
Pharmacokinetics of Non-β-Lactam β-Lactamase Inhibitors



Abstract
:1. Introduction
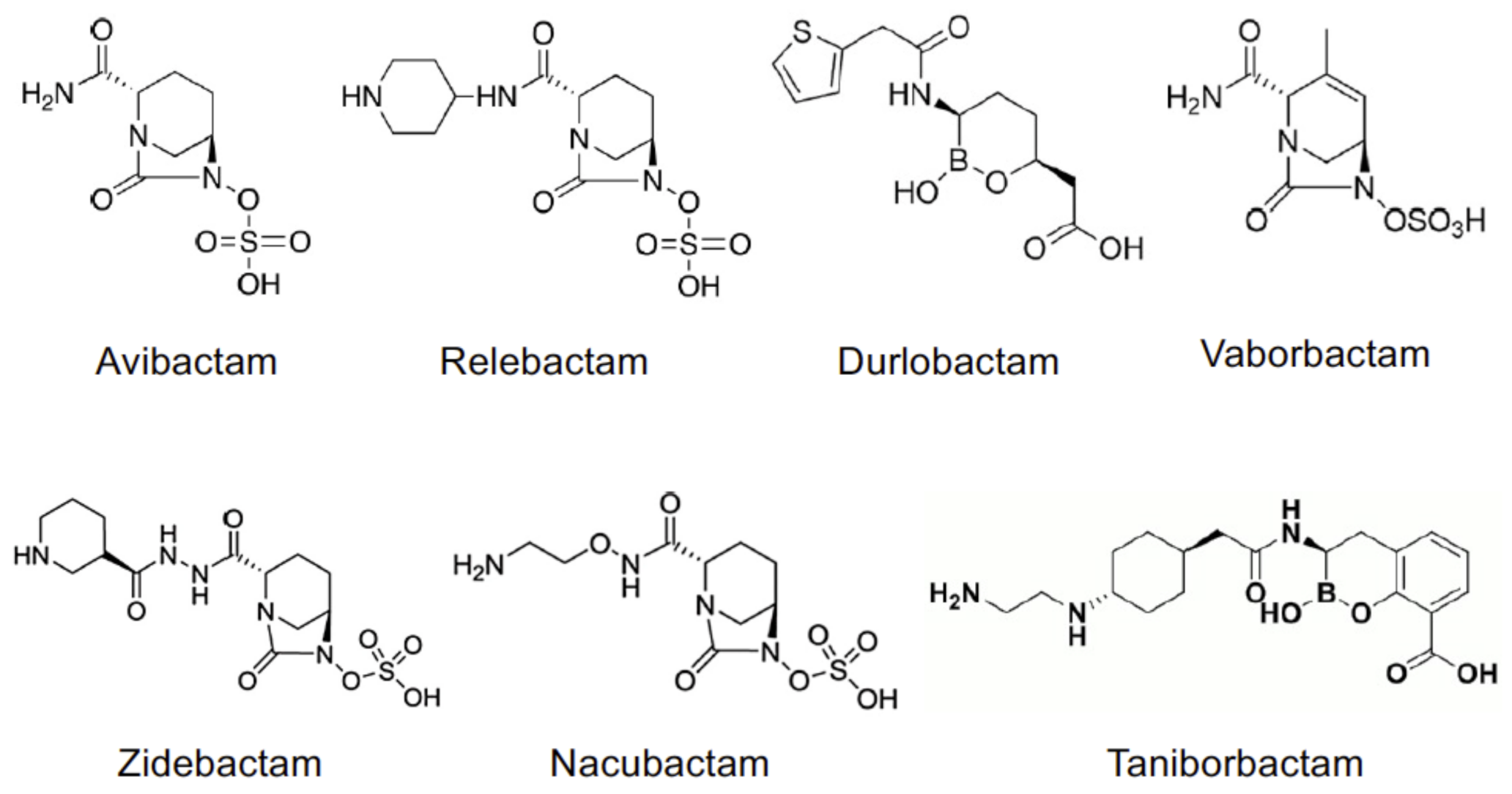
2. Structure and Mechanism of Action
3. Spectrum of Activity of BLIs and Mechanisms of Resistance Structure and Mechanism of Action
4. Pharmacokinetics of BLIs
4.1. Linear Pharmacokinetics
4.2. Distribution
4.3. Biotransformation
4.4. Excretion
5. Pharmacokinetic/Pharmacodynamic Relationships
6. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Determinants of the Activity of Beta-Lactamase Inhibitor Combinations. J. Antimicrob. Chemother. 1993, 31 (Suppl. A), 9–21. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, J.; Melchers, M.J.; van Mil, A.C.; Seyedmousavi, S.; Lagarde, C.M.; Schuck, V.J.; Nichols, W.W.; Mouton, J.W. Pharmacodynamics of Ceftazidime and Avibactam in Neutropenic Mice with Thigh or Lung Infection. Antimicrob. Agents Chemother. 2016, 60, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Aziz, M.H.; Lipman, J.; Akova, M.; Bassetti, M.; De Waele, J.J.; Dimopoulos, G.; Dulhunty, J.; Kaukonen, K.-M.; Koulenti, D.; Martin, C.; et al. Is Prolonged Infusion of Piperacillin/Tazobactam and Meropenem in Critically Ill Patients Associated with Improved Pharmacokinetic/Pharmacodynamic and Patient Outcomes? An Observation from the Defining Antibiotic Levels in Intensive Care Unit Patients (DA. J. Antimicrob. Chemother. 2016, 71, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Vardakas, K.Z.; Voulgaris, G.L.; Maliaros, A.; Samonis, G.; Falagas, M.E. Prolonged versus Short-Term Intravenous Infusion of Antipseudomonal β-Lactams for Patients with Sepsis: A Systematic Review and Meta-Analysis of Randomised Trials. Lancet Infect. Dis. 2018, 18, 108–120. [Google Scholar] [CrossRef]
- Lister, P.D.; Prevan, A.M.; Sanders, C.C. Importance of Beta-Lactamase Inhibitor Pharmacokinetics in the Pharmacodynamics of Inhibitor-Drug Combinations: Studies with Piperacillin-Tazobactam and Piperacillin-Sulbactam. Antimicrob. Agents Chemother. 1997, 41, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Vanscoy, B.; Mendes, R.E.; McCauley, J.; Bhavnani, S.M.; Bulik, C.C.; Okusanya, O.O.; Forrest, A.; Jones, R.N.; Friedrich, L.V.; Steenbergen, J.N.; et al. Pharmacological Basis of β-Lactamase Inhibitor Therapeutics: Tazobactam in Combination with Ceftolozane. Antimicrob. Agents Chemother. 2013, 57, 5924–5930. [Google Scholar] [CrossRef] [Green Version]
- Veiga, R.P.; Paiva, J.-A. Pharmacokinetics-Pharmacodynamics Issues Relevant for the Clinical Use of Beta-Lactam Antibiotics in Critically Ill Patients. Crit. Care 2018, 22, 233. [Google Scholar] [CrossRef] [Green Version]
- Falcone, M.; Daikos, G.L.; Tiseo, G.; Bassoulis, D.; Giordano, C.; Galfo, V.; Leonildi, A.; Tagliaferri, E.; Barnini, S.; Sani, S.; et al. Efficacy of Ceftazidime-Avibactam plus Aztreonam in Patients with Bloodstream Infections Caused by MBL- Producing Enterobacterales. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Interplay between β-Lactamases and New β-Lactamase Inhibitors. Nat. Rev. Microbiol. 2019, 17, 295–306. [Google Scholar] [CrossRef]
- Stachyra, T.; Péchereau, M.-C.; Bruneau, J.-M.; Claudon, M.; Frère, J.-M.; Miossec, C.; Coleman, K.; Black, M.T. Mechanistic Studies of the Inactivation of TEM-1 and P99 by NXL104, a Novel Non-Beta-Lactam Beta-Lactamase Inhibitor. Antimicrob. Agents Chemother. 2010, 54, 5132–5138. [Google Scholar] [CrossRef] [Green Version]
