
Update on the Discovery of Efflux Pump Inhibitors against Critical Priority Gram-Negative Bacteria

 , and
, and 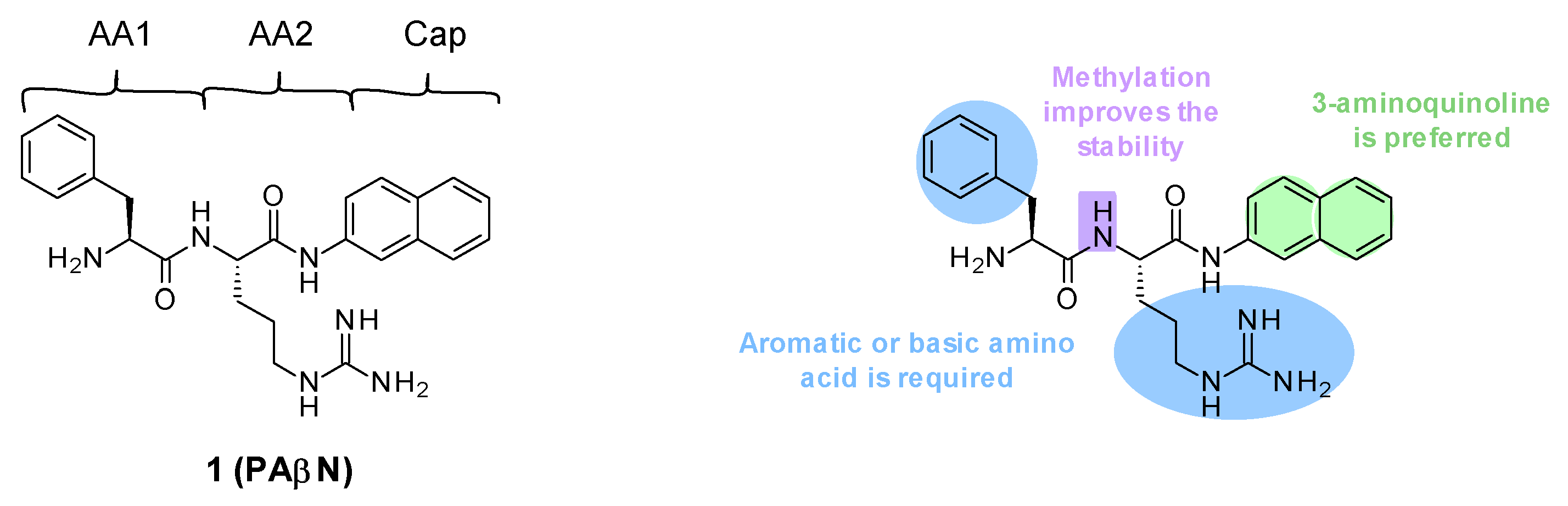{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tripartite RND Multidrug Efflux Pumps
3. Efflux Pump Inhibitors
3.1. PAβN
3.1.1. Structure–Activity Relationships (SARs)
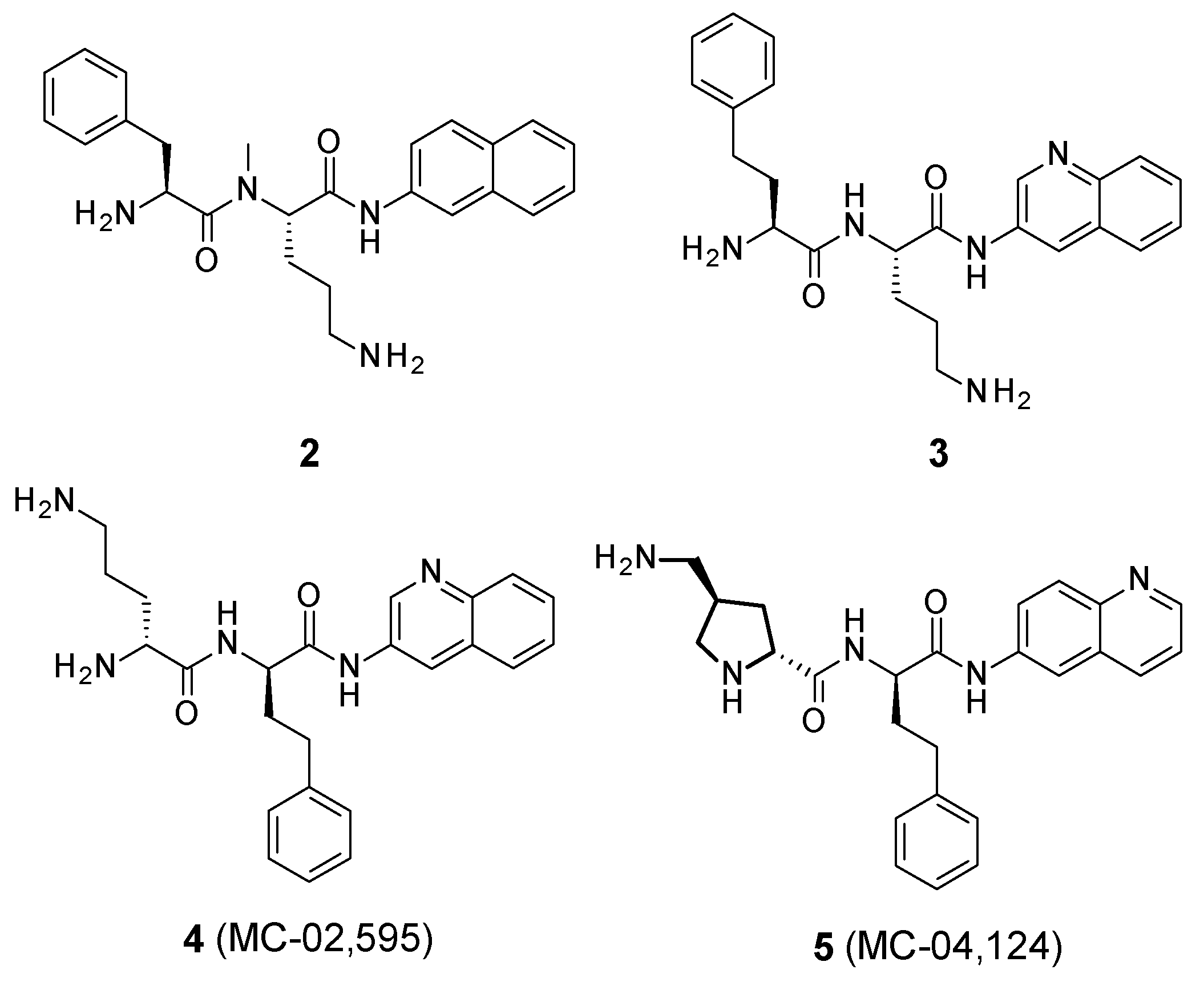
- Replacement of AA1 and AA2: It was described that amino acids 1 and 2 needed to contain both an aromatic and a basic moiety, though the order could be inverted [39,42]. Replacement of L-phenylalanine with an L-homo-phenylalanine led to a 2-fold improvement in EPI potency. In addition, ornithine or an aminomethylproline was accepted as an alternative basic amino side chain [39,43].
- Substitution of the amide bond: Methylation of the amide between AA1 and AA2 led to a slight improvement in compound potency and plasma stability [39].
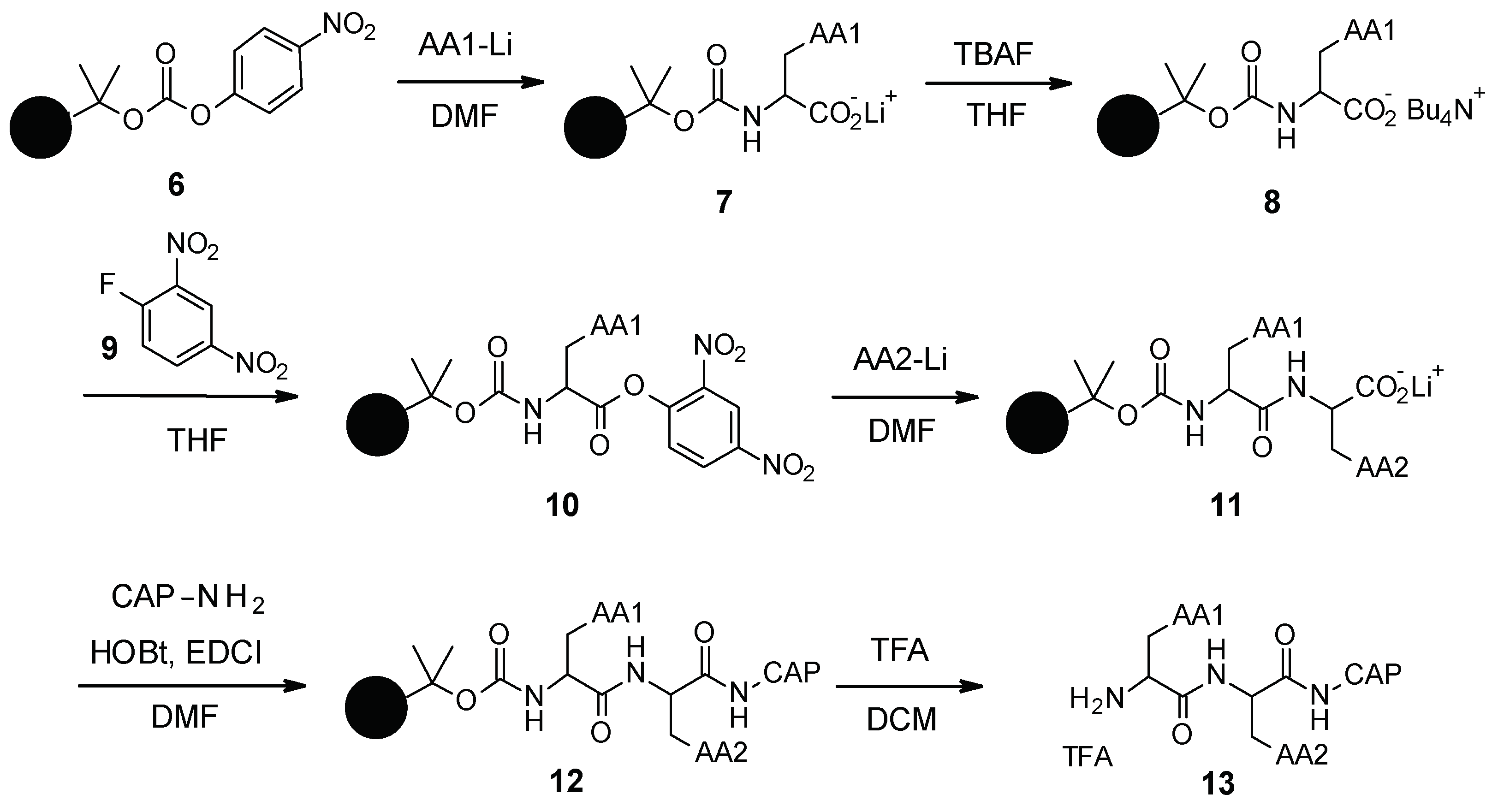
3.1.2. Chemical Synthesis
3.1.3. In Vitro Activity
3.1.4. Pharmacological Properties
3.1.5. In Vivo Activity
3.2. NMP and Arylpiperazines
3.2.1. Structure-Activity Relationships
- The size of the linker between the aromatic ring and the piperazine: Linker elongation led to an improvement in potency.
- The substitution of the phenyl ring: Substitution of the benzene ring with a halogen-containing moiety increased compound potency. In particular, the introduction of a trifluoromethyl group in the meta position was preferred (8-fold improvement in boosting activity).
- The nature of the aromatic ring: Replacement of the phenyl ring by a naphthyl ring led to a 5-fold improvement in potency.
- The substitution of the piperazine: Substitutions of the piperazine ring were not tolerated and led to decreased potency.
