlrc2 | nhl repeat containing 2 | |
| mcm4 | minichromosome maintenance complex component 4 | |
| tac1 | tachykinin 1 | |
| b3gat3 | beta-1,3-glucuronyltransferase 3 | |
| ddost | dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit |
| ID | Term | p-Value | |
|---|---|---|---|
| Up | GO:0016567 | protein ubiquitination | 8.60 × 10−14 |
| GO:0032922 | circadian regulation of gene expression | 9.17 × 10−12 | |
| GO:0009628 | response to abiotic stimulus | 9.19 × 10−8 | |
| GO:0042542 | response to hydrogen peroxide | 2.22 × 10−6 | |
| GO:0034101 | erythrocyte homeostasis | 1.089 × 10−5 | |
| GO:0046031 | ADP metabolic process | 1.34 × 10−5 | |
| GO:0009890 | negative regulation of biosynthetic process | 1.76 × 10−5 | |
| GO:0035162 | embryonic hemopoiesis | 3.25 × 10−5 | |
| GO:0043161 | proteasomal pathway | 3.61 × 10−5 | |
| GO:0006914 | autophagy | 4.51 × 10−5 | |
| Down | GO:0030198 | extracellular matrix organization | 2.00 × 10−7 |
| GO:0001501 | skeletal system development | 4.01 × 10−5 | |
| GO:0007411 | axon guidance | 0.00045 | |
| GO:0048385 | regulation of retinoic acid receptor signaling pathway | 0.00051 | |
| GO:0035567 | noncanonical Wnt signaling pathway | 0.00054 | |
| GO:0009101 | glycoprotein biosynthetic process | 0.0013 | |
| GO:1901137 | carbohydrate derivative biosynthetic process | 0.0014 | |
| GO:0042573 | retinoic acid metabolic process | 0.0015 | |
| GO:0010001 | glial cell differentiation | 0.0016 | |
| GO:0034754 | cellular hormone metabolic process | 0.0020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.-C.; Zhou, Z.-Q.; Chen, Z.-L.; Zhu, R.-K.; Yang, D.; Peng, X.-Y.; Zheng, L.; Tang, C.-F. Identification of Potentially Related Genes and Mechanisms Involved in Skeletal Muscle Atrophy Induced by Excessive Exercise in Zebrafish. Biology 2021, 10, 761. https://doi.org/10.3390/biology10080761
Sun C-C, Zhou Z-Q, Chen Z-L, Zhu R-K, Yang D, Peng X-Y, Zheng L, Tang C-F. Identification of Potentially Related Genes and Mechanisms Involved in Skeletal Muscle Atrophy Induced by Excessive Exercise in Zebrafish. Biology. 2021; 10(8):761. https://doi.org/10.3390/biology10080761
Chicago/Turabian StyleSun, Chen-Chen, Zuo-Qiong Zhou, Zhang-Lin Chen, Run-Kang Zhu, Dong Yang, Xi-Yang Peng, Lan Zheng, and Chang-Fa Tang. 2021. "Identification of Potentially Related Genes and Mechanisms Involved in Skeletal Muscle Atrophy Induced by Excessive Exercise in Zebrafish" Biology 10, no. 8: 761. https://doi.org/10.3390/biology10080761
APA StyleSun, C.-C., Zhou, Z.-Q., Chen, Z.-L., Zhu, R.-K., Yang, D., Peng, X.-Y., Zheng, L., & Tang, C.-F. (2021). Identification of Potentially Related Genes and Mechanisms Involved in Skeletal Muscle Atrophy Induced by Excessive Exercise in Zebrafish. Biology, 10(8), 761. https://doi.org/10.3390/biology10080761