- Morinaka, A.; Tsutsumi, Y.; Yamada, M.; Suzuki, K.; Watanabe, T.; Abe, T.; Furuuchi, T.; Inamura, S.; Sakamaki, Y.; Mitsuhashi, N.; et al. OP0595, a New Diazabicyclooctane: Mode of Action as a Serine β-Lactamase Inhibitor, Antibiotic and β-Lactam “Enhancer”. J. Antimicrob. Chemother. 2015, 70, 2779–2786. [Google Scholar] [CrossRef] [Green Version]
- Moya, B.; Barcelo, I.M.; Bhagwat, S.; Patel, M.; Bou, G.; Papp-Wallace, K.M.; Bonomo, R.A.; Oliver, A. WCK 5107 (Zidebactam) and WCK 5153 Are Novel Inhibitors of PBP2 Showing Potent “β-Lactam Enhancer” Activity against Pseudomonas Aeruginosa, Including Multidrug-Resistant Metallo-β-Lactamase-Producing High-Risk Clones. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Papp-Wallace, K.M.; Nguyen, N.Q.; Jacobs, M.R.; Bethel, C.R.; Barnes, M.D.; Kumar, V.; Bajaksouzian, S.; Rudin, S.D.; Rather, P.N.; Bhavsar, S.; et al. Strategic Approaches to Overcome Resistance against Gram-Negative Pathogens Using β-Lactamase Inhibitors and β-Lactam Enhancers: Activity of Three Novel Diazabicyclooctanes WCK 5153, Zidebactam (WCK 5107), and WCK 4234. J. Med. Chem. 2018, 61, 4067–4086. [Google Scholar] [CrossRef]
- Lepak, A.J.; Zhao, M.; Andes, D.R. WCK 5222 (Cefepime/Zidebactam) Pharmacodynamic Target Analysis against Metallo-β-Lactamase Producing Enterobacteriaceae in the Neutropenic Mouse Pneumonia Model. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef]
- Yahav, D.; Giske, C.G.; Gramatniece, A.; Abodakpi, H.; Tam, V.H.; Leibovici, L. New β-Lactam–β-Lactamase Inhibitor Combinations. Clin. Microbiol. Rev. 2021, 34, 1–61. [Google Scholar] [CrossRef]
- Nukaga, M.; Papp-Wallace, K.M.; Hoshino, T.; Lefurgy, S.T.; Bethel, C.R.; Barnes, M.D.; Zeiser, E.T.; Johnson, J.K.; Bonomo, R.A. Probing the Mechanism of Inactivation of the FOX-4 Cephamycinase by Avibactam. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.R.; Rybak, J.M.; Claeys, K.C. Imipenem-Cilastatin-Relebactam: A Novel β-Lactam-β-Lactamase Inhibitor Combination for the Treatment of Multidrug-Resistant Gram-Negative Infections. Pharmacotherapy 2020, 40, 343–356. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Gao, N.; Jahić, H.; Carter, N.M.; Chen, A.; Miller, A.A. Reversibility of Covalent, Broad-Spectrum Serine β-Lactamase Inhibition by the Diazabicyclooctenone ETX2514. ACS Infect. Dis. 2017, 3, 833–844. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Griffith, D.C.; Dudley, M.N. Vaborbactam: Spectrum of Beta-Lactamase Inhibition and Impact of Resistance Mechanisms on Activity in Enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Trout, R.E.L.; Chu, G.H.; Mcgarry, D.; Jackson, R.W.; Hamrick, J.C.; Daigle, D.M.; Cusick, S.M.; Pozzi, C.; De Luca, F.; et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- And Metallo-β-Lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem. 2020, 63, 2789–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, S.T.; Cain, R.; Wang, D.Y.; Lohans, C.T.; Wareham, D.W.; Oswin, H.P.; Mohammed, J.; Spencer, J.; Fishwick, C.W.G.; McDonough, M.A.; et al. Cyclic Boronates Inhibit All Classes of β-Lactamases. Antimicrob. Agents Chemother. 2017, 61, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crass, R.L.; Pai, M.P. Pharmacokinetics and Pharmacodynamics of β-Lactamase Inhibitors. Pharmacotherapy 2019, 39, 182–195. [Google Scholar] [CrossRef]