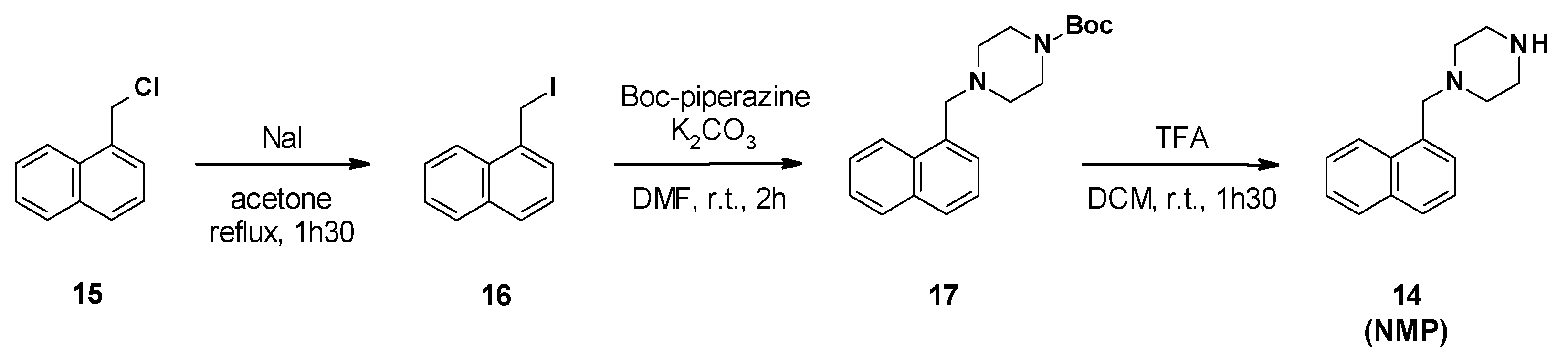
3.2.2. Chemical Synthesis
3.2.3. In Vitro Activity
3.2.4. Pharmacological Properties
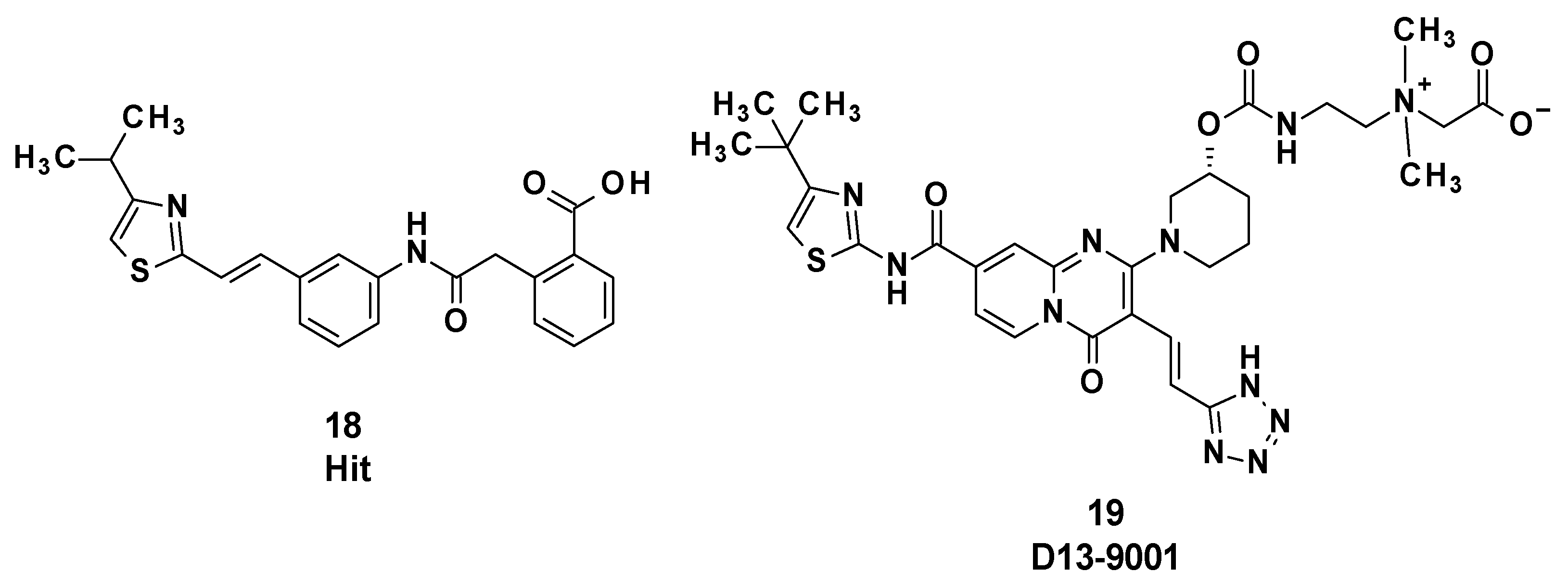
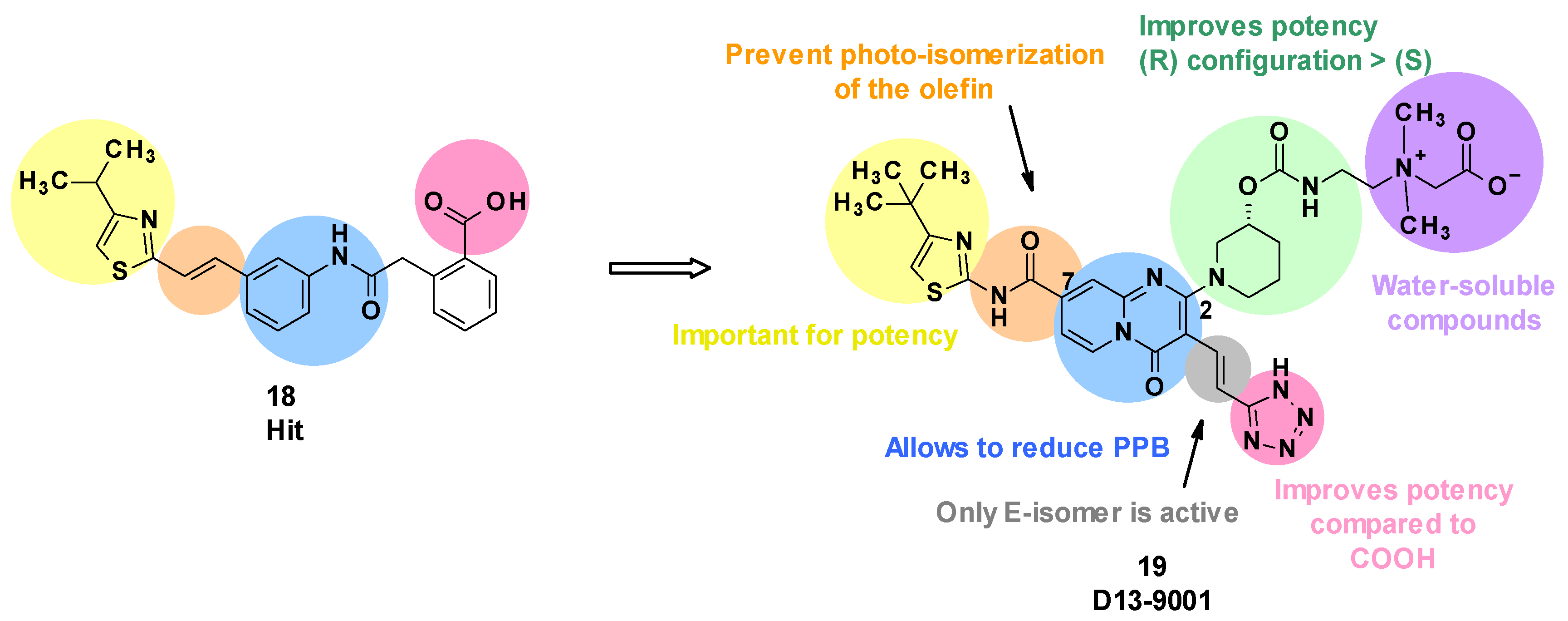
3.3. D13-9001
3.3.1. Structure–Activity Relationships
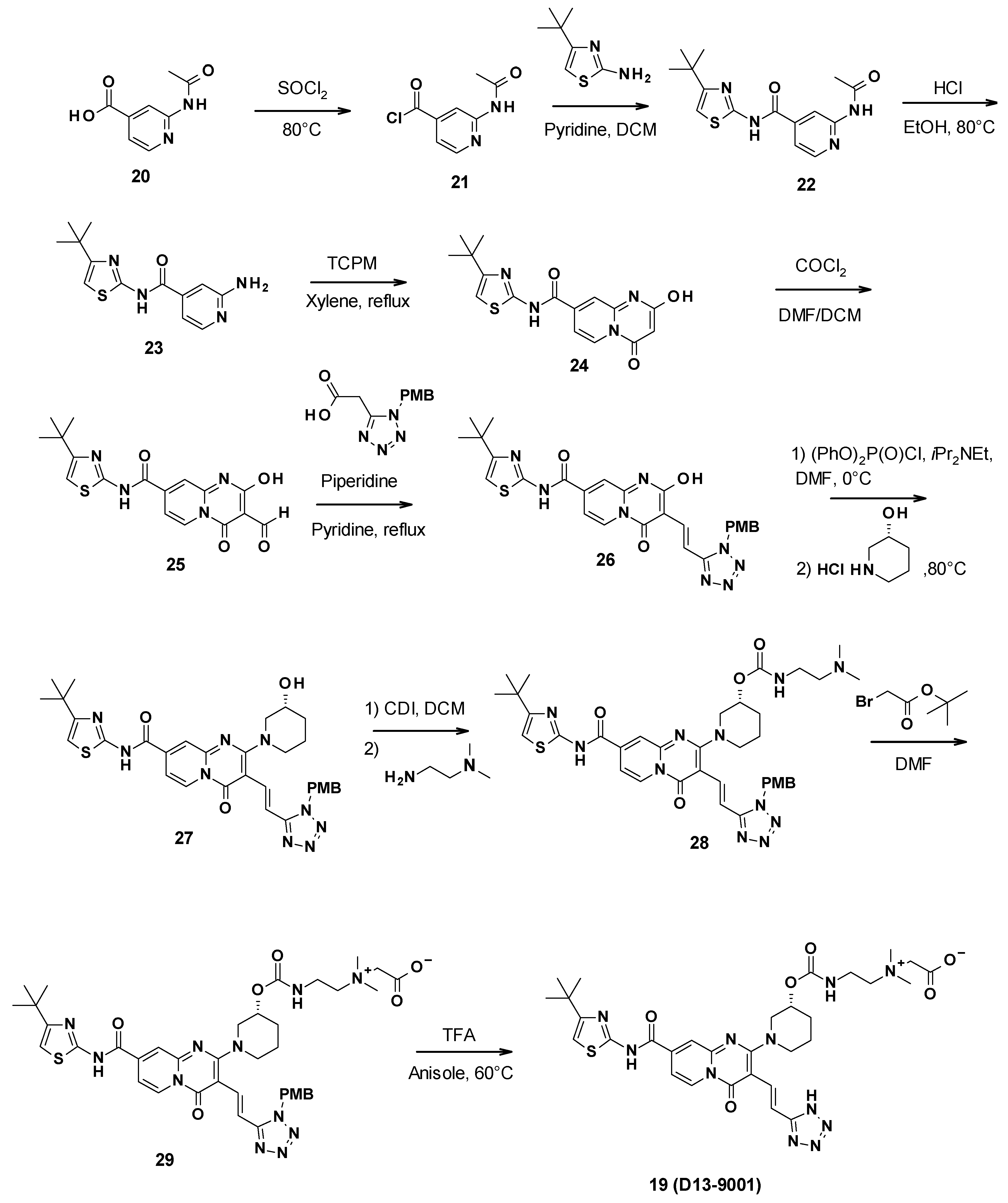
3.3.2. Chemical Synthesis
3.3.3. In Vitro Activity
3.3.4. Pharmacological Properties
3.3.5. In Vivo Activity
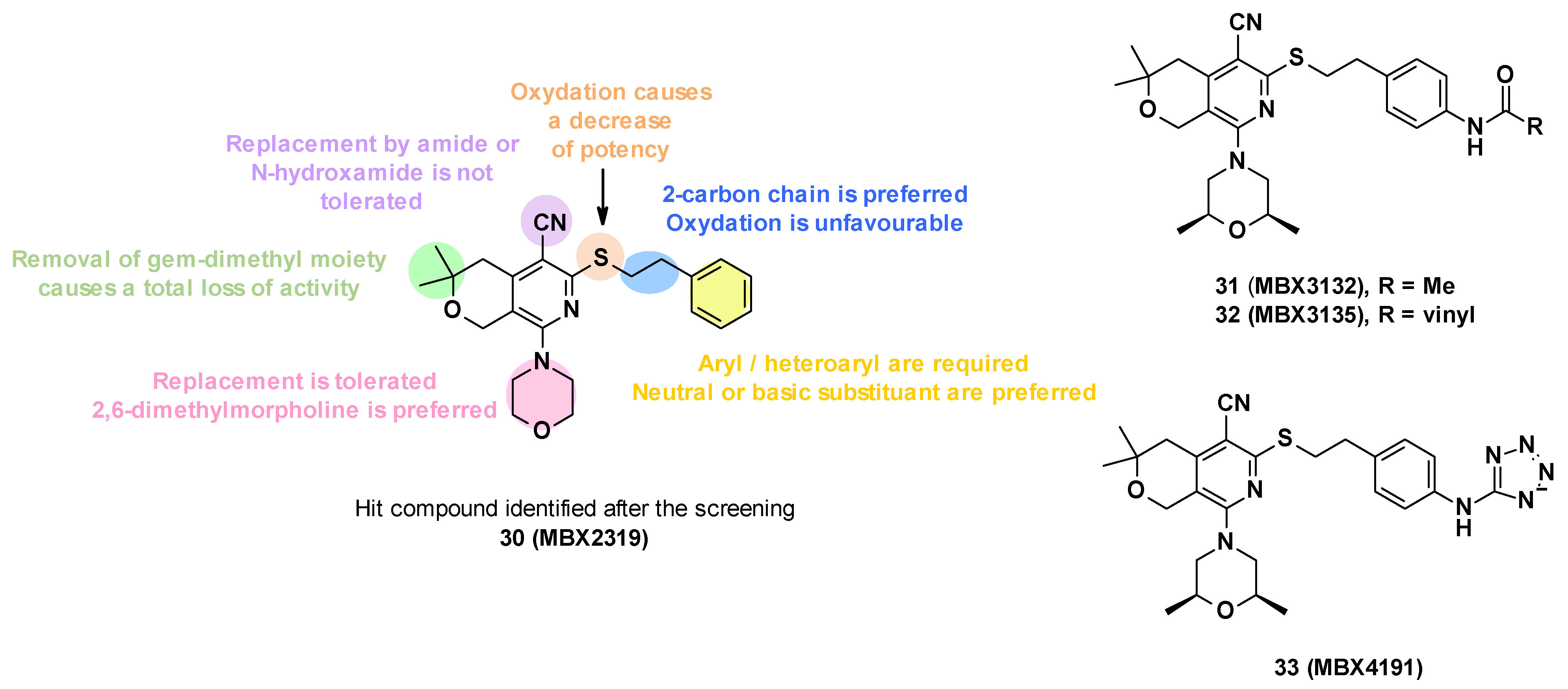
3.4. MBX Compounds
3.4.1. Structure—Activity Relationships
- Replacement of the nitrile group: Only two examples were synthesized due to synthetic problems, one with an N-hydroxyamide and the other with an amide, but these modifications led to a decrease in potency (MPC4 (levofloxacin and piperacillin) > 100 µM).
- The oxidation of the sulfide group to sulfoxide or sulfone also led to a decrease in potency.
- Modification of alkyl linker: The deletion of the chain on the sulfide, the modification of the chain size (one or three carbons), and the oxidation of the chain led to a total loss of activity.
- Modification of gem-dimethyl moiety: Replacement of gem-dimethyl substituent by hydrogen atoms also led to a total loss of activity.
- Modification of the morpholine ring: The morpholine was replaced by a variety of acyclic or cyclic (5,6,7-membered rings) amine. In general, the replacement was tolerated, but the introduction of substituents with basic amines was found to be associated with moderate/strong cytotoxicity. Only the introduction of a 2,6-dimethyl group allowed a good balance between activity and cytotoxicity.
- Substitution/modification of the phenyl ring: The introduction of substituents on the phenyl ring was tolerated, and the activity of the compounds depended on the substituent in the para position. The introduction of a neutral or basic substituent on the phenyl ring seemed to improve the activity.
