
Survival-Based Biomarker Module Identification Associated with Oral Squamous Cell Carcinoma (OSCC)

 , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA RNA-Seq Data Extraction and Differential Expression Analysis
2.2. PPI Network and Its Weighted Form
2.3. Module Detection in Weighted PPI Network
2.4. Survival Analysis of Modules
2.5. OSCC-Specific Hub Gene(s) Detection
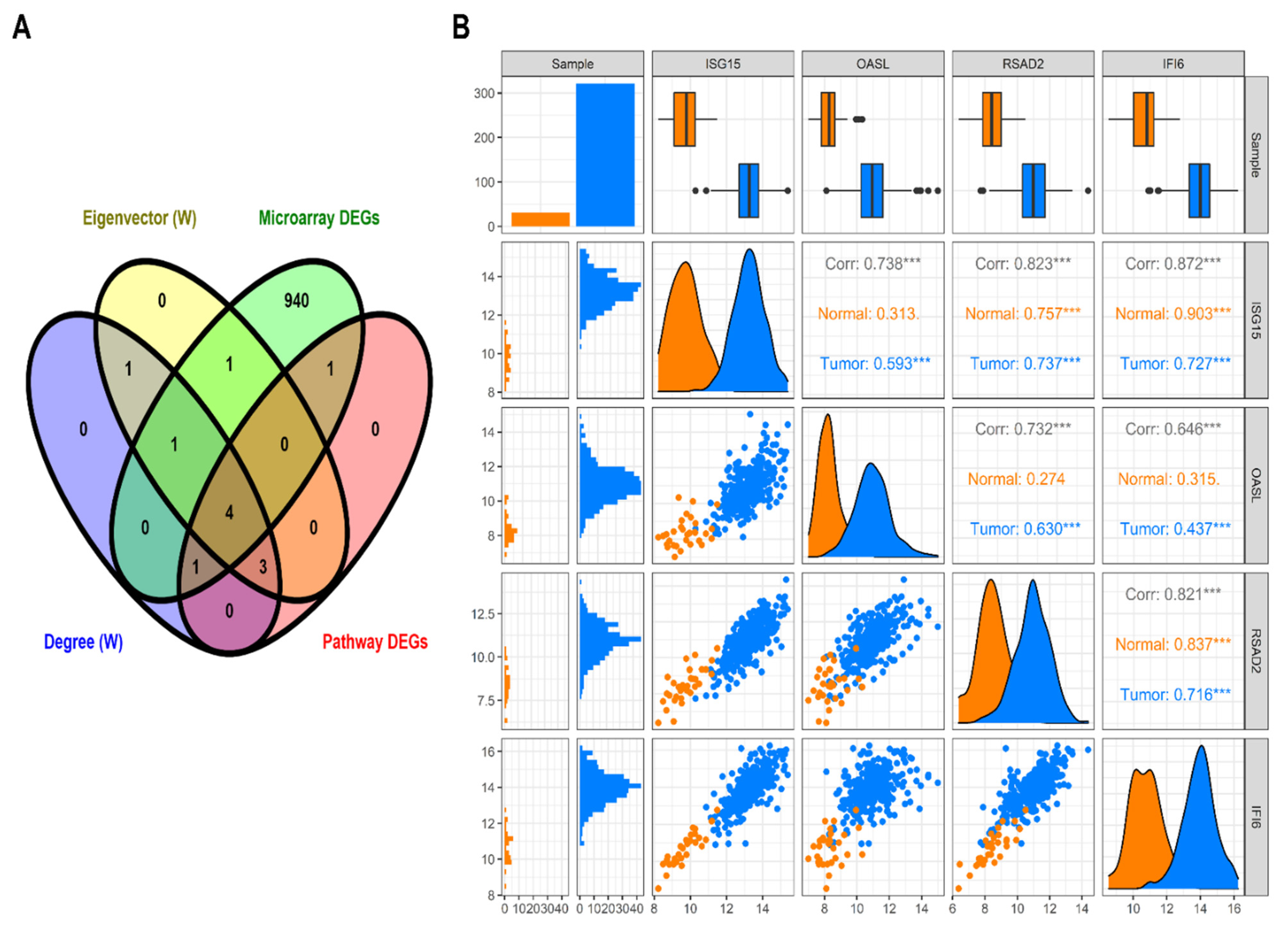
3. Results
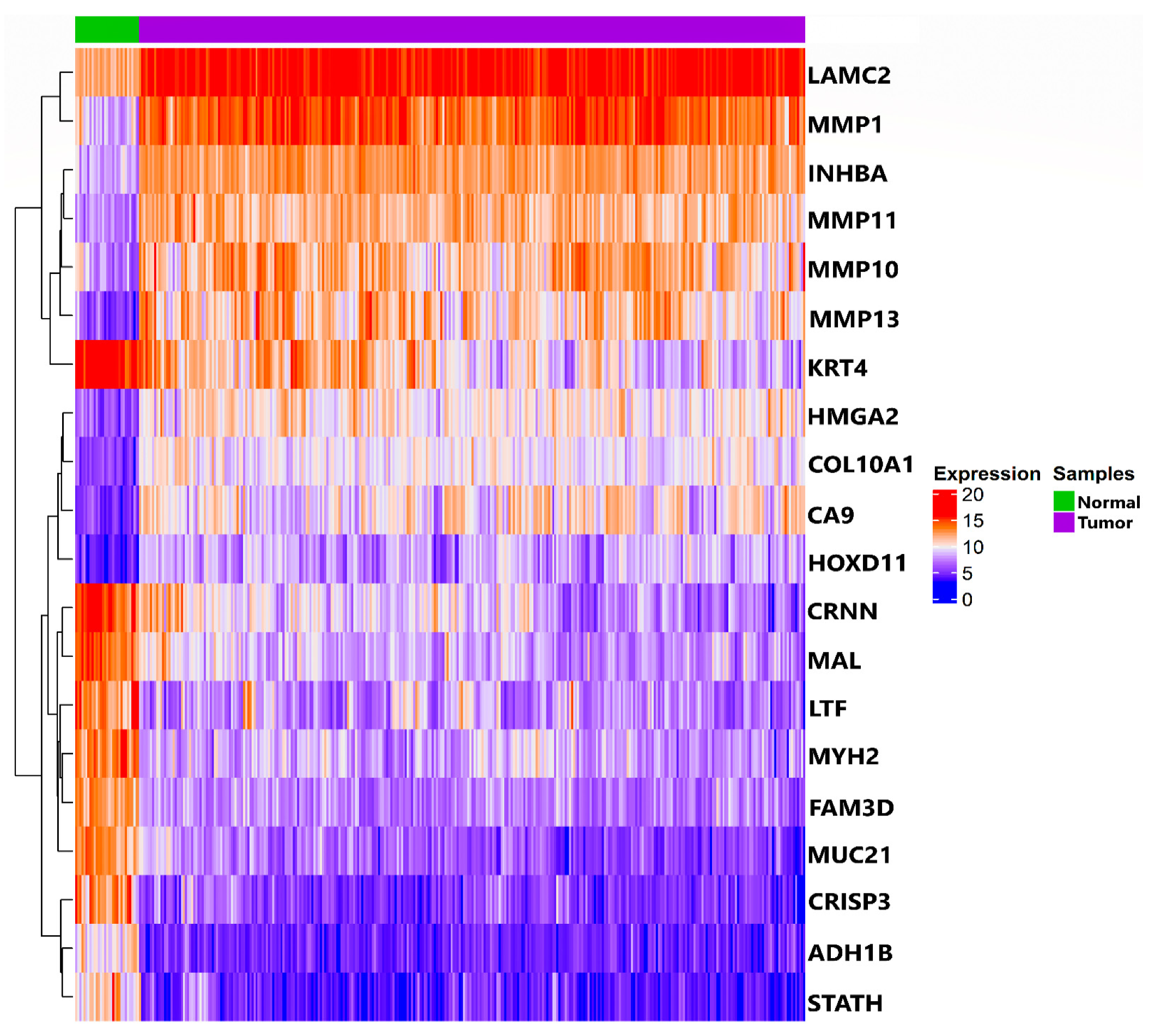
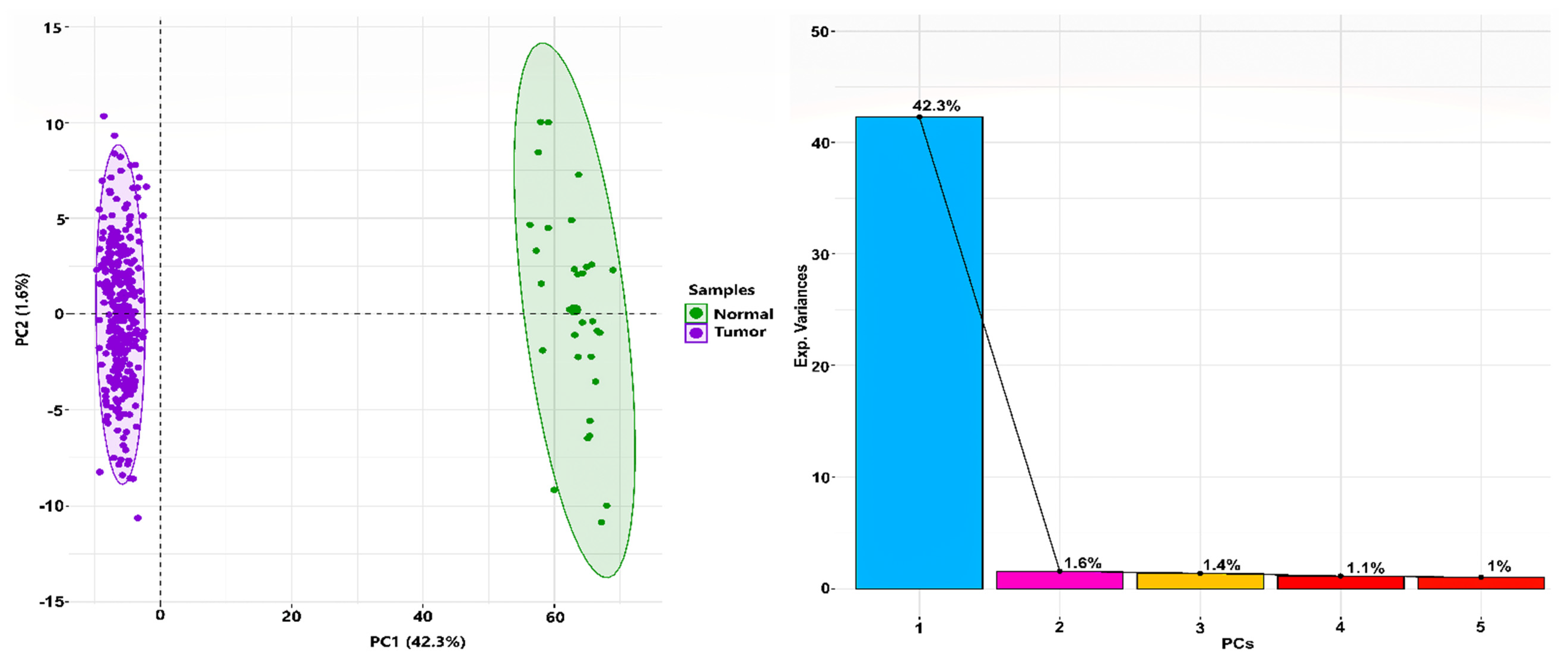
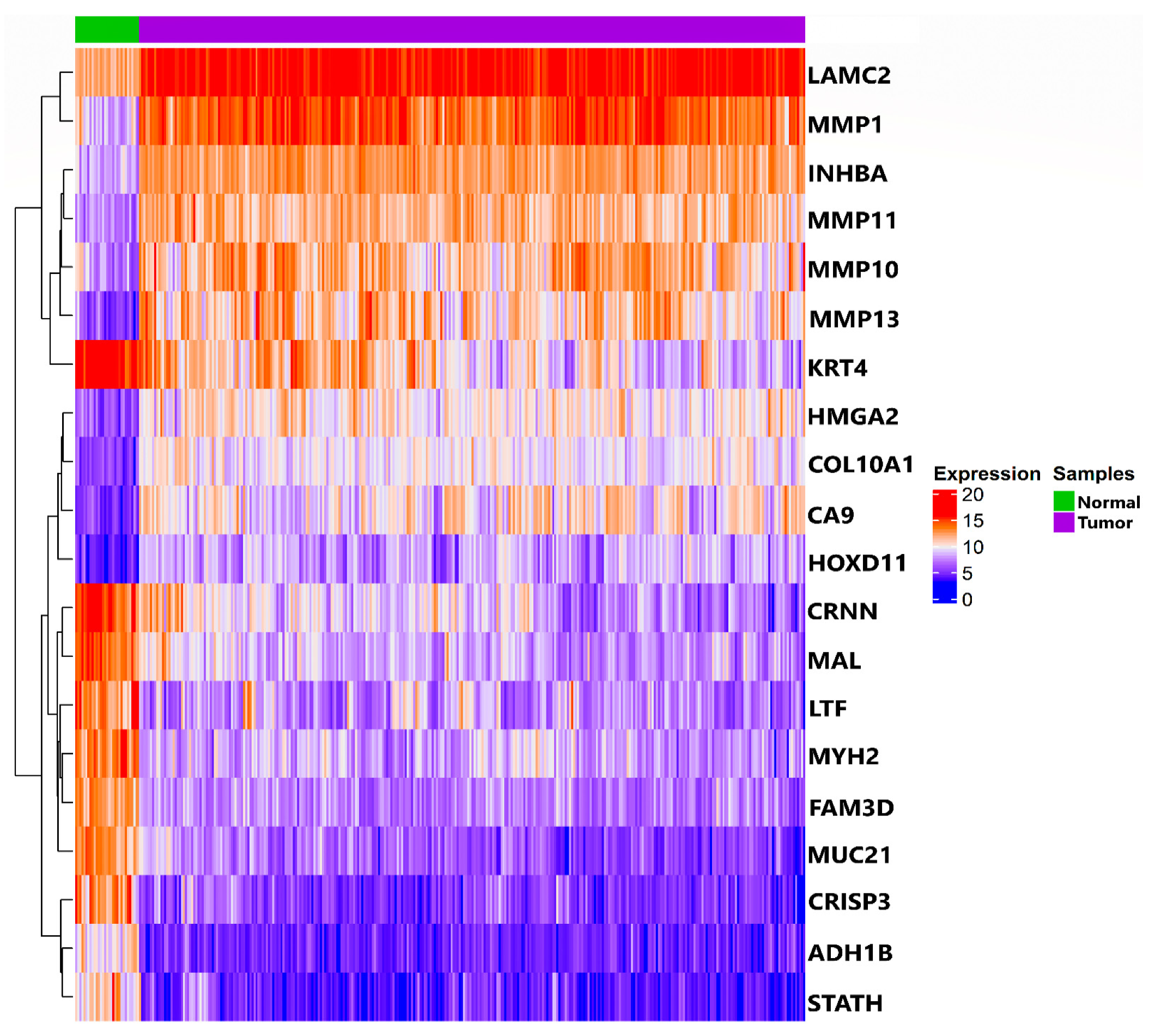
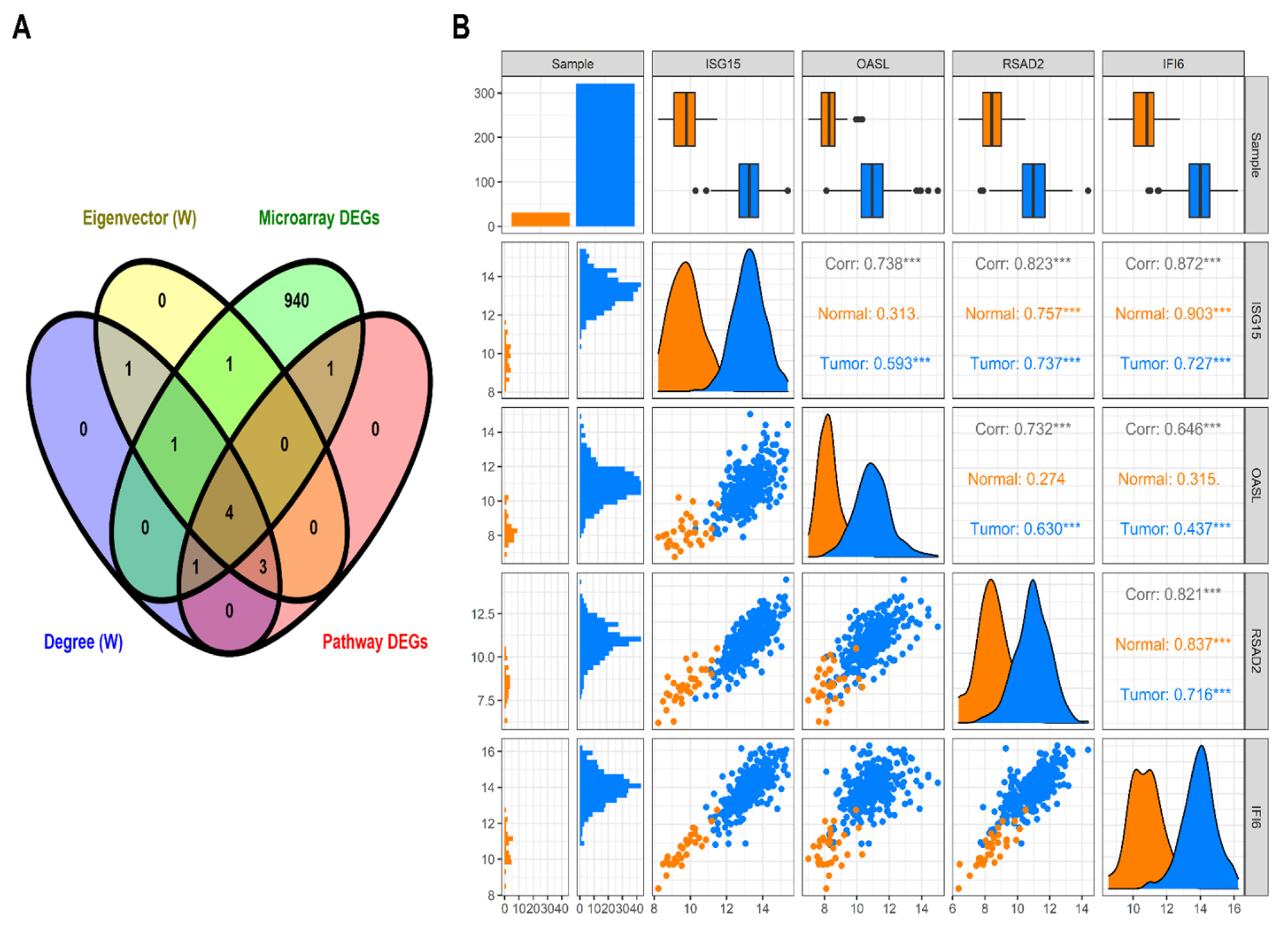
3.1. TCGA RNA-Seq Processing and Differential Expression Analysis
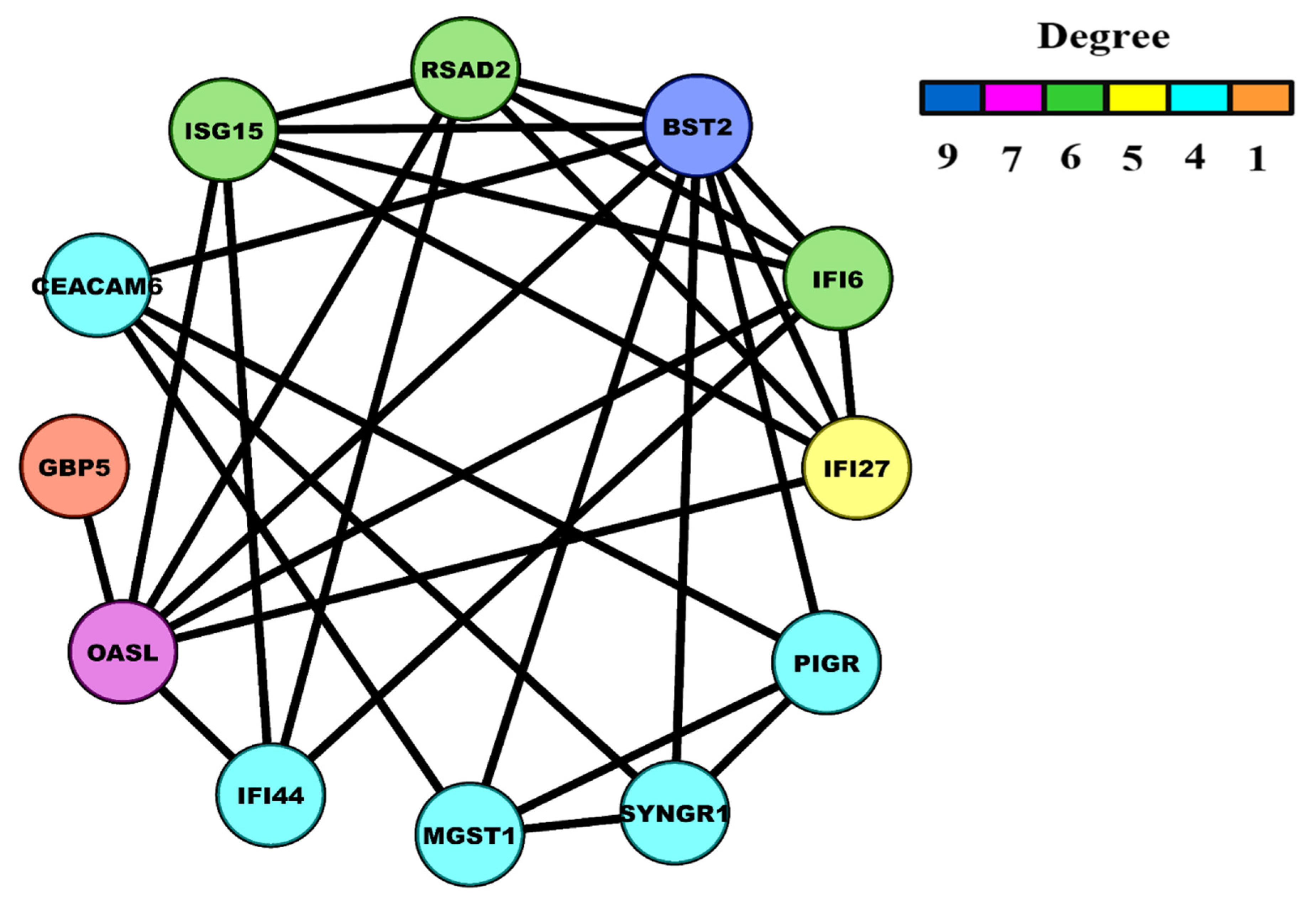
3.2. Construction of Weighted PPI Network and Module(s) Detection
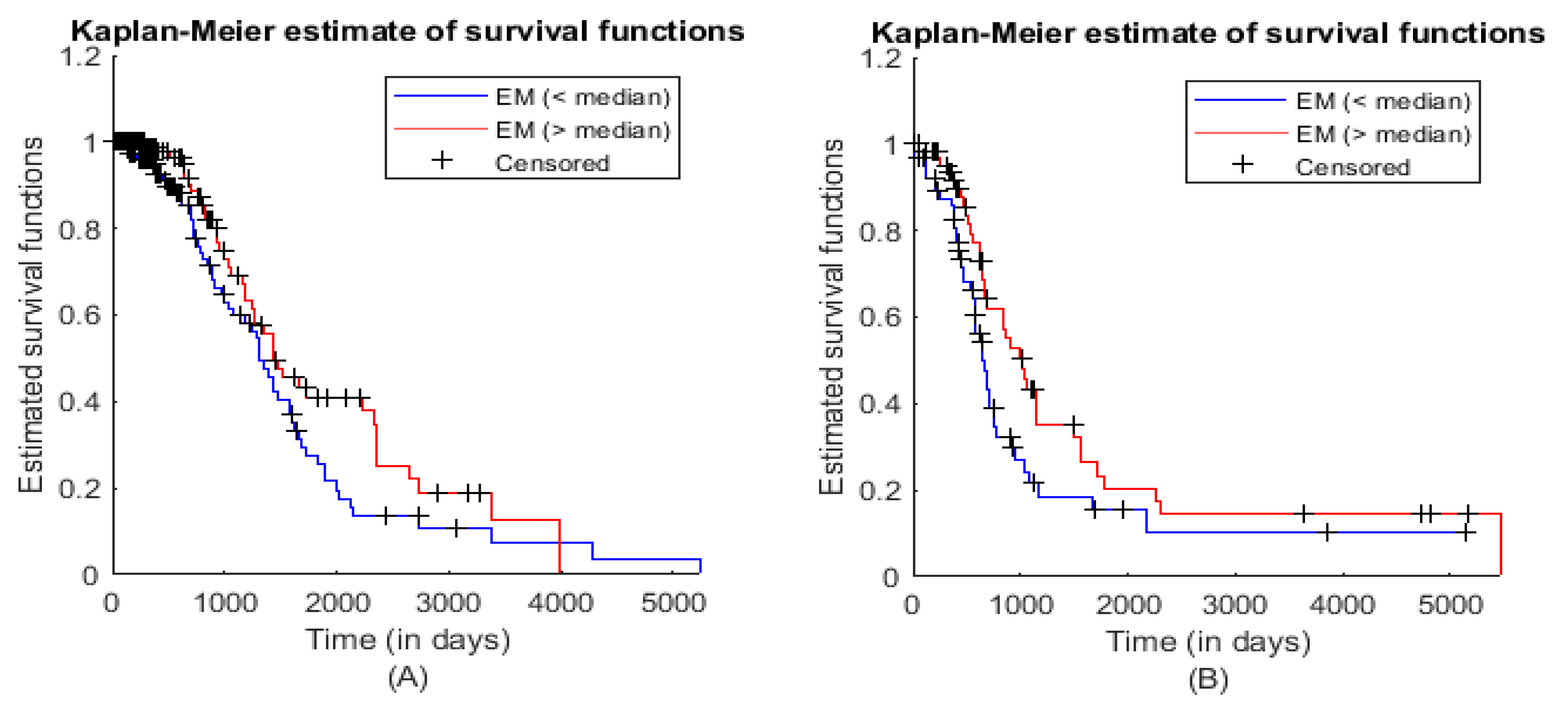
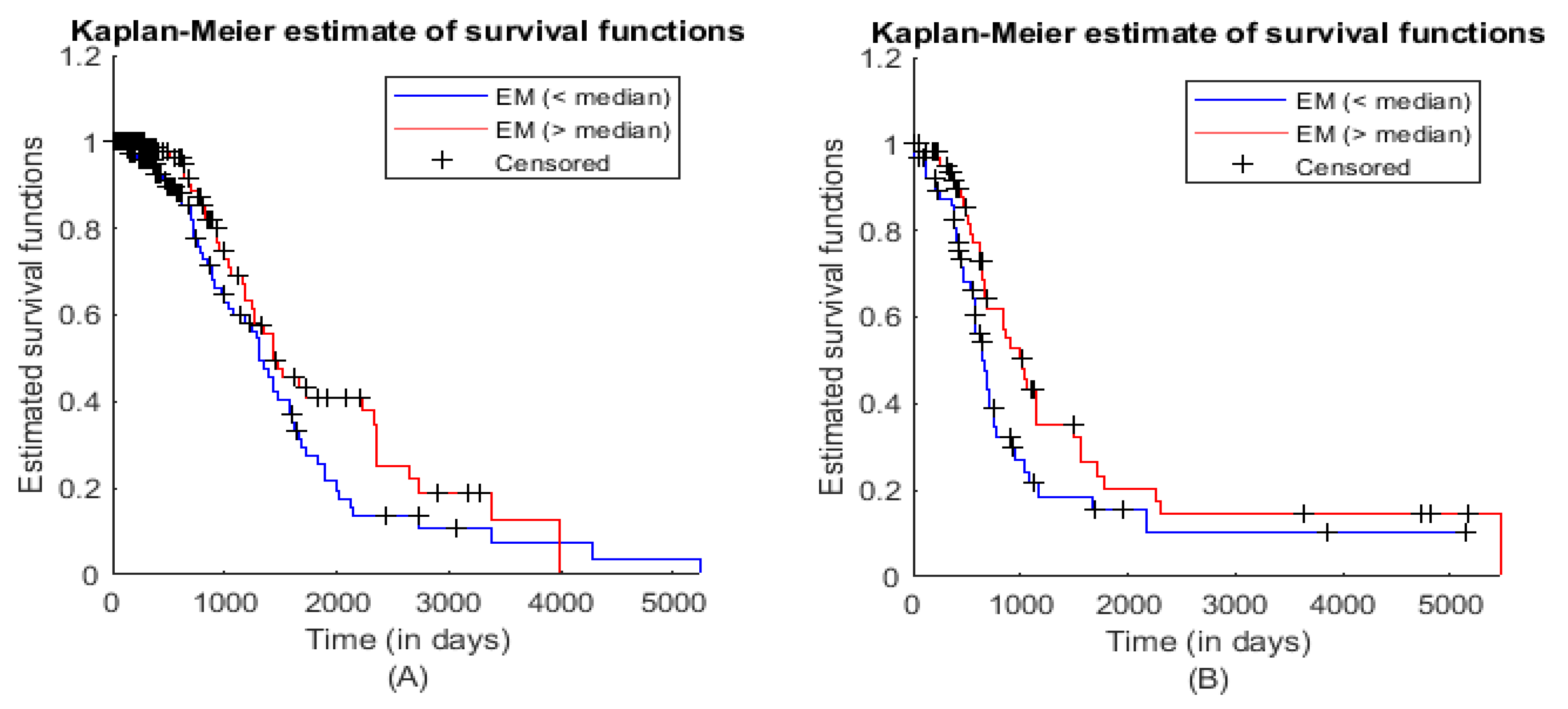
3.3. Module Survival Analysis
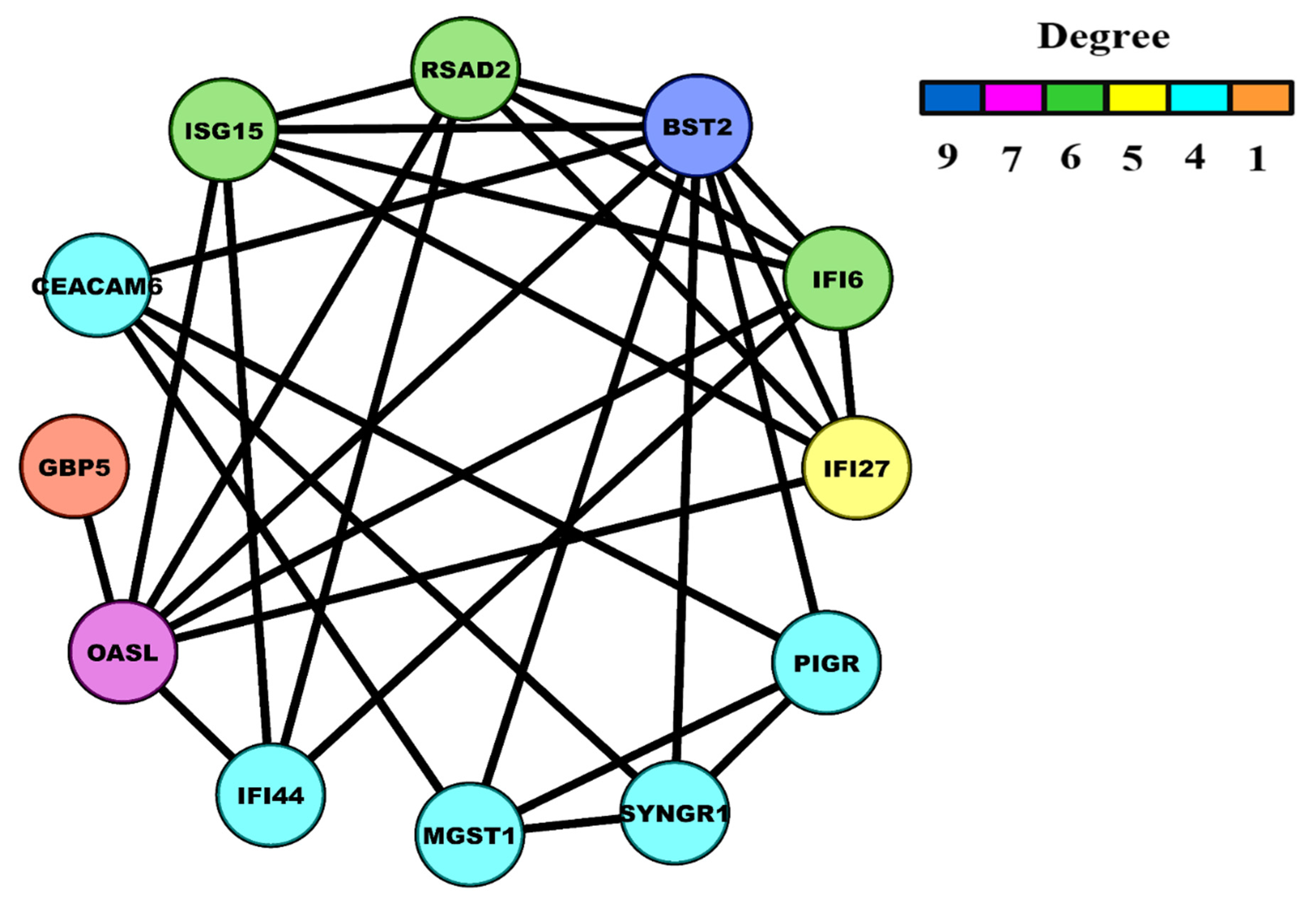
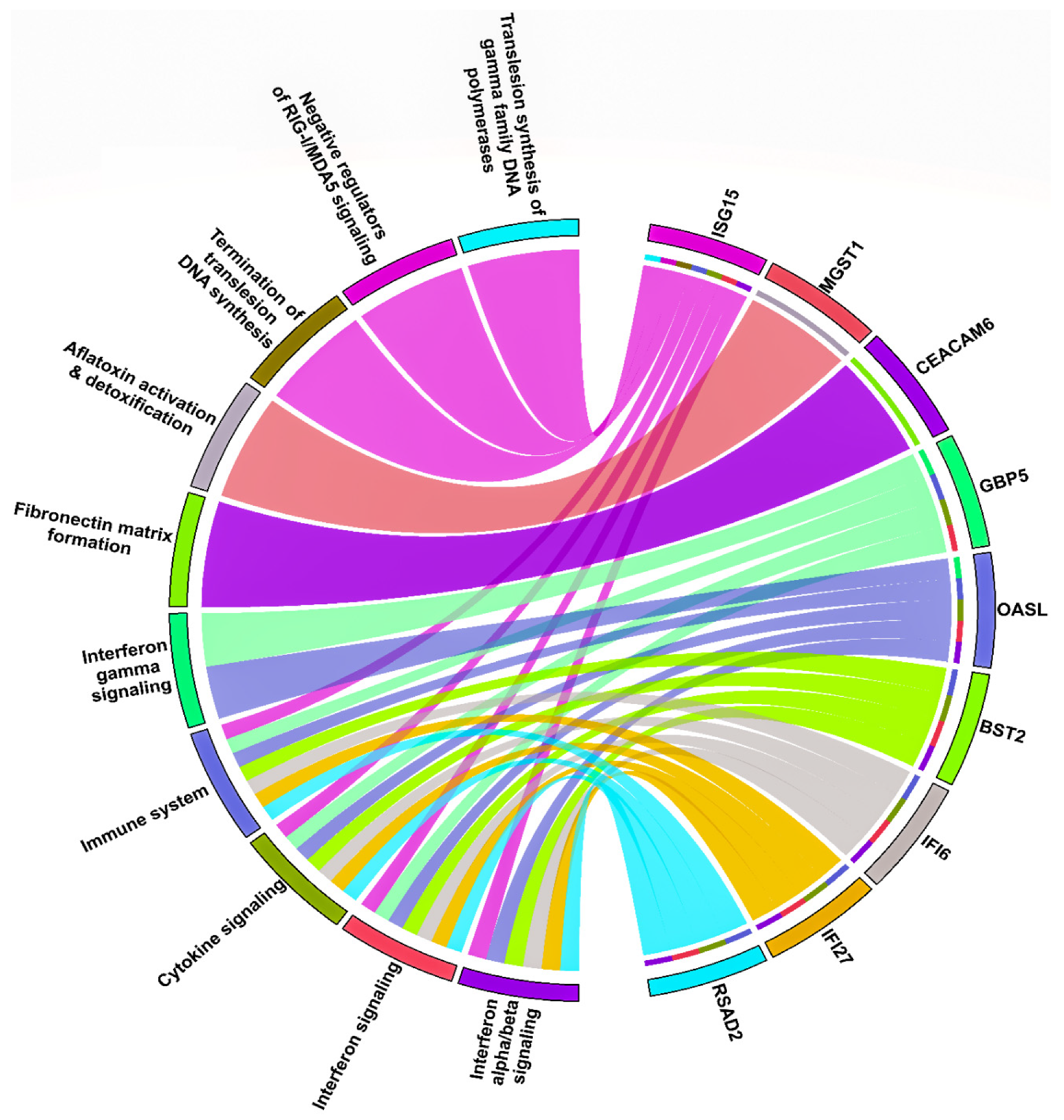
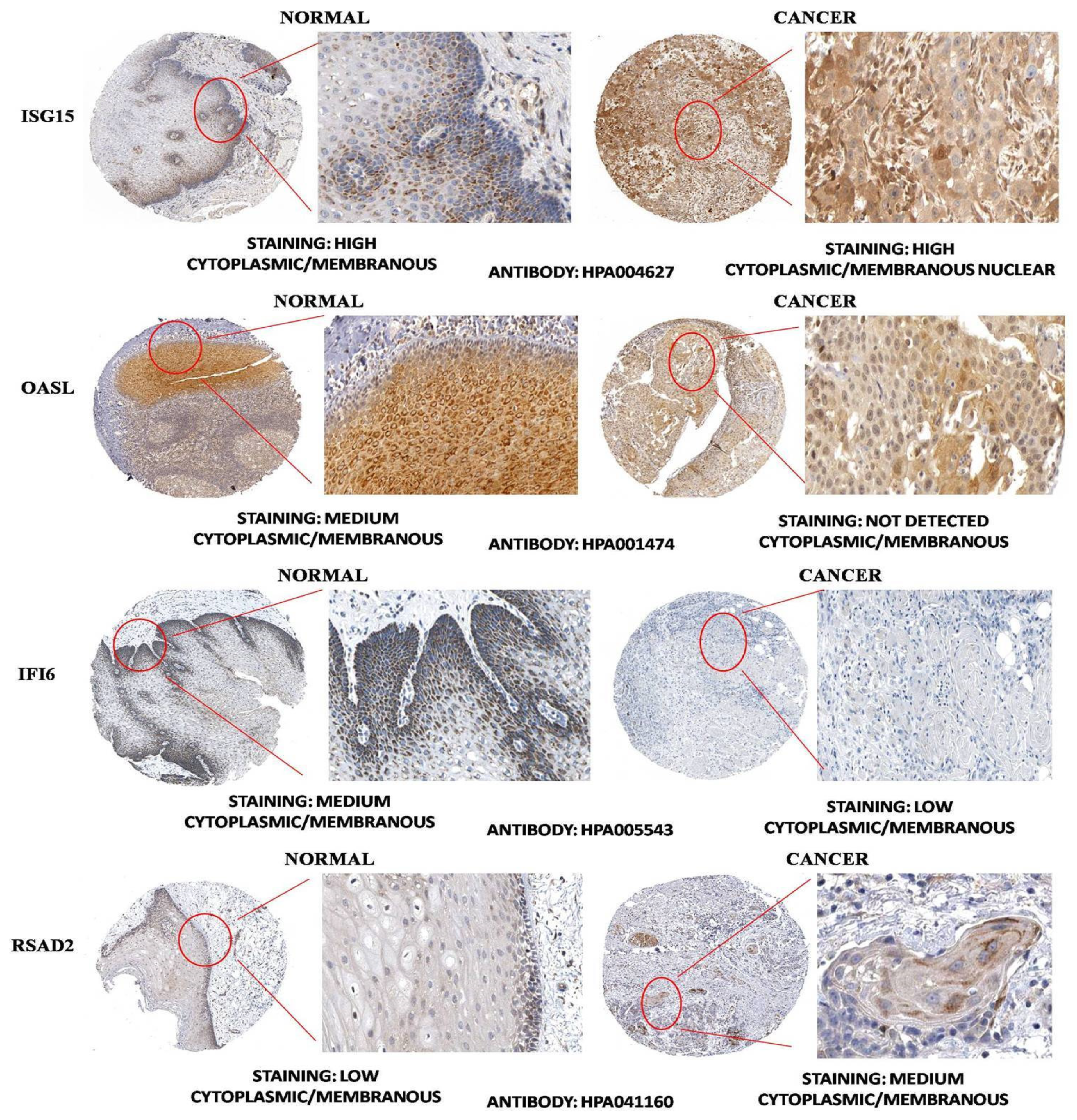
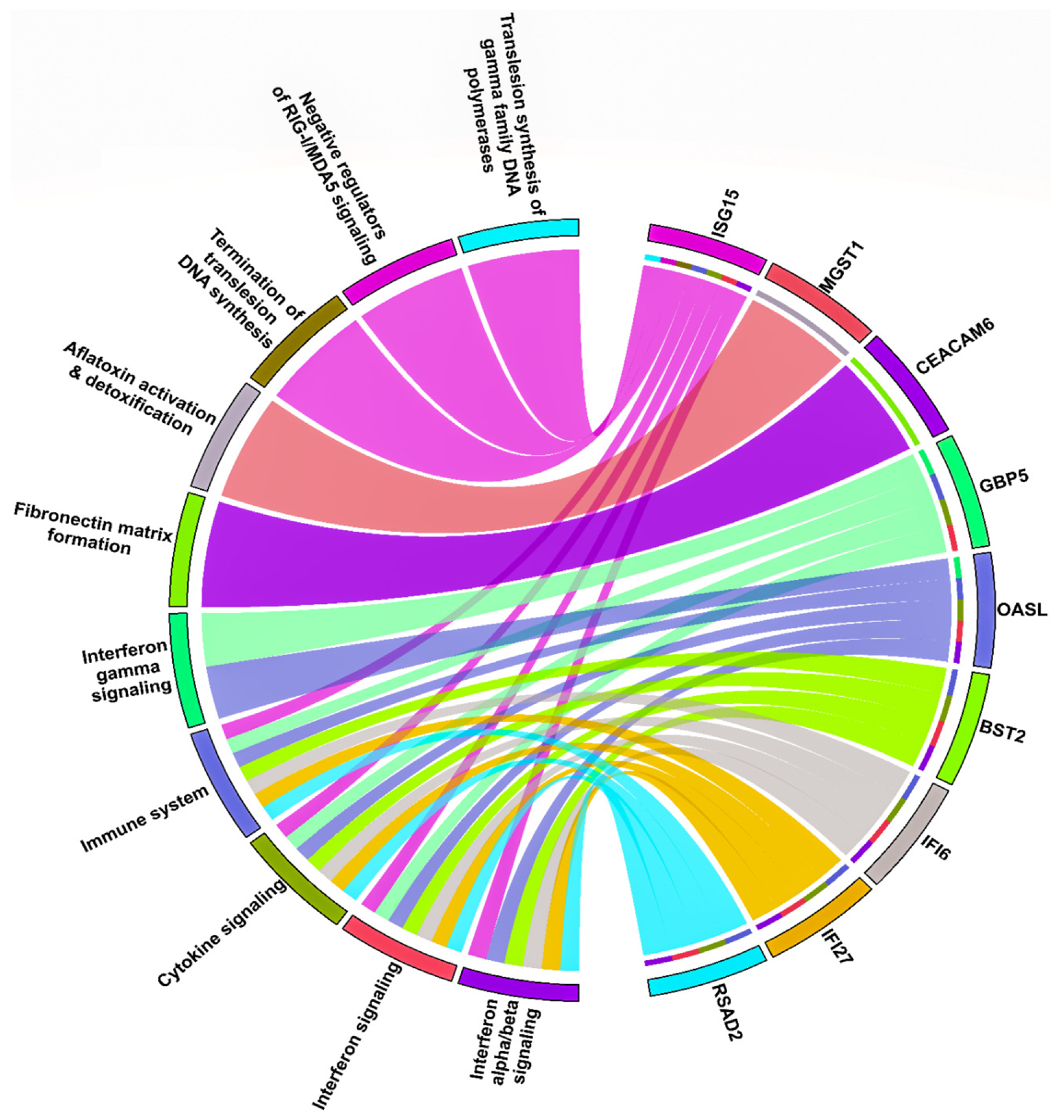
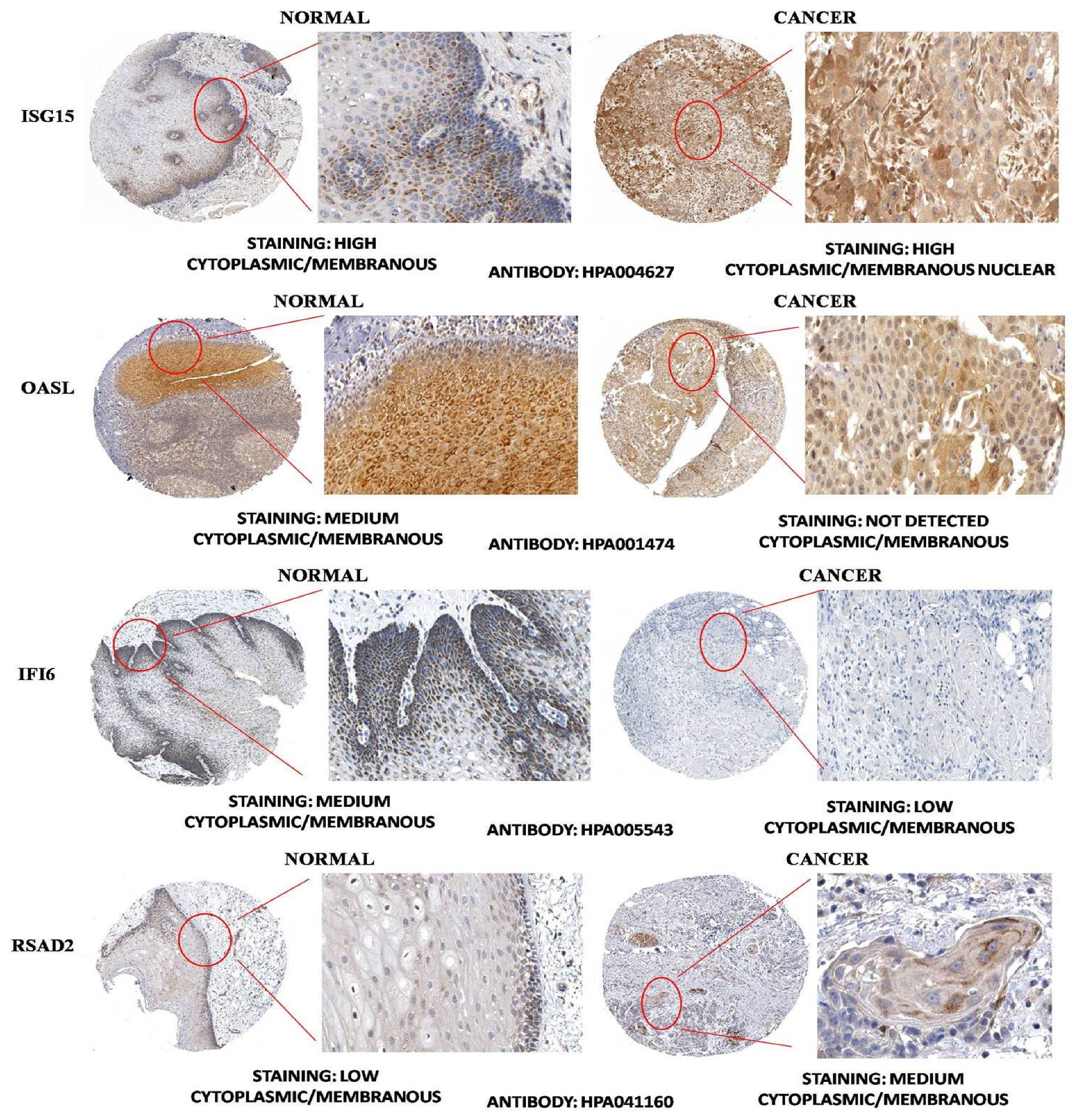
3.4. HNSC-Specific Hub Gene(s) Detection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stein, A.P.; Saha, S.; Kraninger, J.L.; Swick, A.D.; Yu, M.; Lambert, P.F.; Kimple, R.J. Prevalence of Human Papillomavirus in Oropharyngeal Cancer: A Systematic Review. Cancer J. 2015, 21, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Snijders, P.J.; Brakenhoff, R.H. The Molecular Landscape of Head and Neck Cancer. Nat. Rev. Cancer 2018, 18, 269. [Google Scholar] [CrossRef] [PubMed]