- Sheu, C.-C.; Chang, Y.-T.; Lin, S.-Y.; Chen, Y.-H.; Hsueh, P.-R. Infections Caused by Carbapenem-Resistant Enterobacteriaceae: An Update on Therapeutic Options. Front. Microbiol. 2019, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- González-Bello, C.; Rodríguez, D.; Pernas, M.; Rodríguez, Á.; Colchón, E. β-Lactamase Inhibitors To Restore the Efficacy of Antibiotics against Superbugs. J. Med. Chem. 2020, 63, 1859–1881. [Google Scholar] [CrossRef]
- Thomson, K.S.; AbdelGhani, S.; Snyder, J.W.; Thomson, G.K. Activity of Cefepime-Zidebactam against Multidrug-Resistant (MDR) Gram-Negative Pathogens. Antibiotics 2019, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Ucha, J.C.; Arca-Suárez, J.; Bou, G.; Beceiro, A. New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams. Int. J. Mol. Sci. 2020, 21, 9308. [Google Scholar] [CrossRef]
- Krajnc, A.; Brem, J.; Hinchliffe, P.; Calvopiña, K.; Panduwawala, T.D.; Lang, P.A.; Kamps, J.J.A.G.; Tyrrell, J.M.; Widlake, E.; Saward, B.G.; et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-Lactamases. J. Med. Chem. 2019, 62, 8544–8556. [Google Scholar] [CrossRef] [Green Version]
- Bush, K. The ABCD’s of β-Lactamase Nomenclature. J. Infect. Chemother. 2013, 19, 549–559. [Google Scholar] [CrossRef]
- Shahid, M.; Sobia, F.; Singh, A.; Malik, A.; Khan, H.M.; Jonas, D.; Hawkey, P.M. Beta-Lactams and Beta-Lactamase-Inhibitors in Current- or Potential-Clinical Practice: A Comprehensive Update. Crit. Rev. Microbiol. 2009, 35, 81–108. [Google Scholar] [CrossRef]
- Karaiskos, I.; Lagou, S.; Pontikis, K.; Rapti, V.; Poulakou, G. The “Old” and the “New” Antibiotics for MDR Gram-Negative Pathogens: For Whom, When, and How. Front. Public Health 2019, 7, 151. [Google Scholar] [CrossRef] [Green Version]
- Livermore, D.M.; Warner, M.; Mushtaq, S. Activity of MK-7655 Combined with Imipenem against Enterobacteriaceae and Pseudomonas Aeruginosa. J. Antimicrob. Chemother. 2013, 68, 2286–2290. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Lawrence, C.K.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Zhanel, M.; Lagacé-Wiens, P.R.S.; Walkty, A.; Denisuik, A.; Golden, A.; et al. Imipenem-Relebactam and Meropenem-Vaborbactam: Two Novel Carbapenem-β-Lactamase Inhibitor Combinations. Drugs 2018, 78, 65–98. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef]
- Hackel, M.A.; Lomovskaya, O.; Dudley, M.N.; Karlowsky, J.A.; Sahm, D.F. In Vitro Activity of Meropenem-Vaborbactam against Clinical Isolates of KPC-Positive Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Durand-Réville, T.F.; Guler, S.; Comita-Prevoir, J.; Chen, B.; Bifulco, N.; Huynh, H.; Lahiri, S.; Shapiro, A.B.; McLeod, S.M.; Carter, N.M.; et al. ETX2514 Is a Broad-Spectrum β-Lactamase Inhibitor for the Treatment of Drug-Resistant Gram-Negative Bacteria Including Acinetobacter Baumannii. Nat. Microbiol. 2017, 2, 17104. [Google Scholar] [CrossRef]