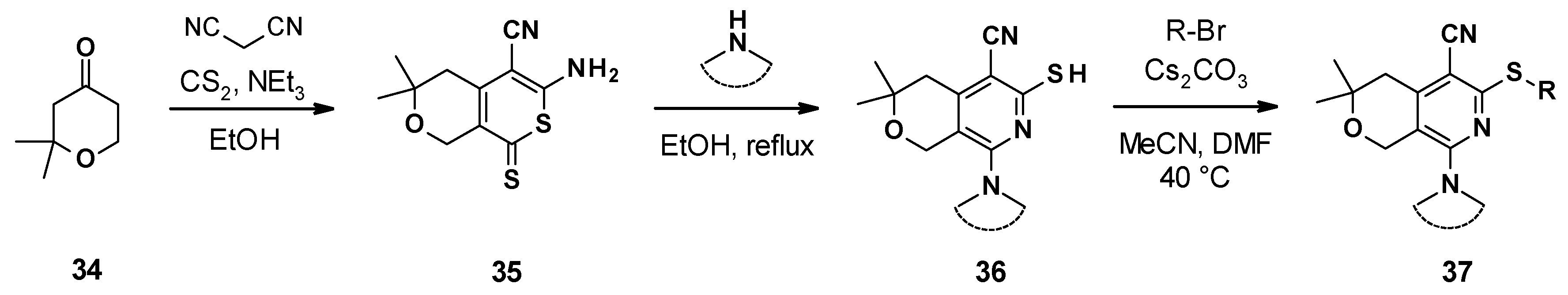
3.4.2. Chemical Synthesis
3.4.3. In Vitro Activity
3.4.4. Mode of Action
3.4.5. Pharmacological Properties
3.4.6. In Vivo Activity
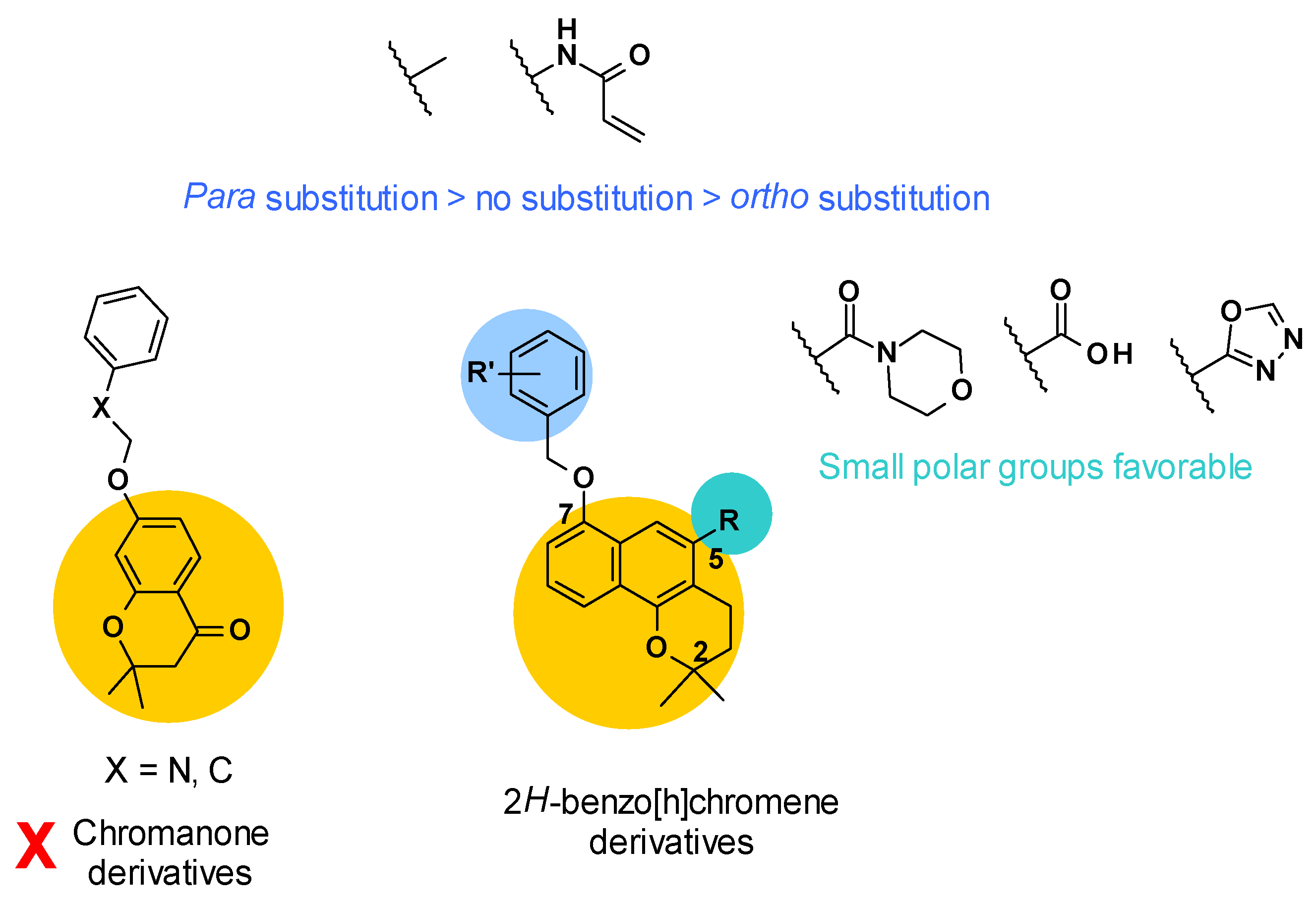
3.5. 2H-Benzo[h]chromene Series
3.5.1. Structure–Activity Relationships
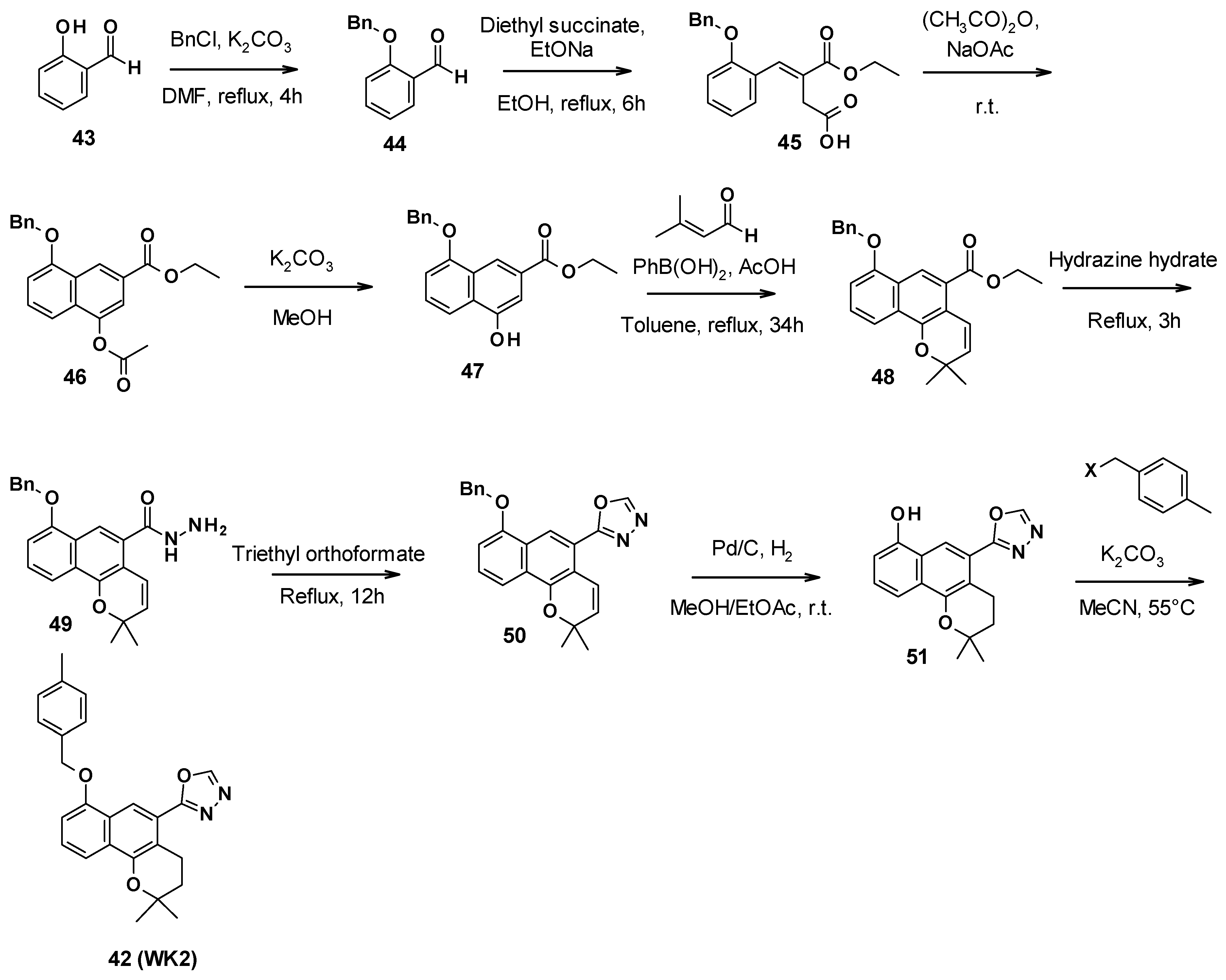
3.5.2. Chemical Synthesis
3.5.3. In Vitro Activity
3.5.4. Pharmacological Properties
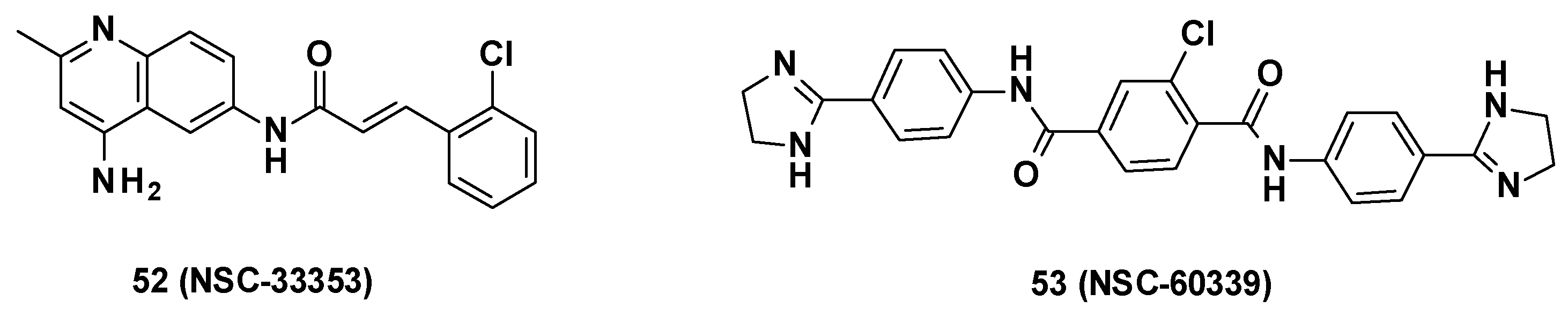
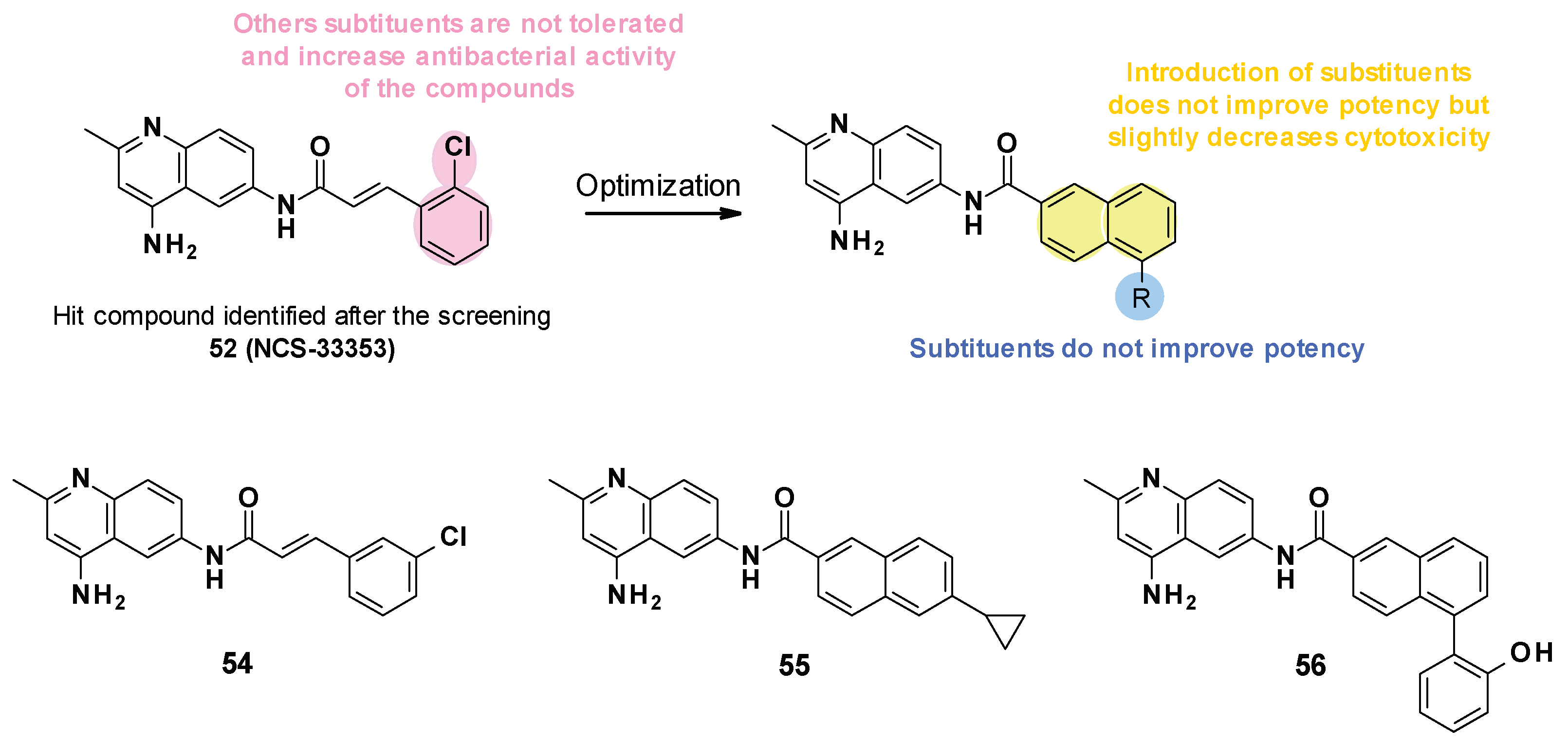
3.6. NSC Series
3.6.1. Structure–Activity Relationships
- Substitution of cinnamoyl moiety: First, the substitution of the cinnamoyl group was explored. The introduction of other electron-withdrawing (Cl, Br, NO2) or electron-donating (isopropyl) groups in position C-2, C-3, or C-4 did not improve the boosting effect of erythromycin (MPC4 = 3.1–200 µM) and novobiocin (MPC4 = 6.25–400 µM). Furthermore, these modifications generally led to an increase in the intrinsic antibacterial activity of the compounds.