- Du, E.; Mazul, A.L.; Farquhar, D.; Brennan, P.; Anantharaman, D.; Abedi-Ardekani, B.; Weissler, M.C.; Hayes, D.N.; Olshan, A.F.; Zevallos, J.P. Long-Term Survival in Head and Neck Cancer: Impact of Site, Stage, Smoking, and Human Papillomavirus Status. Laryngoscope 2019, 129, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Dissanayaka, W.L.; Pitiyage, G.; Kumarasiri, P.V.R.; Liyanage, R.L.P.R.; Dias, K.D.; Tilakaratne, W.M. Clinical and Histopathologic Parameters in Survival of Oral Squamous Cell Carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 113, 518–525. [Google Scholar] [CrossRef]
- Pannone, G.; Santoro, A.; Papagerakis, S.; Muzio, L.L.; De Rosa, G.; Bufo, P. The Role of Human Papillomavirus in the Pathogenesis of Head & Neck Squamous Cell Carcinoma: An Overview. Infect. Agents Cancer 2011, 6, 1–11. [Google Scholar]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.-X.; Zhang, J.; Wang, J.; et al. Exome Sequencing of Head and Neck Squamous Cell Carcinoma Reveals Inactivating Mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Gaykalova, D.A.; Mambo, E.; Choudhary, A.; Houghton, J.; Buddavarapu, K.; Sanford, T.; Darden, W.; Adai, A.; Hadd, A.; Latham, G.; et al. Novel Insight into Mutational Landscape of Head and Neck Squamous Cell Carcinoma. PLoS ONE 2014, 9, e93102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The Clonal and Mutational Evolution Spectrum of Primary Triple-Negative Breast Cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-Spondin Fusions in Colon Cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Johnson, M.E.; Cantalupo, P.G.; Pipas, J.M. Identification of Head and Neck Cancer Subtypes Based on Human Papillomavirus Presence and E2F-Regulated Gene Expression. mSphere 2018, 3, e00580-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lan, Q.; Lin, J. Identification of Key Gene Modules for Human Osteosarcoma by Co-Expression Analysis. World J. Surg. Oncol. 2018, 16, 89. [Google Scholar] [CrossRef] [Green Version]