- Moya, B.; Barcelo, I.M.; Cabot, G.; Torrens, G.; Palwe, S.; Joshi, P.; Umarkar, K.; Takalkar, S.; Periasamy, H.; Bhagwat, S.; et al. In Vitro and In Vivo Activities of β-Lactams in Combination with the Novel β-Lactam Enhancers Zidebactam and WCK 5153 against Multidrug-Resistant Metallo-β-Lactamase-Producing Klebsiella Pneumoniae. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, S.; Vickers, A.; Woodford, N.; Haldimann, A.; Livermore, D.M. Activity of Nacubactam (RG6080/OP0595) Combinations against MBL-Producing Enterobacteriaceae. J. Antimicrob. Chemother. 2019, 74, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Asempa, T.E.; Motos, A.; Abdelraouf, K.; Bissantz, C.; Zampaloni, C.; Nicolau, D.P. Meropenem-Nacubactam Activity against AmpC-Overproducing and KPC-Expressing Pseudomonas Aeruginosa in a Neutropenic Murine Lung Infection Model. Int. J. Antimicrob. Agents 2020, 55, 105838. [Google Scholar] [CrossRef]
- Giddins, M.J.; Macesic, N.; Annavajhala, M.K.; Stump, S.; Khan, S.; McConville, T.H.; Mehta, M.; Gomez-Simmonds, A.; Uhlemann, A.-C. Successive Emergence of Ceftazidime-Avibactam Resistance through Distinct Genomic Adaptations in BlaKPC-2-Harboring Klebsiella Pneumoniae Sequence Type 307 Isolates. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Palzkill, T. Structural and Mechanistic Basis for Extended-Spectrum Drug-Resistance Mutations in Altering the Specificity of TEM, CTX-M, and KPC β-Lactamases. Front. Mol. Biosci. 2018, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.; Nguyen, L.; Trinh, T.; MacDougall, C. Recognizing and Overcoming Resistance to New Beta-Lactam/Beta-Lactamase Inhibitor Combinations. Curr. Infect. Dis. Rep. 2019, 21, 39. [Google Scholar] [CrossRef]
- Nelson, K.; Hemarajata, P.; Sun, D.; Rubio-Aparicio, D.; Tsivkovski, R.; Yang, S.; Sebra, R.; Kasarskis, A.; Nguyen, H.; Hanson, B.M.; et al. Resistance to Ceftazidime-Avibactam Is Due to Transposition of KPC in a Porin-Deficient Strain of Klebsiella Pneumoniae with Increased Efflux Activity. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency (EMA). Summary of Product Characteristics—Ceftazidime/Avibactam—535. Available online: https://www.ema.europa.eu/en/documents/product-information/zavicefta-epar-product-information_en.pdf (accessed on 13 June 2021).
- European Medicines Agency (EMA). Summary of Product Characteristics—Aztreonam—537. Available online: https://www.ema.europa.eu/en/documents/product-information/cayston-epar-product-information_en.pdf (accessed on 13 June 2021).
- Mattie, H. Clinical Pharmacokinetics of Aztreonam. Clin. Pharmacokinet. 1994, 26, 99–106. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Summary of Product Characteristics—Ceftarolin—541. Available online: https://www.ema.europa.eu/en/documents/assessment-report/zinforo-epar-public-assessment-report_en.pdf (accessed on 13 June 2021).
- Patel, T.S.; Pogue, J.M.; Mills, J.P.; Kaye, K.S. Meropenem–Vaborbactam: A New Weapon in the War against Infections Due to Resistant Gram-Negative Bacteria. Future Microbiol. 2018, 13, 971–983. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Summary of Product Characteristics—Vaborbactam/Meropenem—545. Available online: https://www.ema.europa.eu/en/documents/product-information/vaborem-epar-product-information_en.pdf (accessed on 13 June 2021).
- European Medicines Agency (EMA). Summary of Product Characteristics—Relebactam/Imipenem/Cilastatin—547. Available online: https://www.ema.europa.eu/en/documents/product-information/recarbrio-epar-product-information_en.pdf (accessed on 13 June 2021).