- Replacement of cinnamoyl moiety by substituted naphthyl rings: Because the cinnamoyl moiety was thought to be responsible for the high cytotoxicity of NCS-33353, its replacement with a substituted naphthyl was performed. The introduction of electron-withdrawing (Br, CN, CO2Me) or electron-donating (OMe) groups at various positions (C-5, C-6, C-7) allowed a slight decrease in cytotoxicity but did not improve the boosting effect of novobiocin and led to compounds no longer able to boost erythromycin. In order to improve the affinity of the inhibitor for AcrA, aromatic substituents were added in C-5 and C-6 positions, but this led to a decrease or even total loss of the boosting effect on erythromycin and novobiocin.

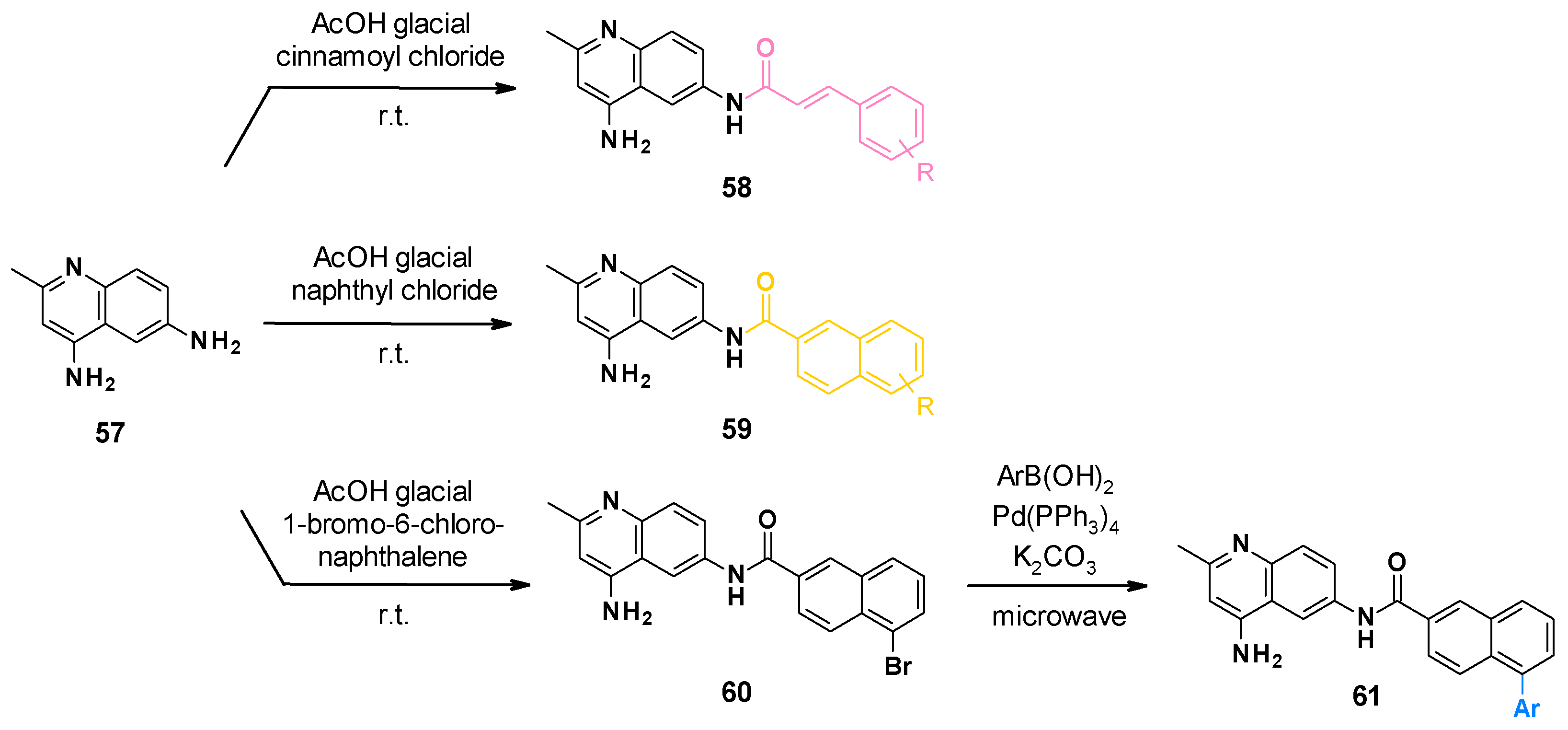
3.6.2. Chemical Synthesis
3.6.3. In Vitro Activity
3.6.4. Mode of Action
3.6.5. Pharmacological Properties
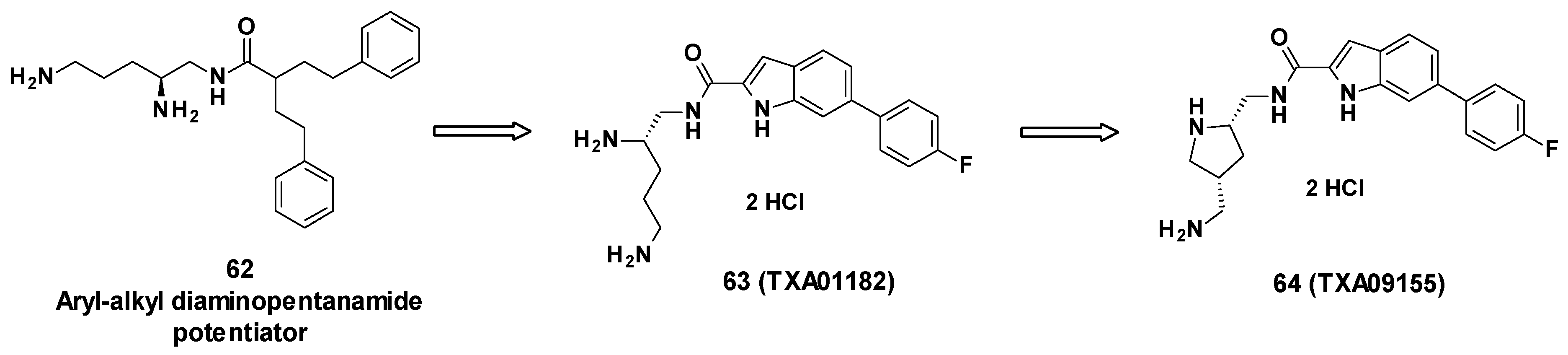
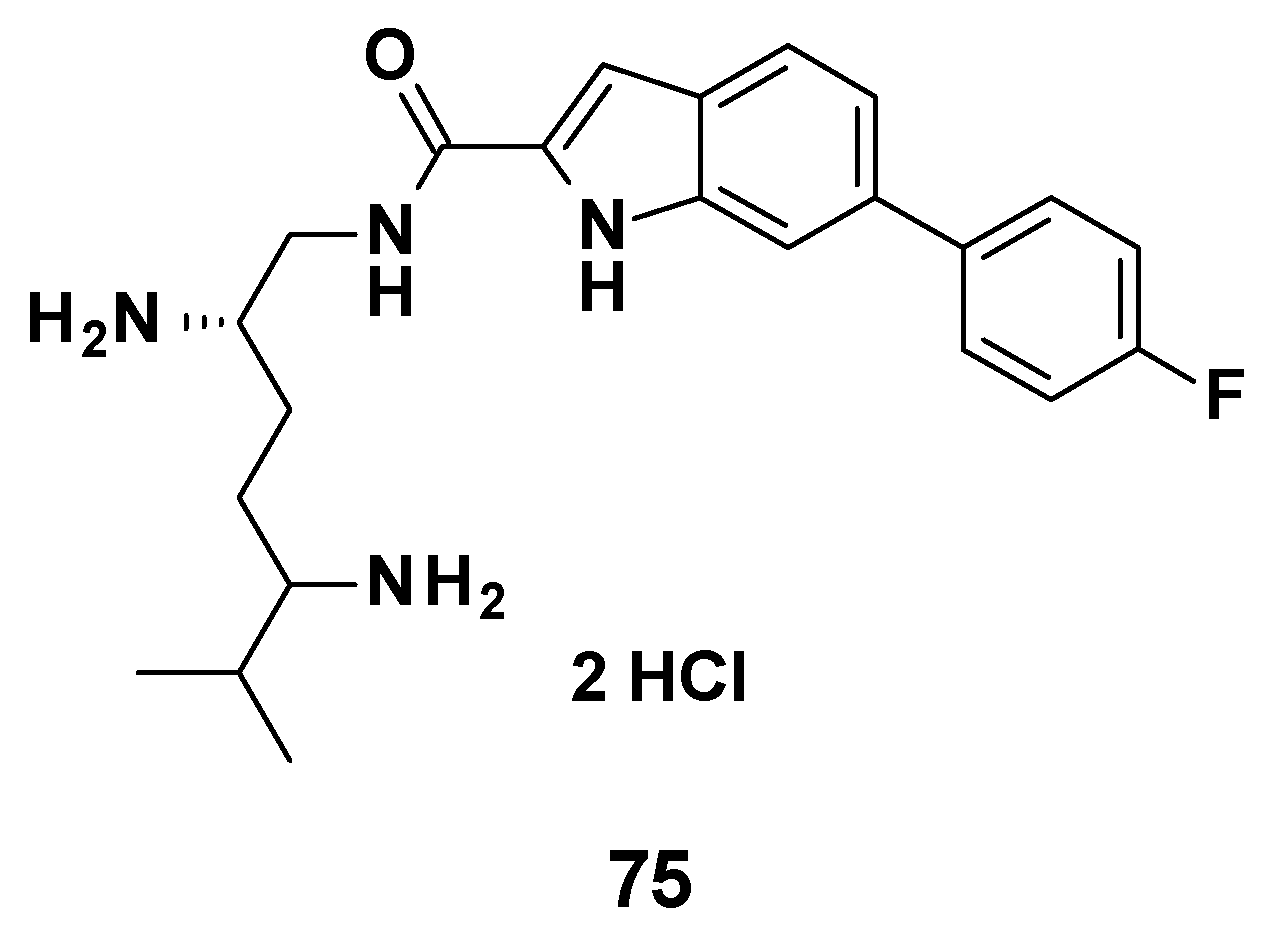
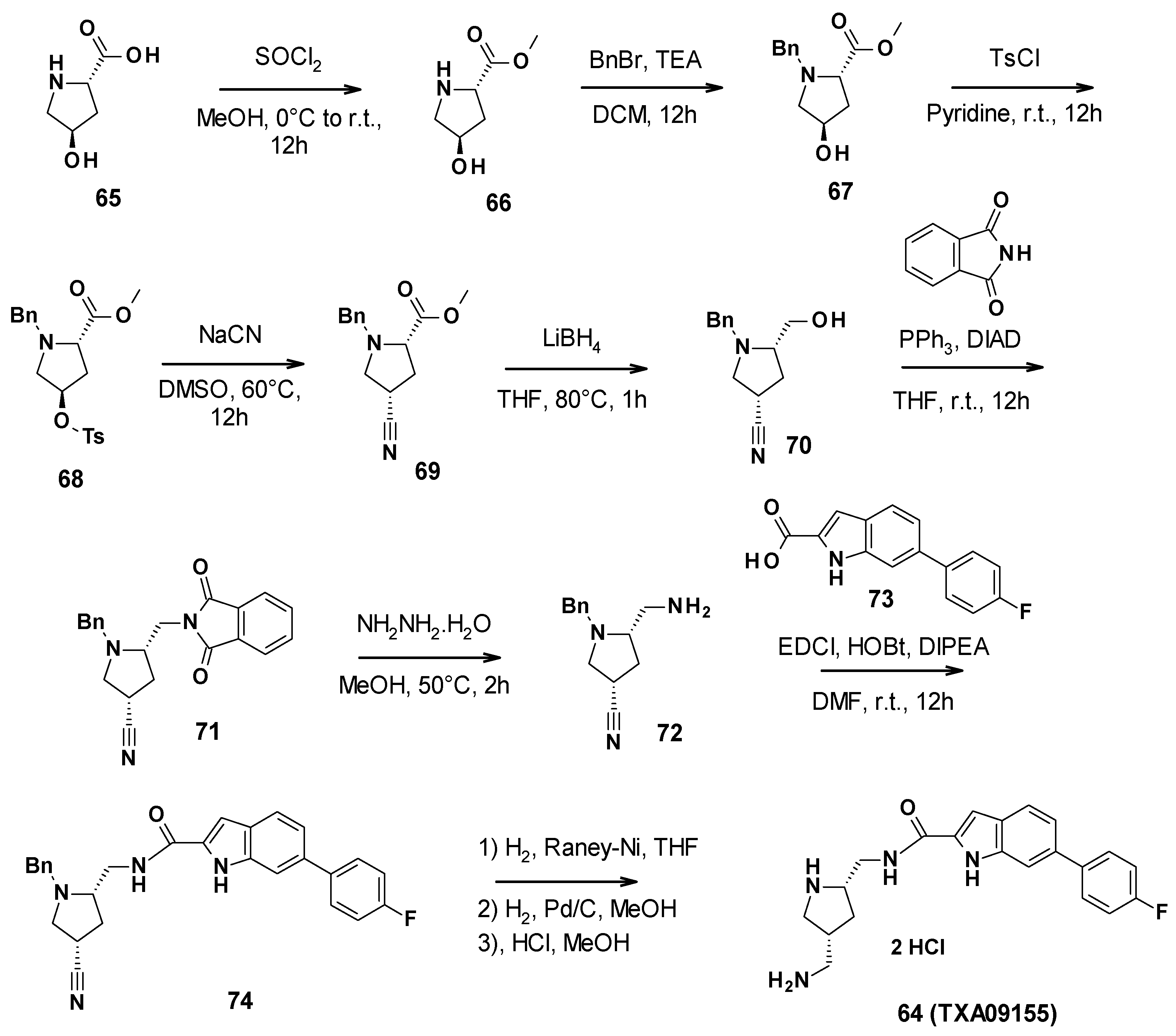
3.7. TXA Compounds
3.7.1. Structure–Activity Relationships
3.7.2. Chemical Synthesis
3.7.3. In Vitro Activity
3.7.4. Pharmacological Properties
3.7.5. In Vivo Activity
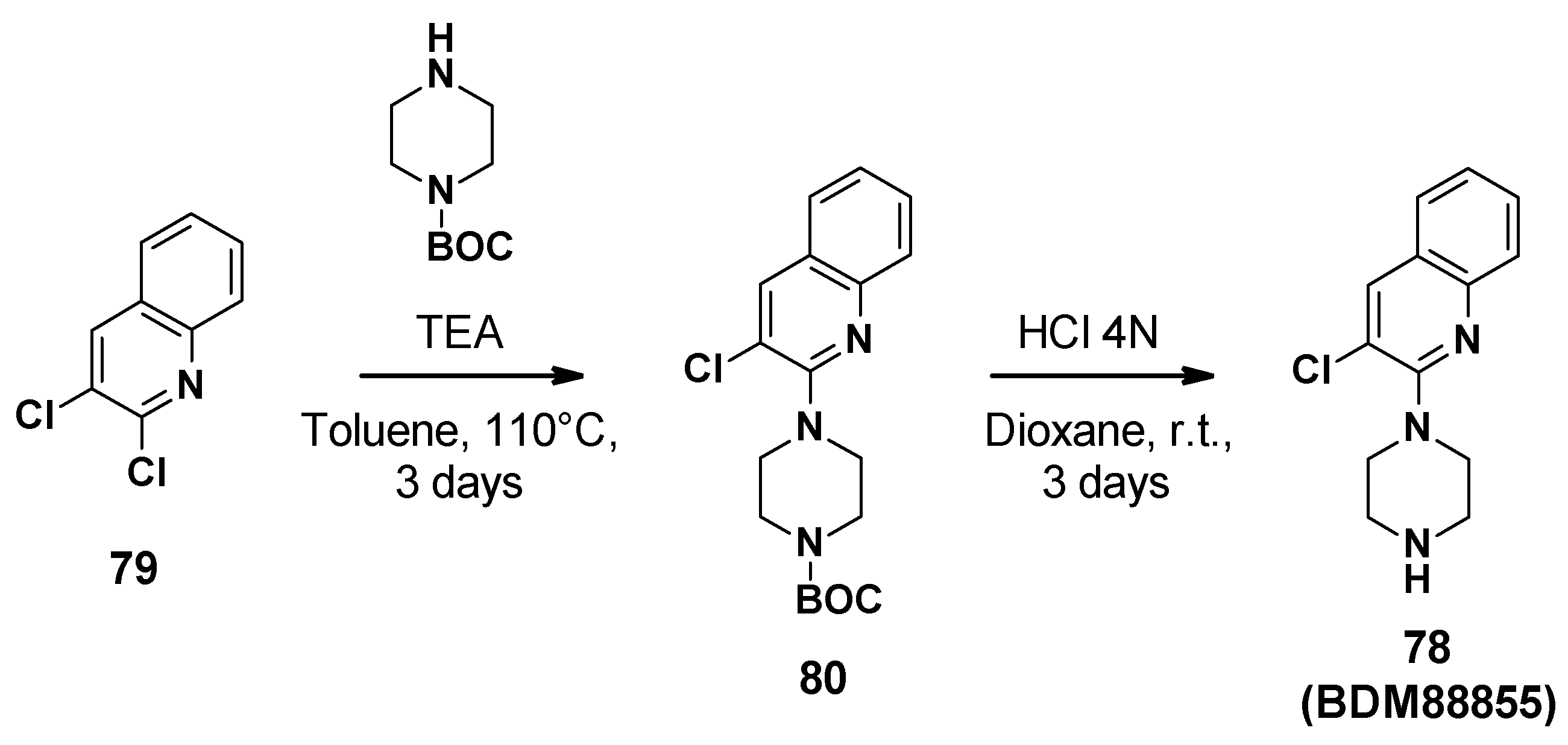
3.8. Pyridylpiperazines (BDM Compounds)
3.8.1. Structure-Activity Relationships
- Modification of the piperazine moiety: Replacement by morpholine or piperidine led to a total loss of activity (EC90 > 500 μM) suggesting that the basic nitrogen was important for potency. In addition, the substitution of the amine with methyl led to a 6-fold decrease in potency.