- Shriwash, N.; Singh, P.; Arora, S.; Ali, S.M.; Ali, S.; Dohare, R. Identification of Differentially Expressed Genes in Small and Non-Small Cell Lung Cancer Based on Meta-Analysis of MRNA. Heliyon 2019, 5, e01707. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Rai, A.; Dohare, R.; Arora, S.; Ali, S.; Parveen, S.; Syed, M.A. Network-Based Identification of Signature Genes KLF6 and SPOCK1 Associated with Oral Submucous Fibrosis. Mol. Clin. Oncol. 2020, 12, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, S.; Singh, K.; Dohare, R. Functional Module Detection in Gene Regulatory Network Associated with Hepatocellular Carcinoma. In Performance Management of Integrated Systems and Its Applications in Software Engineering; Springer: Singapore, 2020; pp. 137–150. [Google Scholar]
- Liu, W.; Li, L.; Li, W. Gene Co-Expression Analysis Identifies Common Modules Related to Prognosis and Drug Resistance in Cancer Cell Lines: Gene Modules Related with Prognosis and Drug Resistance. Int. J. Cancer 2014, 135, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Meiliana, A.; Dewi, N.M.; Wijaya, A. The Immunobiology of Cancer: An Update Review. Indones. Biomed. 2017, 9, 53–72. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data Quality Aware Analysis of Differential Expression in RNA-Seq with NOISeq R/Bioc Package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package BiomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Singh, P.; Sharma, A.; Arora, S.; Shriwash, N.; Rahmani, A.H.; Almatroodi, S.A.; Manda, K.; Dohare, R.; Syed, M.A. Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis. Genes 2019, 10, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Wu, H.; Miao, Z.; Huang, Y.; Hu, Y.; Bi, X.; Wu, D.; Qian, K.; Wang, L.; Wang, C.; et al. Network-Based Survival-Associated Module Biomarker and Its Crosstalk with Cell Death Genes in Ovarian Cancer. Sci. Rep. 2015, 5, 11566. [Google Scholar] [CrossRef]
- Kumar, P.; Dohare, R. A Neighborhood Proximity Based Algorithm for Overlapping Community Structure Detection in Weighted Networks. Front. Comput. Sci. 2019, 13, 1353–1355. [Google Scholar] [CrossRef]
- Newman, M.E.J. The Structure and Function of Complex Networks. SIAM Rev. 2003, 45, 167–256. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Yin, R.-X.; Pan, S.-L.; Yang, S.; Yang, D.-Z.; Lin, W.-X. Weighted Gene Co-Expression Network Analysis Identifies Specific Modules and Hub Genes Related to Hyperlipidemia. Cell. Physiol. Biochem. 2018, 48, 1151–1163. [Google Scholar] [CrossRef]
- Ma, H.-W.; Buer, J.; Zeng, A.-P. Hierarchical Structure and Modules in the Escherichia Coli Transcriptional Regulatory Network Revealed by a New Top-down Approach. BMC Bioinform. 2004, 5, 199. [Google Scholar] [CrossRef] [Green Version]
- Alter, O.; Brown, P.O.; Botstein, D. Singular Value Decomposition for Genome-Wide Expression Data Processing and Modeling. Proc. Natl. Acad. Sci. USA 2000, 97, 10101–10106. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. Clifton NJ 2016, 1418, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.