- Sagan, O.; Yakubsevitch, R.; Yanev, K.; Fomkin, R.; Stone, E.; Hines, D.; O’Donnell, J.; Miller, A.; Isaacs, R.; Srinivasan, S. Pharmacokinetics and Tolerability of Intravenous Sulbactam-Durlobactam with Imipenem-Cilastatin in Hospitalized Adults with Complicated Urinary Tract Infections, Including Acute Pyelonephritis. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [Green Version]
- Foulds, G.; Stankewich, J.P.; Marshall, D.C.; O’Brien, M.M.; Hayes, S.L.; Weidler, D.J.; McMahon, F.G. Pharmacokinetics of Sulbactam in Humans. Antimicrob. Agents Chemother. 1983, 23, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Rodvold, K.A.; Gotfried, M.H.; Chugh, R.; Gupta, M.; Patel, A.; Chavan, R.; Yeole, R.; Friedland, H.D.; Bhatia, A. Plasma and Intrapulmonary Concentrations of Cefepime and Zidebactam Following Intravenous Administration of WCK 5222 to Healthy Adult Subjects. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Mallalieu, N.L.; Winter, E.; Fettner, S.; Patel, K.; Zwanziger, E.; Attley, G.; Rodriguez, I.; Kano, A.; Salama, S.M.; Bentley, D.; et al. Safety and Pharmacokinetic Characterization of Nacubactam, a Novel β-Lactamase Inhibitor, Alone and in Combination with Meropenem, in Healthy Volunteers. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [Green Version]
- Geibel, B.; Dowell, J.A.; Marbury, T.C.; Smith, W.; McGovern, P.C.; Richards, C.; Henkel, T. Pharmacokinetics and Safety of Cefepime-Taniborbactam (Formerly Cefepime/VNRX-5133) in Subjects with Renal Impairment. Open Forum Infect. Dis. 2020, 7, S670. [Google Scholar] [CrossRef]
- Geibel, B.; Dowell, J.; Dickerson, D.; Henkel, T. A Randomized, Double-Blind, Placebo-Controlled Study of the Safety and Pharmacokinetics of Single and Repeat Doses of VNRX-5133 in Healthy Subjects. Open Forum Infect. Dis. 2018, 5, S431. [Google Scholar] [CrossRef]
- Merdjan, H.; Rangaraju, M.; Tarral, A. Safety and Pharmacokinetics of Single and Multiple Ascending Doses of Avibactam Alone and in Combination with Ceftazidime in Healthy Male Volunteers: Results of Two Randomized, Placebo-Controlled Studies. Clin. Drug Investig. 2015, 35, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Lickliter, J.D.; Lawrence, K.; O’Donnell, J.; Isaacs, R. Safety, Pharmacokinetics, and Drug-Drug Interaction Potential of Intravenous Durlobactam, a β-Lactamase Inhibitor, in Healthy Subjects. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [Green Version]
- Griffith, D.C.; Loutit, J.S.; Morgan, E.E.; Durso, S.; Dudley, M.N. Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of the β-Lactamase Inhibitor Vaborbactam (RPX7009) in Healthy Adult Subjects. Antimicrob. Agents Chemother. 2016, 60, 6326–6332. [Google Scholar] [CrossRef] [Green Version]
- Rhee, E.G.; Rizk, M.L.; Calder, N.; Nefliu, M.; Warrington, S.J.; Schwartz, M.S.; Mangin, E.; Boundy, K.; Bhagunde, P.; Colon-Gonzalez, F.; et al. Pharmacokinetics, Safety, and Tolerability of Single and Multiple Doses of Relebactam, a β-Lactamase Inhibitor, in Combination with Imipenem and Cilastatin in Healthy Participants. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lovern, M.; Green, M.L.; Chiu, J.; Zhou, D.; Comisar, C.; Xiong, Y.; Hing, J.; MacPherson, M.; Wright, J.G.; et al. Ceftazidime-Avibactam Population Pharmacokinetic Modeling and Pharmacodynamic Target Attainment Across Adult Indications and Patient Subgroups. Clin. Transl. Sci. 2019, 12, 151–163. [Google Scholar] [CrossRef]
- Tominaga, N.; Edeki, T.; Li, J.; Learoyd, M.; Bouw, M.R.; Das, S. Phase I Study Assessing the Safety, Tolerability, and Pharmacokinetics of Avibactam and Ceftazidime-Avibactam in Healthy Japanese Volunteers. J. Infect. Chemother. 2015, 21, 551–558. [Google Scholar] [CrossRef]
- Nicolau, D.P.; Siew, L.; Armstrong, J.; Li, J.; Edeki, T.; Learoyd, M.; Das, S. Phase 1 Study Assessing the Steady-State Concentration of Ceftazidime and Avibactam in Plasma and Epithelial Lining Fluid Following Two Dosing Regimens. J. Antimicrob. Chemother. 2015, 70, 2862–2869. [Google Scholar] [CrossRef] [Green Version]