- Replacement of the trifluoromethyl group: Substitution with a more polar group (OMe) led to a decrease in potency in contrast to the introduction of hydrophobic substituents. The introduction of halogen atoms was preferred and led to a 5-fold improvement of potency for the compound bearing an iodine atom (77, EC90 = 12 μM).
- Replacement of the pyridine core: Replacement with a quinoline core led to a 15-fold more potent compound (78, EC90 = 3.4 μM).
- Replacement of the chlorine atom: Removal of the chlorine atom led to a significant decrease in potency (EC90 > 250 μM), while replacement with other halogen atoms (Br, I) led to a slight improvement in potency (EC90 = 1.5 μM and 3 μM, respectively).

3.8.2. Chemical Synthesis
3.8.3. In Vitro Activity
3.8.4. Mode of Action
3.8.5. Pharmacological Properties
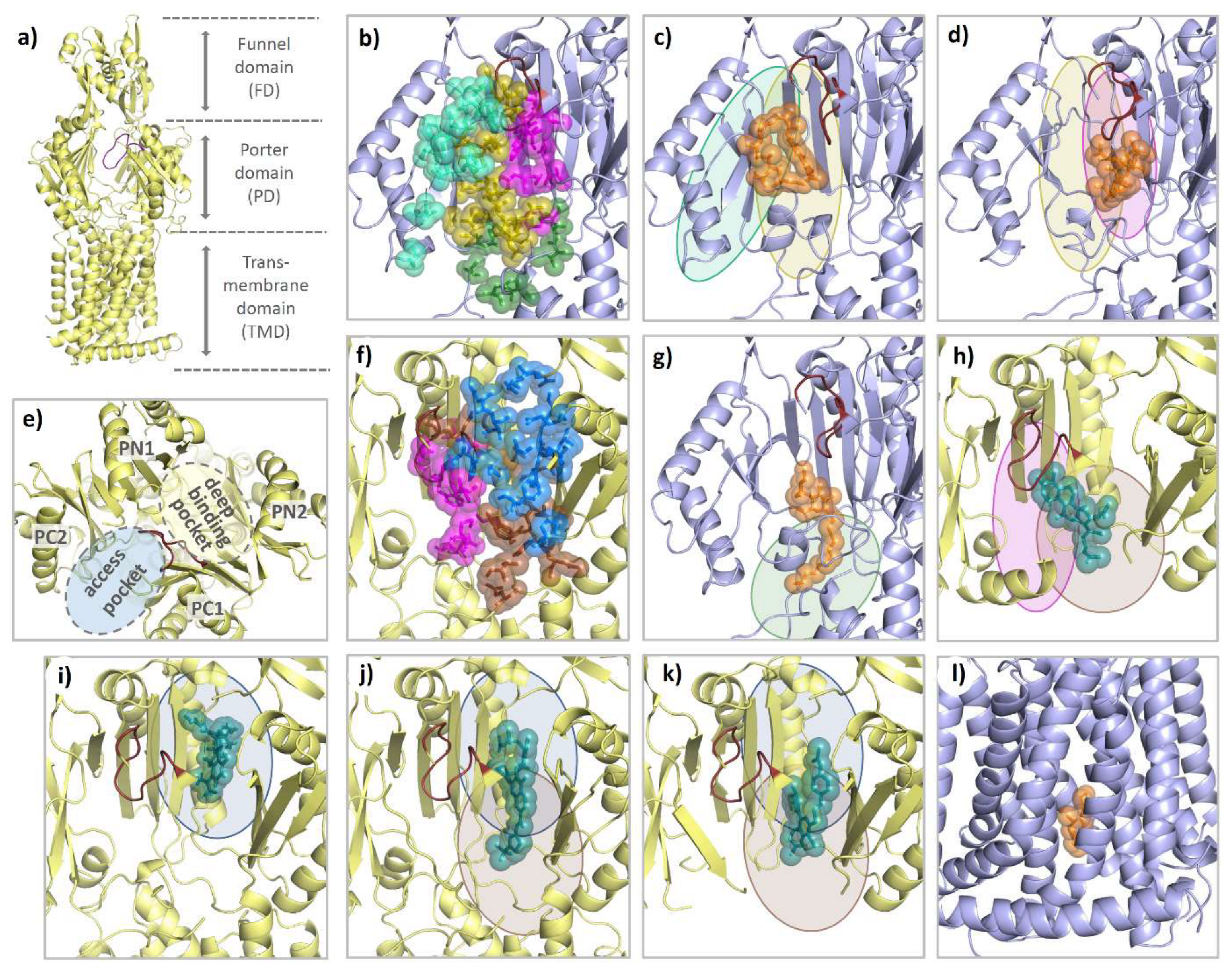
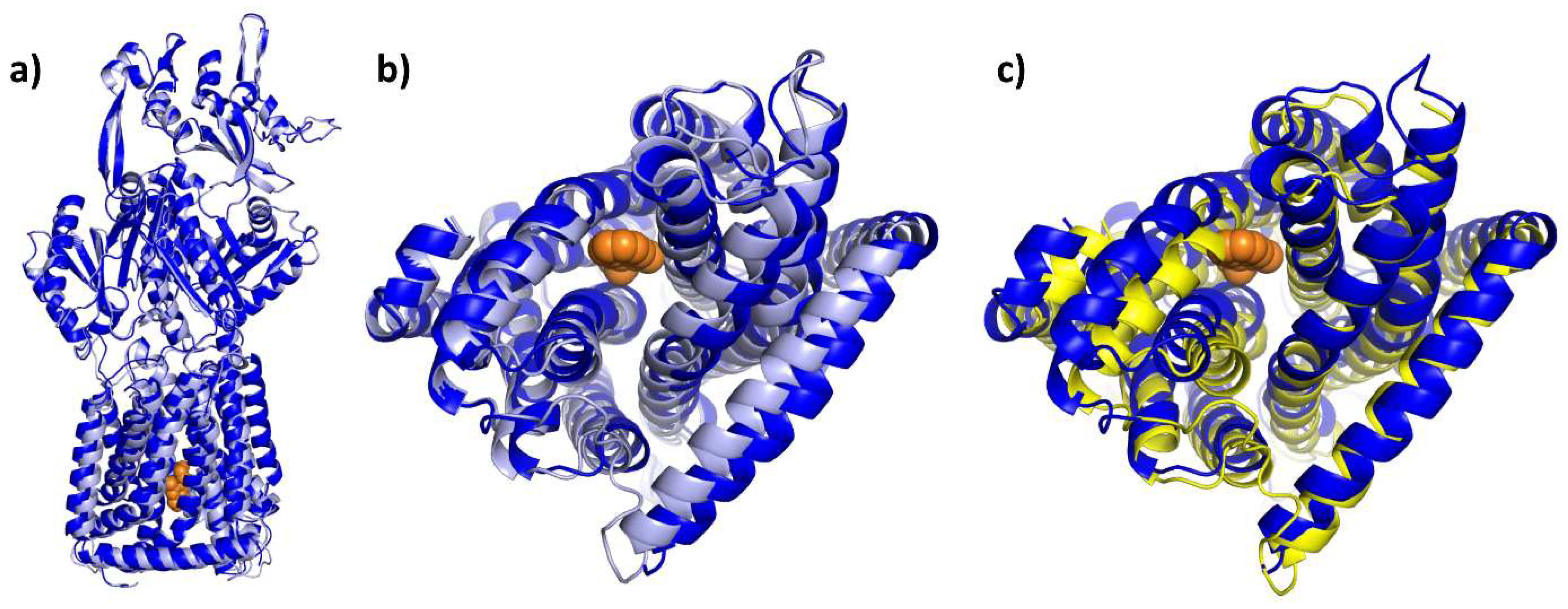
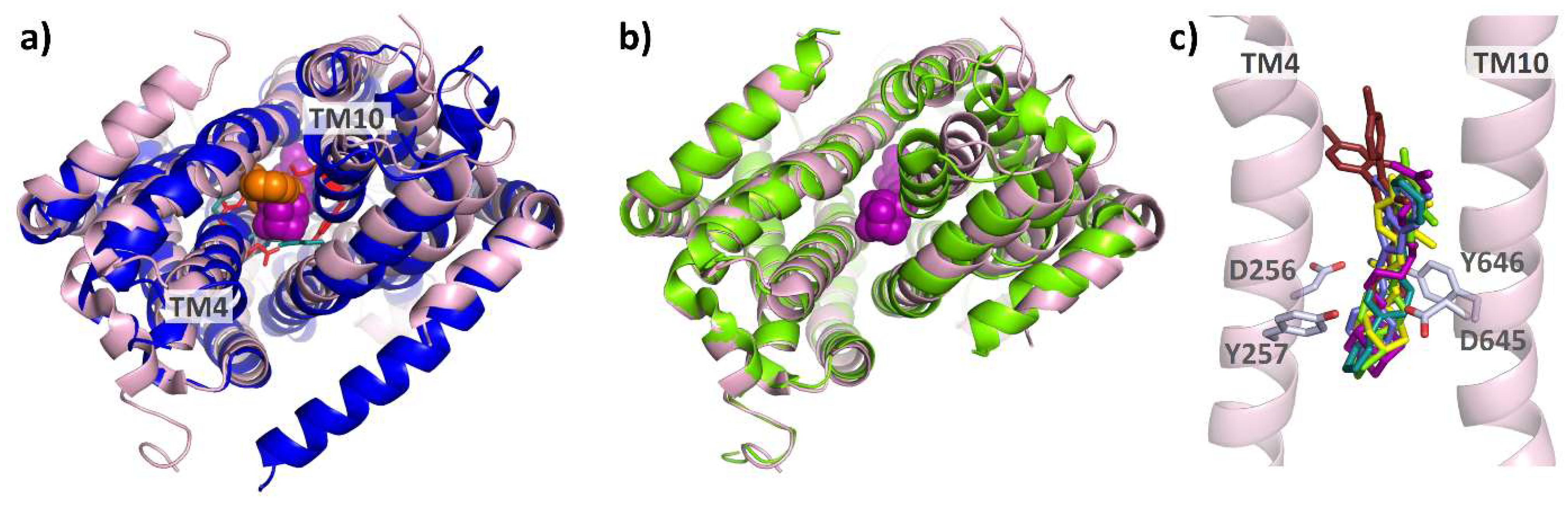
4. Structural Perspective on AcrB Substrate and Inhibitor Binding
4.1. E. coli AcrAB-TolC, the Well-Studied Model System
4.2. Substrate Binding
4.3. Competitive Inhibitor Binding
4.4. Allosteric Inhibitors Affecting Proton Coupling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The Global Preclinical Antibacterial Pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef]
- Venter, H. Reversing Resistance to Counter Antimicrobial Resistance in the World Health Organisation’s Critical Priority of Most Dangerous Pathogens. Biosci. Rep. 2019, 39, BSR20180474. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the Clinical Antibacterial and Antituberculosis Pipeline. Lancet Infect. Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A New Antibiotic Kills Pathogens without Detectable Resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Willyard, C. The Drug-Resistant Bacteria That Pose the Greatest Health Threats. Nature 2017, 543, 15. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.D.; Iinishi, A.; Modaresi, S.M.; Yoo, B.-K.; Curtis, T.D.; Lariviere, P.J.; Liang, L.; Son, S.; Nicolau, S.; Bargabos, R.; et al. Computational Identification of a Systemic Antibiotic for Gram-Negative Bacteria. Nat. Microbiol. 2022, 7, 1661–1672. [Google Scholar] [CrossRef]
- Alav, I.; Kobylka, J.; Kuth, M.S.; Pos, K.M.; Picard, M.; Blair, J.M.A.; Bavro, V.N. Structure, Assembly, and Function of Tripartite Efflux and Type 1 Secretion Systems in Gram-Negative Bacteria. Chem. Rev. 2021, 121, 5479–5596. [Google Scholar] [CrossRef]
- Kobylka, J.; Kuth, M.S.; Müller, R.T.; Geertsma, E.R.; Pos, K.M. AcrB: A Mean, Keen, Drug Efflux Machine. Ann. N. Y. Acad. Sci. 2020, 1459, 38–68. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Richmond, G.E.; Piddock, L.J.V. Multidrug Efflux Pumps in Gram-Negative Bacteria and Their Role in Antibiotic Resistance. Future Microbiol. 2014, 9, 1165–1177. [Google Scholar] [CrossRef]
- Piddock, L.J.V. Clinically Relevant Chromosomally Encoded Multidrug Resistance Efflux Pumps in Bacteria. Clin. Microbiol. Rev. 2006, 19, 382–402. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; Walker, J.K.; Parks, J.M.; Rybenkov, V.V. Multidrug Efflux Pumps and the Two-Faced Janus of Substrates and Inhibitors. Acc. Chem. Res. 2021, 54, 930–939. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; Rybenkov, V.V. Permeability Barriers of Gram-negative Pathogens. Ann. N. Y. Acad. Sci. 2020, 1459, 5–18. [Google Scholar] [CrossRef]
- Bajaj, H.; Acosta Gutierrez, S.; Bodrenko, I.; Malloci, G.; Scorciapino, M.A.; Winterhalter, M.; Ceccarelli, M. Bacterial Outer Membrane Porins as Electrostatic Nanosieves: Exploring Transport Rules of Small Polar Molecules. ACS Nano 2017, 11, 5465–5473. [Google Scholar] [CrossRef] [PubMed]
- Mehla, J.; Malloci, G.; Mansbach, R.; López, C.A.; Tsivkovski, R.; Haynes, K.; Leus, I.V.; Grindstaff, S.B.; Cascella, R.H.; D’Cunha, N.; et al. Predictive Rules of Efflux Inhibition and Avoidance in Pseudomonas Aeruginosa. Mbio 2021, 12, e02785-20. [Google Scholar] [CrossRef]
- El Zahed, S.S.; French, S.; Farha, M.A.; Kumar, G.; Brown, E.D. Physicochemical and Structural Parameters Contributing to the Antibacterial Activity and Efflux Susceptibility of Small-Molecule Inhibitors of Escherichia Coli. Antimicrob. Agents Chemother. 2021, 65, e01925-20. [Google Scholar] [CrossRef] [PubMed]
- Cernicchi, G.; Felicetti, T.; Sabatini, S. Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives. Molecules 2021, 26, 6996. [Google Scholar] [CrossRef]
- Alenazy, R. Drug Efflux Pump Inhibitors: A Promising Approach to Counter Multidrug Resistance in Gram-Negative Pathogens by Targeting AcrB Protein from AcrAB-TolC Multidrug Efflux Pump from Escherichia Coli. Biology 2022, 11, 1328. [Google Scholar] [CrossRef]
- Seukep, A.J.; Mbuntcha, H.G.; Kuete, V.; Chu, Y.; Fan, E.; Guo, M.-Q. What Approaches to Thwart Bacterial Efflux Pumps-Mediated Resistance? Antibiotics 2022, 11, 1287. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Espada, R.; Shahrour, H.; Pitts, B.; Stewart, P.S.; Sánchez-Gómez, S.; Martínez-de-Tejada, G. A Permeability-Increasing Drug Synergizes with Bacterial Efflux Pump Inhibitors and Restores Susceptibility to Antibiotics in Multi-Drug Resistant Pseudomonas Aeruginosa Strains. Sci. Rep. 2019, 9, 3452. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. RND Transporters in the Living World. Res. Microbiol. 2018, 169, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB–TolC Multidrug Efflux Pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, G.; Hryc, C.F.; Blaza, J.N.; Serysheva, I.I.; Schmid, M.F.; Chiu, W.; Luisi, B.F.; Du, D. An Allosteric Transport Mechanism for the AcrAB-TolC Multidrug Efflux Pump. Elife 2017, 6, e24905. [Google Scholar] [CrossRef]
- Glavier, M.; Puvanendran, D.; Salvador, D.; Decossas, M.; Phan, G.; Garnier, C.; Frezza, E.; Cece, Q.; Schoehn, G.; Picard, M.; et al. Antibiotic Export by MexB Multidrug Efflux Transporter Is Allosterically Controlled by a MexA-OprM Chaperone-like Complex. Nat. Commun. 2020, 11, 4948. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Yonehara, R.; Ishizaka-Ikeda, E.; Miyazaki, N.; Maeda, S.; Iwasaki, K.; Nakagawa, A.; Yamashita, E. Structures of the Wild-Type MexAB-OprM Tripartite Pump Reveal Its Complex Formation and Drug Efflux Mechanism. Nat. Commun. 2019, 10, 1520. [Google Scholar] [CrossRef] [PubMed]
- Lamut, A.; Peterlin Mašič, L.; Kikelj, D.; Tomašič, T. Efflux Pump Inhibitors of Clinically Relevant Multidrug Resistant Bacteria. Med. Res. Rev. 2019, 39, 2460–2504. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Adamiak, J.W.; Leus, I.V. Making Sense of Drug-Efflux Transporters in the Physiological Environment. Curr. Opin. Microbiol. 2022, 69, 102179. [Google Scholar] [CrossRef]
- Hernando-Amado, S.; Blanco, P.; Alcalde-Rico, M.; Corona, F.; Reales-Calderón, J.A.; Sánchez, M.B.; Martínez, J.L. Multidrug Efflux Pumps as Main Players in Intrinsic and Acquired Resistance to Antimicrobials. Drug Resist. Updates 2016, 28, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Tzakas, P.; Buckley, A.; Coldham, N.C.; Piddock, L.J.V. Ciprofloxacin-Resistant Salmonella Enterica Serovar Typhimurium Strains Are Difficult To Select in the Absence of AcrB and TolC. Antimicrob. Agents Chemother. 2006, 50, 38–42. [Google Scholar] [CrossRef]
- Nolivos, S.; Cayron, J.; Dedieu, A.; Page, A.; Delolme, F.; Lesterlin, C. Role of AcrAB-TolC Multidrug Efflux Pump in Drug-Resistance Acquisition by Plasmid Transfer. Science 2019, 364, 778–782. [Google Scholar] [CrossRef]
- Pu, Y.; Zhao, Z.; Li, Y.; Zou, J.; Ma, Q.; Zhao, Y.; Ke, Y.; Zhu, Y.; Chen, H.; Baker, M.A.B.; et al. Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol. Cell 2016, 62, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.; Passador, L.; Srikumar, R.; Tsang, E.; Nezezon, J.; Poole, K. Influence of the MexAB-OprM Multidrug Efflux System on Quorum Sensing in Pseudomonas Aeruginosa. J. Bacteriol. 1998, 180, 5443–5447. [Google Scholar] [CrossRef] [PubMed]
- Aendekerk, S.; Diggle, S.P.; Song, Z.; Høiby, N.; Cornelis, P.; Williams, P.; Cámara, M. The MexGHI-OpmD Multidrug Efflux Pump Controls Growth, Antibiotic Susceptibility and Virulence in Pseudomonas Aeruginosa via 4-Quinolone-Dependent Cell-to-Cell Communication. Microbiology 2005, 151, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Alav, I.; Sutton, J.M.; Rahman, K.M. Role of Bacterial Efflux Pumps in Biofilm Formation. J. Antimicrob. Chemother. 2018, 73, 2003–2020. [Google Scholar] [CrossRef]