-X. CytoNCA: A Cytoscape Plugin for Centrality Analysis and Evaluation of Protein Interaction Networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A Subcellular Map of the Human Proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a Knowledge-Based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Berglund, L.; Björling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Al-Khalili Szigyarto, C.; Persson, A.; Ottosson, J.; Wernérus, H.; Nilsson, P.; et al. A Genecentric Human Protein Atlas for Expression Profiles Based on Antibodies. Mol. Cell. Proteom. 2008, 7, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.-K.; Amini, B.; Andersen, E.; Andersson, A.-C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A Human Protein Atlas for Normal and Cancer Tissues Based on Antibody Proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.D. ISG15: A Double Edged Sword in Cancer. OncoImmunology 2015, 4, e1052935. [Google Scholar] [CrossRef] [Green Version]
- Han, H.G.; Moon, H.W.; Jeon, Y.J. ISG15 in Cancer: Beyond Ubiquitin-like Protein. Cancer Lett. 2018, 438, 52–62. [Google Scholar] [CrossRef]
- Laljee, R.P.; Muddaiah, S.; Salagundi, B.; Cariappa, P.M.; Indra, A.S.; Sanjay, V.; Ramanathan, A. Interferon Stimulated Gene—ISG15 is a Potential Diagnostic Biomarker in Oral Squamous Cell Carcinomas. Asian Pac. J. Cancer Prev. 2013, 14, 1147–1150. [Google Scholar] [CrossRef] [Green Version]
- Sumino, J.; Uzawa, N.; Okada, N.; Miyaguchi, K.; Mogushi, K.; Takahashi, K.-I.; Sato, H.; Michikawa, C.; Nakata, Y.; Tanaka, H.; et al. Gene expression changes in initiation and progression of oral squamous cell carcinomas revealed by laser microdissection and oligonucleotide microarray analysis. Int. J. Cancer 2013, 132, 540–548. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Y.; Ghosh, A.; Cuevas, R.A.; Forero, A.; Dhar, J.; Ibsen, M.S.; Schmid-Burgk, J.L.; Schmidt, T.; Ganapathiraju, M.K.; et al. Antiviral Activity of Human OASL Protein Is Mediated by Enhancing Signaling of the RIG-I RNA Sensor. Immunity 2014, 40, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Wang, L.; Shen, H.; Wang, X. Regulatory Roles of OASL in Lung Cancer Cell Sensitivity to Actinidia Chinensis Planch Root Extract (AcRoots). Cell Biol. Toxicol. 2018, 34, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, C. Prognostic Characterization of OAS1/OAS2/OAS3/OASL in Breast Cancer. BMC Cancer 2020, 20, 575. [Google Scholar] [CrossRef]
- Lohavanichbutr, P.; Méndez, E.; Holsinger, F.C.; Rue, T.C.; Zhang, Y.; Houck, J.; Upton, M.P.; Futran, N.D.; Schwartz, S.; Wang, P.; et al. A 13-gene signature prognostic of HPV-negative OSCC: Discovery and external validation. Clin. Cancer Res. 2013, 19, 1197–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheriyath, V.; Kaur, J.; Davenport, A.; Khalel, A.; Chowdhury, N.; Gaddipati, L. G1P3 (IFI6), a Mitochondrial Localised Antiapoptotic Protein, Promotes Metastatic Potential of Breast Cancer Cells through MtROS. Br. J. Cancer 2018, 119, 52–64. [Google Scholar] [CrossRef]
- Li, G.; Li, X.; Yang, M.; Xu, L.; Deng, S.; Ran, L. Prediction of Biomarkers of Oral Squamous Cell Carcinoma Using Microarray Technology. Sci. Rep. 2017, 7, 42105. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zu, T.; Li, T.; Li, M.; Mi, J.; Bai, F.; Liu, G.; Wen, J.; Li, H.; Brakebusch, C.; et al. ATF3 downmodulates its new targets IFI6 and IFI27 to suppress the growth and migration of tongue squamous cell carcinoma cells. PLoS Genet. 2017, 17, e1009283. [Google Scholar] [CrossRef]
- Cheriyath, V.; Glaser, K.B.; Waring, J.F.; Baz, R.; Hussein, M.A.; Borden, E.C. G1P3, an IFN-Induced Survival Factor, Antagonizes TRAIL-Induced Apoptosis in Human Myeloma Cells. J. Clin. Investig. 2007, 117, 3107–3117. [Google Scholar] [CrossRef] [Green Version]