- Dimelow, R.; Wright, J.G.; MacPherson, M.; Newell, P.; Das, S. Population Pharmacokinetic Modelling of Ceftazidime and Avibactam in the Plasma and Epithelial Lining Fluid of Healthy Volunteers. Drugs R D 2018, 18, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Bensman, T.J.; Wang, J.; Jayne, J.; Fukushima, L.; Rao, A.P.; D’Argenio, D.Z.; Beringer, P.M. Pharmacokinetic-Pharmacodynamic Target Attainment Analyses To Determine Optimal Dosing of Ceftazidime-Avibactam for the Treatment of Acute Pulmonary Exacerbations in Patients with Cystic Fibrosis. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Falcone, M.; Menichetti, F.; Cattaneo, D.; Tiseo, G.; Baldelli, S.; Galfo, V.; Leonildi, A.; Tagliaferri, E.; Di Paolo, A.; Pai, M.P. Pragmatic Options for Dose Optimization of Ceftazidime/Avibactam with Aztreonam in Complex Patients. J. Antimicrob. Chemother. 2021, 76, 1025–1031. [Google Scholar] [CrossRef]
- Stein, G.E.; Smith, C.L.; Scharmen, A.; Kidd, J.M.; Cooper, C.; Kuti, J.; Mitra, S.; Nicolau, D.P.; Havlichek, D.H. Pharmacokinetic and Pharmacodynamic Analysis of Ceftazidime/Avibactam in Critically Ill Patients. Surg. Infect. (Larchmt) 2019, 20, 55–61. [Google Scholar] [CrossRef]
- Wenzler, E.; Gotfried, M.H.; Loutit, J.S.; Durso, S.; Griffith, D.C.; Dudley, M.N.; Rodvold, K.A. Meropenem-RPX7009 Concentrations in Plasma, Epithelial Lining Fluid, and Alveolar Macrophages of Healthy Adult Subjects. Antimicrob. Agents Chemother. 2015, 59, 7232–7239. [Google Scholar] [CrossRef] [Green Version]
- Rodvold, K.A.; Gotfried, M.H.; Isaacs, R.D.; O’Donnell, J.P.; Stone, E. Plasma and Intrapulmonary Concentrations of ETX2514 and Sulbactam Following Intravenous Administration of ETX2514SUL to Healthy Adult Subjects. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Bhagunde, P.; Patel, P.; Lala, M.; Watson, K.; Copalu, W.; Xu, M.; Kulkarni, P.; Young, K.; Rizk, M.L. Population Pharmacokinetic Analysis for Imipenem-Relebactam in Healthy Volunteers and Patients with Bacterial Infections. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Rizk, M.L.; Rhee, E.G.; Jumes, P.A.; Gotfried, M.H.; Zhao, T.; Mangin, E.; Bi, S.; Chavez-Eng, C.M.; Zhang, Z.; Butterton, J.R. Intrapulmonary Pharmacokinetics of Relebactam, a Novel β-Lactamase Inhibitor, Dosed in Combination with Imipenem-Cilastatin in Healthy Subjects. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, A.; Gori, G.; Tascini, C.; Danesi, R.; Del Tacca, M. Clinical Pharmacokinetics of Antibacterials in Cerebrospinal Fluid. Clin. Pharmacokinet. 2013, 52, 511–542. [Google Scholar] [CrossRef]
- Lee, Y.R.; Baker, N.T. Meropenem-Vaborbactam: A Carbapenem and Beta-Lactamase Inhibitor with Activity against Carbapenem-Resistant Enterobacteriaceae. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1411–1419. [Google Scholar] [CrossRef]
- Novelli, A.; Del Giacomo, P.; Rossolini, G.M.; Tumbarello, M. Meropenem/Vaborbactam: A next Generation β-Lactam β-Lactamase Inhibitor Combination. Expert Rev. Anti. Infect. Ther. 2020, 18, 643–655. [Google Scholar] [CrossRef]
- Giri, P.; Patel, H.; Srinivas, N.R. Review of Clinical Pharmacokinetics of Avibactam, A Newly Approved Non-β Lactam β-Lactamase Inhibitor Drug, In Combination Use With Ceftazidime. Drug Res. (Stuttg.) 2019, 69, 245–255. [Google Scholar] [CrossRef]
- Vishwanathan, K.; Mair, S.; Gupta, A.; Atherton, J.; Clarkson-Jones, J.; Edeki, T.; Das, S. Assessment of the Mass Balance Recovery and Metabolite Profile of Avibactam in Humans and in Vitro Drug-Drug Interaction Potential. Drug Metab. Dispos. 2014, 42, 932–942. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B. A Brief Review of a New Antibiotic: Meropenem-Vaborbactam. Sr. Care Pharm. 2019, 34, 187–191. [Google Scholar]
- Rubino, C.M.; Bhavnani, S.M.; Loutit, J.S.; Lohse, B.; Dudley, M.N.; Griffith, D.C. Single-Dose Pharmacokinetics and Safety of Meropenem-Vaborbactam in Subjects with Chronic Renal Impairment. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Rubino, C.M.; Bhavnani, S.M.; Loutit, J.S.; Morgan, E.E.; White, D.; Dudley, M.N.; Griffith, D.C. Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of Vaborbactam and Meropenem Alone and in Combination Following Single and Multiple Doses in Healthy Adult Subjects. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.; Preston, R.A.; Mamikonyan, G.; Stone, E.; Isaacs, R. Pharmacokinetics, Safety, and Tolerability of Intravenous Durlobactam and Sulbactam in Subjects with Renal Impairment and Healthy Matched Control Subjects. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.H.; Nicolau, D.P. Augmented Renal Clearance and How to Augment Antibiotic Dosing. Antibiotics 2020, 9, 393. [Google Scholar] [CrossRef]