- Sun, J.; Deng, Z.; Yan, A. Bacterial Multidrug Efflux Pumps: Mechanisms, Physiology and Pharmacological Exploitations. Biochem. Biophys. Res. Commun. 2014, 453, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Jesin, J.A.; Stone, T.A.; Mitchell, C.J.; Reading, E.; Deber, C.M. Peptide-Based Approach to Inhibition of the Multidrug Resistance Efflux Pump AcrB. Biochemistry 2020, 59, 3973–3981. [Google Scholar] [CrossRef]
- Abdali, N.; Parks, J.M.; Haynes, K.M.; Chaney, J.L.; Green, A.T.; Wolloscheck, D.; Walker, J.K.; Rybenkov, V.V.; Baudry, J.; Smith, J.C.; et al. Reviving Antibiotics: Efflux Pump Inhibitors That Interact with AcrA, a Membrane Fusion Protein of the AcrAB-TolC Multidrug Efflux Pump. ACS Infect. Dis. 2017, 3, 89–98. [Google Scholar] [CrossRef]
- Chamberland, S.; Hecker, S.J.; Lee, V.J.; Trias, J. Inhibiteurs de pompe d’efflux. WO 1996033285A1, 19 April 1996. [Google Scholar]
- Renau, T.E.; Léger, R.; Flamme, E.M.; Sangalang, J.; She, M.W.; Yen, R.; Gannon, C.L.; Griffith, D.; Chamberland, S.; Lomovskaya, O.; et al. Inhibitors of Efflux Pumps in Pseudomonas a Eruginosa Potentiate the Activity of the Fluoroquinolone Antibacterial Levofloxacin. J. Med. Chem. 1999, 42, 4928–4931. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Bostian, K.A. Practical Applications and Feasibility of Efflux Pump Inhibitors in the Clinic—A Vision for Applied Use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and Characterization of Inhibitors of Multidrug Resistance Efflux Pumps in Pseudomonas Aeruginosa: Novel Agents for Combination Therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Renau, T.E.; Gannon, C.L.; Mathias, K.M.; Lomovskaya, O.; Chamberland, S.; Lee, V.J.; Ohta, T.; Nakayama, K.; Ishida, Y. Addressing the Stability of C-Capped Dipeptide E‚ux Pump Inhibitors That Potentiate the Activity of Levofloxacin In. Bioorg. Med. Chem. Lett. 2001, 5, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Renau, T.E.; Léger, R.; Filonova, L.; Flamme, E.M.; Wang, M.; Yen, R.; Madsen, D.; Griffith, D.; Chamberland, S.; Dudley, M.N.; et al. Conformationally-Restricted Analogues of Efflux Pump Inhibitors That Potentiate the Activity of Levofloxacin in Pseudomonas Aeruginosa. Bioorganic Med. Chem. Lett. 2003, 13, 2755–2758. [Google Scholar] [CrossRef]
- Léger, R.; Yen, R.; She, M.W.; Lee, V.J.; Hecker, S.J. N-Linked Solid Phase Peptide Synthesis. Tetrahedron Lett. 1998, 39, 4171–4174. [Google Scholar] [CrossRef]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The Efflux Inhibitor Phenylalanine-Arginine Beta-Naphthylamide (PAβN) Permeabilizes the Outer Membrane of Gram-Negative Bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef]
- Lin, K.-H.; Lo, C.-C.; Chou, M.-C.; Yeh, T.-H.; Chen, K.-L.; Liao, W.-Y.; Lo, H.-R. Synergistic Actions of Benzyl Isothiocyanate with Ethylenediaminetetraacetic Acid and Efflux Pump Inhibitor Phenylalanine-Arginine β-Naphthylamide Against Multidrug-Resistant Escherichia Coli. Microb. Drug Resist. 2020, 26, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Cordova, J.; Kumar, A. Activity of the Efflux Pump Inhibitor Phenylalanine-Arginine β-Naphthylamide against the AdeFGH Pump of Acinetobacter Baumannii. Int. J. Antimicrob. Agents 2011, 37, 420–424. [Google Scholar] [CrossRef]
- Watkins, W.J.; Landaverry, Y.; Léger, R.; Litman, R.; Renau, T.E.; Williams, N.; Yen, R.; Zhang, J.Z.; Chamberland, S.; Madsen, D.; et al. The Relationship between Physicochemical Properties, In Vitro Activity and Pharmacokinetic Profiles of Analogues of Diamine-Containing Efflux Pump Inhibitors. Bioorganic Med. Chem. Lett. 2003, 13, 4241–4244. [Google Scholar] [CrossRef]
- Bohnert, J.A.; Kern, W.V. Selected Arylpiperazines Are Capable of Reversing Multidrug Resistance in Escherichia Coli Overexpressing RND Efflux Pumps. Antimicrob. Agents Chemother. 2005, 49, 849–852. [Google Scholar] [CrossRef]
- Chupak, L.S.; Ding, M.; Gentles, R.G.; Huang, Y.; Martin, S.W.; Mcdonald, I.M.; Mercer, S.E.; Olson, R.E.; Velaparthi, U.; Wichroski, M.; et al. Naphthyridinone Compounds Useful as t Cell Activators. WO 2020006016A1, 2 January 2020. [Google Scholar]
- Kern, W.V.; Steinke, P.; Schumacher, A.; Schuster, S.; Baum, H.; von Bohnert, J.A. Effect of 1-(1-Naphthylmethyl)-Piperazine, a Novel Putative Efflux Pump Inhibitor, on Antimicrobial Drug Susceptibility in Clinical Isolates of Escherichia Coli. J. Antimicrob. Chemother. 2006, 57, 339–343. [Google Scholar] [CrossRef]
- Schumacher, A.; Steinke, P.; Bohnert, J.A.; Akova, M.; Jonas, D.; Kern, W.V. Effect of 1-(1-Naphthylmethyl)-Piperazine, a Novel Putative Efflux Pump Inhibitor, on Antimicrobial Drug Susceptibility in Clinical Isolates of Enterobacteriaceae Other than Escherichia Coli. J. Antimicrob. Chemother. 2006, 57, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Pannek, S.; Higgins, P.G.; Steinke, P.; Jonas, D.; Akova, M.; Bohnert, J.A.; Seifert, H.; Kern, W.V. Multidrug Efflux Inhibition in Acinetobacter Baumannii: Comparison between 1-(1-Naphthylmethyl)-Piperazine and Phenyl-Arginine-β-Naphthylamide. J. Antimicrob. Chemother. 2006, 57, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Bolla, J.-M.; Alibert-Franco, S.; Handzlik, J.; Chevalier, J.; Mahamoud, A.; Boyer, G.; Kieć-Kononowicz, K.; Pagès, J.-M. Strategies for Bypassing the Membrane Barrier in Multidrug Resistant Gram-Negative Bacteria. FEBS Lett. 2011, 585, 1682–1690. [Google Scholar] [CrossRef]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Hayashi, K.; Nagata, C.; Hoshino, K.; Onodera, Y.; Nishino, K.; Yamaguchi, A. Structural Basis for the Inhibition of Bacterial Multidrug Exporters. Nature 2013, 500, 102–106. [Google Scholar] [CrossRef]
- Nakayama, K.; Ishida, Y.; Ohtsuka, M.; Kawato, H.; Yoshida, K.; Yokomizo, Y.; Hosono, S.; Ohta, T.; Hoshino, K.; Ishida, H.; et al. MexAB-OprM-Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 1: Discovery and Early Strategies for Lead Optimization. Bioorganic Med. Chem. Lett. 2003, 13, 4201–4204. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Ishida, Y.; Ohtsuka, M.; Kawato, H.; Yoshida, K.; Yokomizo, Y.; Ohta, T.; Hoshino, K.; Otani, T.; Kurosaka, Y.; et al. MexAB-OprM Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 2: Achieving Activity in vivo through the Use of Alternative Scaffolds. Bioorganic Med. Chem. Lett. 2003, 13, 4205–4208. [Google Scholar] [CrossRef]
- Nakayama, K.; Kawato, H.; Watanabe, J.; Ohtsuka, M.; Yoshida, K.; Yokomizo, Y.; Sakamoto, A.; Kuru, N.; Ohta, T.; Hoshino, K.; et al. MexAB-OprM Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 3: Optimization of Potency in the Pyridopyrimidine Series through the Application of a Pharmacophore Model. Bioorganic Med. Chem. Lett. 2004, 14, 475–479. [Google Scholar] [CrossRef]
- Nakayama, K.; Kuru, N.; Ohtsuka, M.; Yokomizo, Y.; Sakamoto, A.; Kawato, H.; Yoshida, K.; Ohta, T.; Hoshino, K.; Akimoto, K.; et al. MexAB-OprM Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 4: Addressing the Problem of Poor Stability Due to Photoisomerization of an Acrylic Acid Moiety. Bioorganic Med. Chem. Lett. 2004, 14, 2493–2497. [Google Scholar] [CrossRef]
- Yoshida, K.; Nakayama, K.; Kuru, N.; Kobayashi, S.; Ohtsuka, M.; Takemura, M.; Hoshino, K.; Kanda, H.; Zhang, J.Z.; Lee, V.J.; et al. MexAB-OprM Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 5: Carbon-Substituted Analogues at the C-2 Position. Bioorganic Med. Chem. 2006, 14, 1993–2004. [Google Scholar] [CrossRef]
- Yoshida, K.; Nakayama, K.; Yokomizo, Y.; Ohtsuka, M.; Takemura, M.; Hoshino, K.; Kanda, H.; Namba, K.; Nitanai, H.; Zhang, J.Z.; et al. MexAB-OprM Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 6: Exploration of Aromatic Substituents. Bioorganic Med. Chem. 2006, 14, 8506–8518. [Google Scholar] [CrossRef]
- Yoshida, K.; Nakayama, K.; Ohtsuka, M.; Kuru, N.; Yokomizo, Y.; Sakamoto, A.; Takemura, M.; Hoshino, K.; Kanda, H.; Nitanai, H.; et al. MexAB-OprM Specific Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Part 7: Highly Soluble and in vivo Active Quaternary Ammonium Analogue D13-9001, a Potential Preclinical Candidate. Bioorganic Med. Chem. 2007, 15, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Rathi, E.; Kumar, A.; Kini, S.G. Computational Approaches in Efflux Pump Inhibitors: Current Status and Prospects. Drug Discov. Today 2020, 25, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Kwasny, S.M.; Kim, H.-S.; Nguyen, S.T.; Houseweart, C.; D’Souza, S.; Walker, G.C.; Peet, N.P.; Nikaido, H.; Bowlin, T.L. Characterization of a Novel Pyranopyridine Inhibitor of the AcrAB Efflux Pump of Escherichia Coli. Antimicrob. Agents Chemother. 2014, 58, 722–733. [Google Scholar] [CrossRef]