- Tahara, E.; Tahara, H.; Kanno, M.; Naka, K.; Takeda, Y.; Matsuzaki, T.; Yamazaki, R.; Ishihara, H.; Yasui, W.; Barrett, J.C.; et al. G1P3, an Interferon Inducible Gene 6-16, Is Expressed in Gastric Cancers and Inhibits Mitochondrial-Mediated Apoptosis in Gastric Cancer Cell Line TMK-1 Cell. Cancer Immunol. Immunother. CII 2005, 54, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Yu, T.; Temam, S.; Ziober, B.L.; Wang, J.; Schwartz, J.L.; Mao, L.; Wong, D.T.; Zhou, X. Transcriptomic Dissection of Tongue Squamous Cell Carcinoma. BMC Genom. 2008, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Sezin, T.; Vorobyev, A.; Sadik, C.D.; Zillikens, D.; Gupta, Y.; Ludwig, R.J. Gene Expression Analysis Reveals Novel Shared Gene Signatures and Candidate Molecular Mechanisms between Pemphigus and Systemic Lupus Erythematosus in CD4+ T Cells. Front. Immunol. 2018, 8, 1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Yang, Q.; Cui, Q.; Zhang, D.; Kong, D.; Liao, X.; Ren, J.; Gong, Y.; Wu, G. Weighted Gene Correlation Network Analysis Identifies RSAD2, HERC5, and CCL8 as Prognostic Candidates for Breast Cancer. J. Cell. Physiol. 2020, 235, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, H.; Zheng, L. Biological information analysis of differentially expressed genes in oral squamous cell carcinoma tissues in GEO database. J. BUON 2018, 23, 1662–1670. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistical Characteristics | Samples for ME (<Median) | Samples for ME (>Median) |
|---|---|---|
| Hazard Rate | 0.0047 | 0.0040 |
| 95% Confidence Interval | 0.9615 (Lower limit) | 2.1424 (Upper limit) |