- Preston, R.A.; Mamikonyan, G.; DeGraff, S.; Chiou, J.; Kemper, C.J.; Xu, A.; Mastim, M.; Yeole, R.; Chavan, R.; Patel, A.; et al. Single-Center Evaluation of the Pharmacokinetics of WCK 5222 (Cefepime-Zidebactam Combination) in Subjects with Renal Impairment. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Merdjan, H.; Tarral, A.; Das, S.; Li, J. Phase 1 Study Assessing the Pharmacokinetic Profile and Safety of Avibactam in Patients With Renal Impairment. J. Clin. Pharmacol. 2017, 57, 211–218. [Google Scholar] [CrossRef]
- Wenzler, E.; Bunnell, K.L.; Bleasdale, S.C.; Benken, S.; Danziger, L.H.; Rodvold, K.A. Pharmacokinetics and Dialytic Clearance of Ceftazidime-Avibactam in a Critically Ill Patient on Continuous Venovenous Hemofiltration. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Kufel, W.D.; Eranki, A.P.; Paolino, K.M.; Call, A.; Miller, C.D.; Mogle, B.T. In Vivo Pharmacokinetic Analysis of Meropenem/Vaborbactam during Continuous Venovenous Haemodialysis. J. Antimicrob. Chemother. 2019, 74, 2117–2118. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Yu, Y.; Wei, X.; Florian, J.; Jang, S.H.; Reynolds, K.S.; Wang, Y. Evaluation of Hemodialysis Effect on Pharmacokinetics of Meropenem/Vaborbactam in End-Stage Renal Disease Patients Using Modeling and Simulation. J. Clin. Pharmacol. 2020, 60, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Vena, A.; Giacobbe, D.R.; Castaldo, N.; Cattelan, A.; Mussini, C.; Luzzati, R.; De Rosa, F.G.; Del Puente, F.; Mastroianni, C.M.; Cascio, A.; et al. Clinical Experience with Ceftazidime-Avibactam for the Treatment of Infections Due to Multidrug-Resistant Gram-Negative Bacteria Other than Carbapenem-Resistant Enterobacterales. Antibiotics 2020, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soukup, P.; Faust, A.C.; Edpuganti, V.; Putnam, W.C.; McKinnell, J.A. Steady-State Ceftazidime-Avibactam Serum Concentrations and Dosing Recommendations in a Critically Ill Patient Being Treated for Pseudomonas Aeruginosa Pneumonia and Undergoing Continuous Venovenous Hemodiafiltration. Pharmacotherapy 2019, 39, 1216–1222. [Google Scholar] [CrossRef]
- Sime, F.B.; Pandey, S.; Karamujic, N.; Parker, S.; Alexander, E.; Loutit, J.; Durso, S.; Griffith, D.; Lipman, J.; Wallis, S.C.; et al. Ex Vivo Characterization of Effects of Renal Replacement Therapy Modalities and Settings on Pharmacokinetics of Meropenem and Vaborbactam. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.; Houle, R.; Lin, M.; Yabut, J.; Cox, K.; Wu, J.; Chu, X. Role of Transporters in the Disposition of a Novel β-Lactamase Inhibitor: Relebactam (MK-7655). J. Antimicrob. Chemother. 2019, 74, 1894–1903. [Google Scholar] [CrossRef]
- Nichols, W.W.; Newell, P.; Critchley, I.A.; Riccobene, T.; Das, S. Avibactam Pharmacokinetic/Pharmacodynamic Targets. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, F.; Fucile, C.; Del Bono, V.; Marini, V.; Parisini, A.; Molin, A.; Zuccoli, M.L.; Milano, G.; Danesi, R.; Marchese, A.; et al. Population Pharmacokinetics and Probability of Target Attainment of Meropenem in Critically Ill Patients. Eur. J. Clin. Pharmacol. 2016, 72, 839–848. [Google Scholar] [CrossRef]
- Pillar, C.M.; Stoneburner, A.; Shinabarger, D.L.; Krause, K.M.; Nichols, W.W. The Postantibiotic Effect and Post-β-Lactamase-Inhibitor Effect of Ceftazidime, Ceftaroline and Aztreonam in Combination with Avibactam against Target Gram-Negative Bacteria. Lett. Appl. Microbiol. 2016, 63, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Coleman, K.; Levasseur, P.; Girard, A.-M.; Borgonovi, M.; Miossec, C.; Merdjan, H.; Drusano, G.; Shlaes, D.; Nichols, W.W. Activities of Ceftazidime and Avibactam against β-Lactamase-Producing Enterobacteriaceae in a Hollow-Fiber Pharmacodynamic Model. Antimicrob. Agents Chemother. 2014, 58, 3366–3372. [Google Scholar] [CrossRef] [Green Version]