- Nguyen, S.T.; Kwasny, S.M.; Ding, X.; Cardinale, S.C.; McCarthy, C.T.; Kim, H.-S.; Nikaido, H.; Peet, N.P.; Williams, J.D.; Bowlin, T.L.; et al. Structure–Activity Relationships of a Novel Pyranopyridine Series of Gram-Negative Bacterial Efflux Pump Inhibitors. Bioorganic Med. Chem. 2015, 23, 2024–2034. [Google Scholar] [CrossRef]
- Sjuts, H.; Vargiu, A.V.; Kwasny, S.M.; Nguyen, S.T.; Kim, H.-S.; Ding, X.; Ornik, A.R.; Ruggerone, P.; Bowlin, T.L.; Nikaido, H.; et al. Molecular Basis for Inhibition of AcrB Multidrug Efflux Pump by Novel and Powerful Pyranopyridine Derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, 3509–3514. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Kwasny, S.M.; Barber, R.; Cardinale, S.C.; Carter, K.; Pulse, M.; Weiss, W.J.; Bowlin, T.L.; Aron, Z. In Vivo Proof of Principle for MBX-4191, A Pyranopyridine Efflux Pump Inhibitor. Poster 564 presented at the American Society of Microbiology (ASM) Microbe 2018 Conference, Atlanta, Georgia, 7–11 June 2018. [Google Scholar]
- Moir, D.T.; Opperman, T.J.; Aron, Z.D.; Bowlin, T.L. Adjunctive Therapy for Multidrug-Resistant Bacterial Infections: Type III Secretion System and Efflux Inhibitors. Drug Discov. Today 2021, 26, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Alenazy, R.; Gu, X.; Polyak, S.W.; Zhang, P.; Sykes, M.J.; Zhang, N.; Venter, H.; Ma, S. Design and Structural Optimization of Novel 2H-Benzo[h]Chromene Derivatives That Target AcrB and Reverse Bacterial Multidrug Resistance. Eur. J. Med. Chem. 2021, 213, 113049. [Google Scholar] [CrossRef]
- Wang, Y.; Mowla, R.; Guo, L.; Ogunniyi, A.D.; Rahman, T.; De Barros Lopes, M.A.; Ma, S.; Venter, H. Evaluation of a Series of 2-Napthamide Derivatives as Inhibitors of the Drug Efflux Pump AcrB for the Reversal of Antimicrobial Resistance. Bioorganic Med. Chem. Lett. 2017, 27, 733–739. [Google Scholar] [CrossRef]
- Wang, Y.; Mowla, R.; Ji, S.; Guo, L.; De Barros Lopes, M.A.; Jin, C.; Song, D.; Ma, S.; Venter, H. Design, Synthesis and Biological Activity Evaluation of Novel 4-Subtituted 2-Naphthamide Derivatives as AcrB Inhibitors. Eur. J. Med. Chem. 2018, 143, 699–709. [Google Scholar] [CrossRef]
- Jin, C.; Alenazy, R.; Wang, Y.; Mowla, R.; Qin, Y.; Tan, J.Q.E.; Modi, N.D.; Gu, X.; Polyak, S.W.; Venter, H.; et al. Design, Synthesis and Evaluation of a Series of 5-Methoxy-2,3-Naphthalimide Derivatives as AcrB Inhibitors for the Reversal of Bacterial Resistance. Bioorganic Med. Chem. Lett. 2019, 29, 882–889. [Google Scholar] [CrossRef]
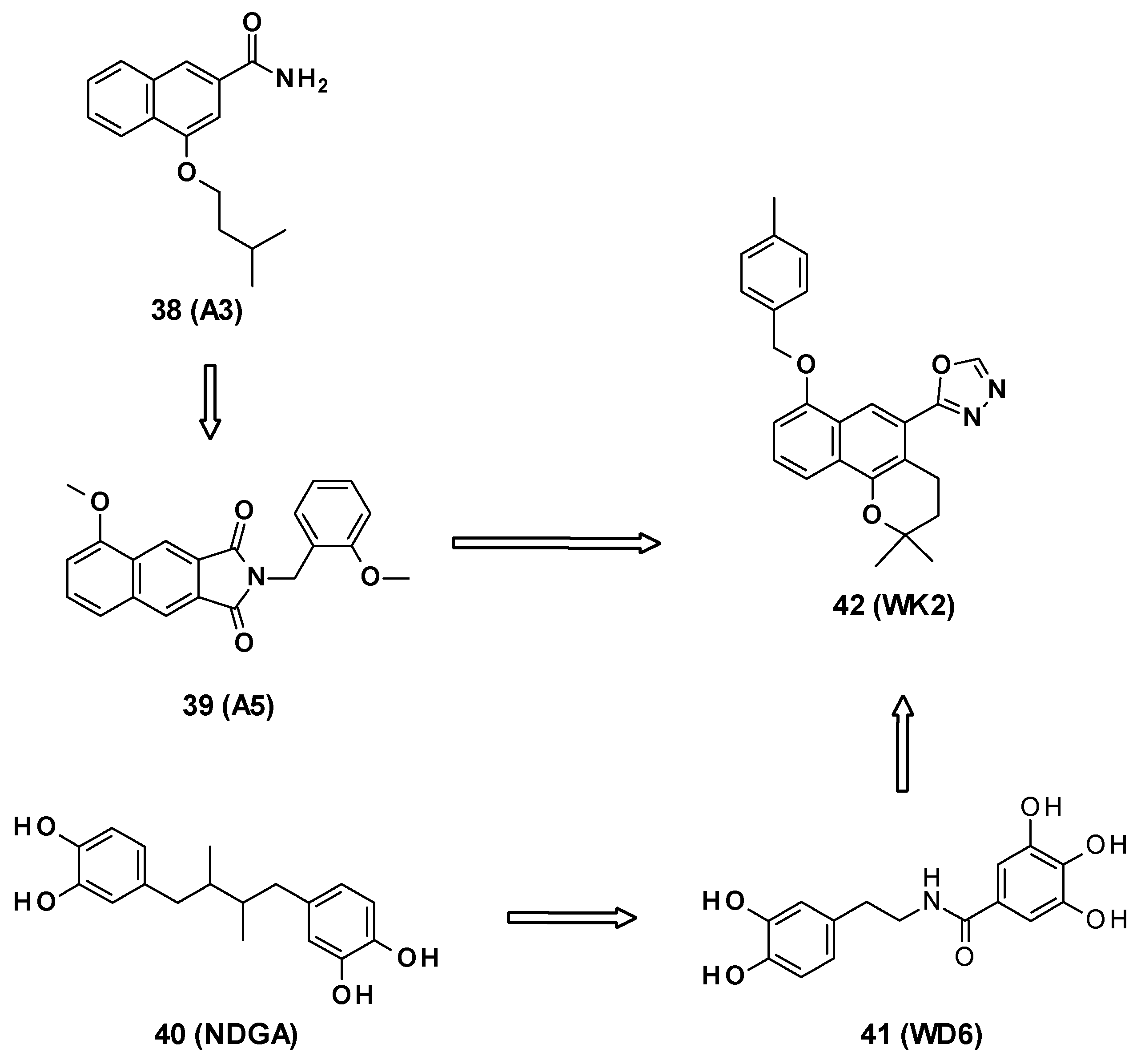
- Wang, Y.; Alenzy, R.; Song, D.; Liu, X.; Teng, Y.; Mowla, R.; Ma, Y.; Polyak, S.W.; Venter, H.; Ma, S. Structural Optimization of Natural Product Nordihydroguaretic Acid to Discover Novel Analogues as AcrB Inhibitors. Eur. J. Med. Chem. 2020, 186, 111910. [Google Scholar] [CrossRef] [PubMed]
- Ohene-Agyei, T.; Mowla, R.; Rahman, T.; Venter, H. Phytochemicals Increase the Antibacterial Activity of Antibiotics by Acting on a Drug Efflux Pump. MicrobiologyOpen 2014, 3, 885–896. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.M. Identification of Binding Sites for Efflux Pump Inhibitors of the AcrAB-TolC Component AcrA. Biophys. J. 2019, 116, 648–658. [Google Scholar] [CrossRef]
- D’Cunha, N.; Moniruzzaman, M.; Haynes, K.; Malloci, G.; Cooper, C.J.; Margiotta, E.; Vargiu, A.V.; Uddin, M.R.; Leus, I.V.; Cao, F.; et al. Mechanistic Duality of Bacterial Efflux Substrates and Inhibitors: Example of Simple Substituted Cinnamoyl and Naphthyl Amides. ACS Infect. Dis. 2021, 7, 2650–2665. [Google Scholar] [CrossRef]
- Blankson, G.; Parhi, A.K.; Kaul, M.; Pilch, D.S.; LaVoie, E.J. Structure-Activity Relationships of Potentiators of the Antibiotic Activity of Clarithromycin against Escherichia Coli. Eur. J. Med. Chem. 2019, 178, 30–38. [Google Scholar] [CrossRef]
- Yuan, Y.; Rosado-Lugo, J.D.; Zhang, Y.; Datta, P.; Sun, Y.; Cao, Y.; Banerjee, A.; Parhi, A.K. Evaluation of Heterocyclic Carboxamides as Potential Efflux Pump Inhibitors in Pseudomonas Aeruginosa. Antibiotics 2021, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rosado-Lugo, J.D.; Datta, P.; Sun, Y.; Cao, Y.; Banerjee, A.; Yuan, Y.; Parhi, A.K. Evaluation of a Conformationally Constrained Indole Carboxamide as a Potential Efflux Pump Inhibitor in Pseudomonas Aeruginosa. Antibiotics 2022, 11, 716. [Google Scholar] [CrossRef] [PubMed]
- Dötsch, A.; Becker, T.; Pommerenke, C.; Magnowska, Z.; Jänsch, L.; Häussler, S. Genomewide Identification of Genetic Determinants of Antimicrobial Drug Resistance in Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2009, 53, 2522–2531. [Google Scholar] [CrossRef] [PubMed]
- Hirvas, L.; Coleman, J.; Koski, P.; Vaara, M. Bacterial ‘Histone-like Protein I’ (HLP-I) Is an Outer Membrane Constituent? FEBS Lett. 1990, 262, 123–126. [Google Scholar] [CrossRef]
- Lavoie, E.J.; Parhi, A.; Yuan, Y.; Zhang, Y.; SUN, Y. Indole Derivatives as Efflux Pump Inhibitors. WO 2018165611A1, 13 September 2018. [Google Scholar]
- Plé, C.; Tam, H.-K.; Vieira Da Cruz, A.; Compagne, N.; Jiménez-Castellanos, J.-C.; Müller, R.T.; Pradel, E.; Foong, W.E.; Malloci, G.; Ballée, A.; et al. Pyridylpiperazine-Based Allosteric Inhibitors of RND-Type Multidrug Efflux Pumps. Nat. Commun. 2022, 13, 115. [Google Scholar] [CrossRef]
- Teelucksingh, T.; Thompson, L.K.; Zhu, S.; Kuehfuss, N.M.; Goetz, J.A.; Gilbert, S.E.; MacNair, C.R.; Geddes-McAlister, J.; Brown, E.D.; Cox, G. A Genetic Platform to Investigate the Functions of Bacterial Drug Efflux Pumps. Nat. Chem. Biol. 2022, 18, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Brandstätter, L.; Sokolova, L.; Eicher, T.; Seeger, M.A.; Briand, C.; Cha, H.; Cernescu, M.; Bohnert, J.; Kern, W.V.; Brutschy, B.; et al. Analysis of AcrB and AcrB/DARPin Ligand Complexes by LILBID MS. Biochim. Et Biophys. Acta (BBA)—Biomembr. 2011, 1808, 2189–2196. [Google Scholar] [CrossRef]
- Yu, L.; Lu, W.; Wei, Y. AcrB Trimer Stability and Efflux Activity, Insight from Mutagenesis Studies. PLoS ONE 2011, 6, e28390. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.A.; Schiefner, A.; Eicher, T.; Verrey, F.; Diederichs, K.; Pos, K.M. Structural Asymmetry of AcrB Trimer Suggests a Peristaltic Pump Mechanism. Science 2006, 313, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Matsumoto, T.; Yamaguchi, A. Crystal Structures of a Multidrug Transporter Reveal a Functionally Rotating Mechanism. Nature 2006, 443, 173–179. [Google Scholar] [CrossRef]
- Sennhauser, G.; Amstutz, P.; Briand, C.; Storchenegger, O.; Grütter, M.G. Drug Export Pathway of Multidrug Exporter AcrB Revealed by DARPin Inhibitors. PLoS Biol. 2006, 5, e7. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.A.; von Ballmoos, C.; Eicher, T.; Brandstätter, L.; Verrey, F.; Diederichs, K.; Pos, K.M. Engineered Disulfide Bonds Support the Functional Rotation Mechanism of Multidrug Efflux Pump AcrB. Nat. Struct. Mol. Biol. 2008, 15, 199–205. [Google Scholar] [CrossRef]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Nishino, K.; Yamaguchi, A. Structures of the Multidrug Exporter AcrB Reveal a Proximal Multisite Drug-Binding Pocket. Nature 2011, 480, 565–569. [Google Scholar] [CrossRef]
- Eicher, T.; Cha, H.; Seeger, M.A.; Brandstätter, L.; El-Delik, J.; Bohnert, J.A.; Kern, W.V.; Verrey, F.; Grütter, M.G.; Diederichs, K.; et al. Transport of Drugs by the Multidrug Transporter AcrB Involves an Access and a Deep Binding Pocket That Are Separated by a Switch-Loop. Proc. Natl. Acad. Sci. USA 2012, 109, 5687–5692. [Google Scholar] [CrossRef]
- Tam, H.-K.; Foong, W.E.; Oswald, C.; Herrmann, A.; Zeng, H.; Pos, K.M. Allosteric Drug Transport Mechanism of Multidrug Transporter AcrB. Nat. Commun. 2021, 12, 3889. [Google Scholar] [CrossRef]
- Zwama, M.; Yamasaki, S.; Nakashima, R.; Sakurai, K.; Nishino, K.; Yamaguchi, A. Multiple Entry Pathways within the Efflux Transporter AcrB Contribute to Multidrug Recognition. Nat. Commun. 2018, 9, 124. [Google Scholar] [CrossRef]