| Hazard Ratio | 1.4353 | |
| z-value | 1.66584 | |
| p-value (two-tailed test) | 0.09575 | |
| Statistical Characteristics | Samples for ME (<Median) | Samples for ME (>Median) |
|---|---|---|
| Hazard Rate | 0.0072 | 0.0065 |
| 95% Confidence Interval | 1.0084 (Lower limit) | 2.4600 (Upper limit) |
| Hazard Ratio | 1.5750 | |
| z-value | 1.88315 | |
| p-value (two-tailed test) | 0.05968 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.; Rai, A.; Verma, A.K.; Alsahli, M.A.; Rahmani, A.H.; Almatroodi, S.A.; Alrumaihi, F.; Dev, K.; Sinha, A.; Sankhwar, S.; et al. Survival-Based Biomarker Module Identification Associated with Oral Squamous Cell Carcinoma (OSCC). Biology 2021, 10, 760. https://doi.org/10.3390/biology10080760
Singh P, Rai A, Verma AK, Alsahli MA, Rahmani AH, Almatroodi SA, Alrumaihi F, Dev K, Sinha A, Sankhwar S, et al. Survival-Based Biomarker Module Identification Associated with Oral Squamous Cell Carcinoma (OSCC). Biology. 2021; 10(8):760. https://doi.org/10.3390/biology10080760
Chicago/Turabian StyleSingh, Prithvi, Arpita Rai, Amit Kumar Verma, Mohammed A. Alsahli, Arshad Husain Rahmani, Saleh A. Almatroodi, Faris Alrumaihi, Kapil Dev, Anuradha Sinha, Shweta Sankhwar, and et al. 2021. "Survival-Based Biomarker Module Identification Associated with Oral Squamous Cell Carcinoma (OSCC)" Biology 10, no. 8: 760. https://doi.org/10.3390/biology10080760
APA StyleSingh, P., Rai, A., Verma, A. K., Alsahli, M. A., Rahmani, A. H., Almatroodi, S. A., Alrumaihi, F., Dev, K., Sinha, A., Sankhwar, S., & Dohare, R. (2021). Survival-Based Biomarker Module Identification Associated with Oral Squamous Cell Carcinoma (OSCC). Biology, 10(8), 760. https://doi.org/10.3390/biology10080760