- Tam, H.-K.; Malviya, V.N.; Foong, W.-E.; Herrmann, A.; Malloci, G.; Ruggerone, P.; Vargiu, A.V.; Pos, K.M. Binding and Transport of Carboxylated Drugs by the Multidrug Transporter AcrB. J. Mol. Biol. 2020, 432, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Eicher, T.; Seeger, M.A.; Anselmi, C.; Zhou, W.; Brandstätter, L.; Verrey, F.; Diederichs, K.; Faraldo-Gómez, J.D.; Pos, K.M. Coupling of Remote Alternating-Access Transport Mechanisms for Protons and Substrates in the Multidrug Efflux Pump AcrB. Elife 2014, 3, e03145. [Google Scholar] [CrossRef]
- Yue, Z.; Chen, W.; Zgurskaya, H.I.; Shen, J. Constant PH Molecular Dynamics Reveals How Proton Release Drives the Conformational Transition of a Transmembrane Efflux Pump. J. Chem. Theory Comput. 2017, 13, 6405–6414. [Google Scholar] [CrossRef]
- Fischer, N.; Kandt, C. Porter Domain Opening and Closing Motions in the Multi-Drug Efflux Transporter AcrB. Biochim. Et Biophys. Acta (BBA)—Biomembr. 2013, 1828, 632–641. [Google Scholar] [CrossRef]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal Structure of Bacterial Multidrug Efflux Transporter AcrB. Nature 2002, 419, 587–593. [Google Scholar] [CrossRef]
- Cha, H.; Müller, R.T.; Pos, K.M. Switch-Loop Flexibility Affects Transport of Large Drugs by the Promiscuous AcrB Multidrug Efflux Transporter. Antimicrob. Agents Chemother. 2014, 58, 4767–4772. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Tamura, N.; van Veen, H.W.; Yamaguchi, A.; Murakami, S. β-Lactam Selectivity of Multidrug Transporters AcrB and AcrD Resides in the Proximal Binding Pocket. J. Biol. Chem. 2014, 289, 10680–10690. [Google Scholar] [CrossRef] [PubMed]
- Elkins, C.A.; Nikaido, H. Substrate Specificity of the RND-Type Multidrug Efflux Pumps AcrB and AcrD of Escherichia coli Is Determined Predominately by Two Large Periplasmic Loops. J. Bacteriol. 2002, 184, 6490–6498. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multidrug Efflux Pumps of Gram-Negative Bacteria. J. Bacteriol. 1996, 178, 5853–5859. [Google Scholar] [CrossRef]
- Pos, K.M. Drug Transport Mechanism of the AcrB Efflux Pump. Biochim. Biophys. Acta 2009, 1794, 782–793. [Google Scholar] [CrossRef]
- Zwama, M.; Nishino, K. Ever-Adapting RND Efflux Pumps in Gram-Negative Multidrug-Resistant Pathogens: A Race against Time. Antibiotics 2021, 10, 774. [Google Scholar] [CrossRef] [PubMed]
- Ornik-Cha, A.; Wilhelm, J.; Kobylka, J.; Sjuts, H.; Vargiu, A.V.; Malloci, G.; Reitz, J.; Seybert, A.; Frangakis, A.S.; Pos, K.M. Structural and Functional Analysis of the Promiscuous AcrB and AdeB Efflux Pumps Suggests Different Drug Binding Mechanisms. Nat. Commun. 2021, 12, 6919. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Vavra, M.; Kern, W.V. Evidence of a Substrate-Discriminating Entrance Channel in the Lower Porter Domain of the Multidrug Resistance Efflux Pump AcrB. Antimicrob. Agents Chemother. 2016, 60, 4315–4323. [Google Scholar] [CrossRef]
- Ababou, A.; Koronakis, V. Structures of Gate Loop Variants of the AcrB Drug Efflux Pump Bound by Erythromycin Substrate. PLoS ONE 2016, 11, e0159154. [Google Scholar] [CrossRef]
- Oswald, C.; Tam, H.-K.; Pos, K.M. Transport of Lipophilic Carboxylates Is Mediated by Transmembrane Helix 2 in Multidrug Transporter AcrB. Nat. Commun. 2016, 7, 13819. [Google Scholar] [CrossRef]
- Reading, E.; Ahdash, Z.; Fais, C.; Ricci, V.; Wang-Kan, X.; Grimsey, E.; Stone, J.; Malloci, G.; Lau, A.M.; Findlay, H.; et al. Perturbed Structural Dynamics Underlie Inhibition and Altered Efflux of the Multidrug Resistance Pump AcrB. Nat. Commun. 2020, 11, 5565. [Google Scholar] [CrossRef]
- Takatsuka, Y.; Chen, C.; Nikaido, H. Mechanism of Recognition of Compounds of Diverse Structures by the Multidrug Efflux Pump AcrB of Escherichia Coli. Proc. Natl. Acad. Sci. USA 2010, 107, 6559–6565. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Nikaido, H. Multidrug Binding Properties of the AcrB Efflux Pump Characterized by Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 20637–20642. [Google Scholar] [CrossRef]
- Aparna, V.; Dineshkumar, K.; Mohanalakshmi, N.; Velmurugan, D.; Hopper, W. Identification of Natural Compound Inhibitors for Multidrug Efflux Pumps of Escherichia Coli and Pseudomonas Aeruginosa Using In Silico High-Throughput Virtual Screening and In Vitro Validation. PLoS ONE 2014, 9, e101840. [Google Scholar] [CrossRef]
- Kinana, A.D.; Vargiu, A.V.; May, T.; Nikaido, H. Aminoacyl β-Naphthylamides as Substrates and Modulators of AcrB Multidrug Efflux Pump. Proc. Natl. Acad. Sci. USA 2016, 113, 1405–1410. [Google Scholar] [CrossRef]
- Zuo, Z.; Weng, J.; Wang, W. Insights into the Inhibitory Mechanism of D13-9001 to the Multidrug Transporter AcrB through Molecular Dynamics Simulations. J. Phys. Chem. B 2016, 120, 2145–2154. [Google Scholar] [CrossRef]
- Dey, D.; Kavanaugh, L.G.; Conn, G.L. Antibiotic Substrate Selectivity of Pseudomonas Aeruginosa MexY and MexB Efflux Systems Is Determined by a Goldilocks Affinity. Antimicrob. Agents Chemother. 2020, 64, e00496-e20. [Google Scholar] [CrossRef]
- Bohnert, J.A.; Schuster, S.; Seeger, M.A.; Fähnrich, E.; Pos, K.M.; Kern, W.V. Site-Directed Mutagenesis Reveals Putative Substrate Binding Residues in the Escherichia Coli RND Efflux Pump AcrB. J. Bacteriol. 2008, 190, 8225–8229. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Nakashima, K.; Nishino, K.; Kotani, K.; Tomida, J.; Inoue, M.; Kawamura, Y. Berberine Is a Novel Type Efflux Inhibitor Which Attenuates the MexXY-Mediated Aminoglycoside Resistance in Pseudomonas Aeruginosa. Front. Microbiol. 2016, 7, 1223. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Nakae, T. Identification of Essential Charged Residues in Transmembrane Segments of the Multidrug Transporter MexB of Pseudomonas Aeruginosa. J. Bacteriol. 2001, 183, 1734–1739. [Google Scholar] [CrossRef]
- Seeger, M.A.; von Ballmoos, C.; Verrey, F.; Pos, K.M. Crucial Role of Asp408 in the Proton Translocation Pathway of Multidrug Transporter AcrB: Evidence from Site-Directed Mutagenesis and Carbodiimide Labeling. Biochemistry 2009, 48, 5801–5812. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, Y.; Nikaido, H. Threonine-978 in the Transmembrane Segment of the Multidrug Efflux Pump AcrB of Escherichia Coli Is Crucial for Drug Transport as a Probable Component of the Proton Relay Network. J. Bacteriol. 2006, 188, 7284–7289. [Google Scholar] [CrossRef]
- Zhang, B.; Li, J.; Yang, X.; Wu, L.; Zhang, J.; Yang, Y.; Zhao, Y.; Zhang, L.; Yang, X.; Yang, X.; et al. Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell 2019, 176, 636–648. [Google Scholar] [CrossRef]
- Yang, X.; Hu, T.; Yang, X.; Xu, W.; Yang, H.; Guddat, L.W.; Zhang, B.; Rao, Z. Structural Basis for the Inhibition of Mycobacterial MmpL3 by NITD-349 and SPIRO. J. Mol. Biol. 2020, 432, 4426–4434. [Google Scholar] [CrossRef]
- Schuster, S.; Kohler, S.; Buck, A.; Dambacher, C.; König, A.; Bohnert, J.A.; Kern, W.V. Random Mutagenesis of the Multidrug Transporter AcrB from Escherichia Coli for Identification of Putative Target Residues of Efflux Pump Inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6870–6878. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, J.M.; Yazidi, A.; Yang, L.; Fabre, L.; Li, W.; Jacques, B.; Angala, S.k.; Rouiller, I.; Zgurskaya, H.I.; Sygusch, J.; et al. Structure–Function Profile of MmpL3, the Essential Mycolic Acid Transporter from Mycobacterium Tuberculosis. ACS Infect. Dis. 2016, 2, 702–713. [Google Scholar] [CrossRef] [PubMed]






















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Compagne, N.; Vieira Da Cruz, A.; Müller, R.T.; Hartkoorn, R.C.; Flipo, M.; Pos, K.M. Update on the Discovery of Efflux Pump Inhibitors against Critical Priority Gram-Negative Bacteria. Antibiotics 2023, 12, 180. https://doi.org/10.3390/antibiotics12010180
Compagne N, Vieira Da Cruz A, Müller RT, Hartkoorn RC, Flipo M, Pos KM. Update on the Discovery of Efflux Pump Inhibitors against Critical Priority Gram-Negative Bacteria. Antibiotics. 2023; 12(1):180. https://doi.org/10.3390/antibiotics12010180
Chicago/Turabian StyleCompagne, Nina, Anais Vieira Da Cruz, Reinke T. Müller, Ruben C. Hartkoorn, Marion Flipo, and Klaas M. Pos. 2023. "Update on the Discovery of Efflux Pump Inhibitors against Critical Priority Gram-Negative Bacteria" Antibiotics 12, no. 1: 180. https://doi.org/10.3390/antibiotics12010180
APA StyleCompagne, N., Vieira Da Cruz, A., Müller, R. T., Hartkoorn, R. C., Flipo, M., & Pos, K. M. (2023). Update on the Discovery of Efflux Pump Inhibitors against Critical Priority Gram-Negative Bacteria. Antibiotics, 12(1), 180. https://doi.org/10.3390/antibiotics12010180