- Ehmann, D.E.; Jahic, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of Avibactam Inhibition against Class A, C, and D β-Lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef] [Green Version]
- Bhagunde, P.; Zhang, Z.; Racine, F.; Carr, D.; Wu, J.; Young, K.; Rizk, M.L. A Translational Pharmacokinetic/Pharmacodynamic Model to Characterize Bacterial Kill in the Presence of Imipenem-Relebactam. Int. J. Infect. Dis. 2019, 89, 55–61. [Google Scholar] [CrossRef]
- Griffith, D.C.; Sabet, M.; Tarazi, Z.; Lomovskaya, O.; Dudley, M.N. Pharmacokinetics/Pharmacodynamics of Vaborbactam, a Novel Beta-Lactamase Inhibitor, in Combination with Meropenem. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Boidin, C.; Moshiri, P.; Dahyot-Fizelier, C.; Goutelle, S.; Lefeuvre, S. Pharmacokinetic Variability of Beta-Lactams in Critically Ill Patients: A Narrative Review. Anaesth. Crit. Care Pain Med. 2020, 39, 87–109. [Google Scholar] [CrossRef]
- Motsch, J.; Murta de Oliveira, C.; Stus, V.; Köksal, I.; Lyulko, O.; Boucher, H.W.; Kaye, K.S.; File, T.M.; Brown, M.L.; Khan, I.; et al. RESTORE-IMI 1: A Multicenter, Randomized, Double-Blind Trial Comparing Efficacy and Safety of Imipenem/Relebactam vs Colistin Plus Imipenem in Patients With Imipenem-Nonsusceptible Bacterial Infections. Clin. Infect. Dis. 2020, 70, 1799–1808. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.L.; Motsch, J.; Kaye, K.S.; File, T.M.; Boucher, H.W.; Vendetti, N.; Aggrey, A.; Joeng, H.-K.; Tipping, R.W.; Du, J.; et al. Evaluation of Renal Safety Between Imipenem/Relebactam and Colistin Plus Imipenem in Patients with Imipenem-Nonsusceptible Bacterial Infections in the Randomized, Phase 3 RESTORE-IMI 1 Study. Open Forum Infect. Dis. 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Kaye, K.S.; Boucher, H.W.; Brown, M.L.; Aggrey, A.; Khan, I.; Joeng, H.-K.; Tipping, R.W.; Du, J.; Young, K.; Butterton, J.R.; et al. Comparison of Treatment Outcomes between Analysis Populations in the RESTORE-IMI 1 Phase 3 Trial of Imipenem-Cilastatin-Relebactam versus Colistin plus Imipenem-Cilastatin in Patients with Imipenem-Nonsusceptible Bacterial Infections. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- McLeod, S.M.; Moussa, S.H.; Hackel, M.A.; Miller, A.A. In Vitro Activity of Sulbactam-Durlobactam against Acinetobacter Baumannii—Calcoaceticus Complex Isolates Collected Globally in 2016 and 2017. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- PEW Trusts. Available online: https://www.pewtrusts.org/it/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development (accessed on 13 June 2021).
- Abdul-Aziz, M.H.; Alffenaar, J.-W.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.-A.; Pea, F.; Sjovall, F.; et al. Antimicrobial Therapeutic Drug Monitoring in Critically Ill Adult Patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Sillén, H.; Mitchell, R.; Sleigh, R.; Mainwaring, G.; Catton, K.; Houghton, R.; Glendining, K. Determination of Avibactam and Ceftazidime in Human Plasma Samples by LC-MS. Bioanalysis 2015, 7, 1423–1434. [Google Scholar] [CrossRef]
- Abdulla, A.; Bahmany, S.; Wijma, R.A.; van der Nagel, B.C.H.; Koch, B.C.P. Simultaneous Determination of Nine β-Lactam Antibiotics in Human Plasma by an Ultrafast Hydrophilic-Interaction Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1060, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.A.; Nicolau, D.P. Development of an HPLC Method for the Determination of Meropenem/Vaborbactam in Biological and Aqueous Matrixes. J. Chromatogr. Sci. 2020, 58, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Sarli, V.; Ciofi, L.; Lastella, M.; Muscatello, B.; Pisaturo, F.; Paolilli, O.; Luci, G.; Cucchiara, F.; Pellegrini, G.; Bocci, G.; et al. Appropriateness of Repetitive Therapeutic Drug Monitoring and Laboratory Turnaround Time. Clin. Chem. Lab. Med. 2019, 57, e331–e333. [Google Scholar] [CrossRef] [PubMed]
- Sprague, D.A.; Ensom, M.H.H. Limited-Sampling Strategies for Anti-Infective Agents: Systematic Review. Can. J. Hosp. Pharm. 2009, 62, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, A.; Tascini, C.; Polillo, M.; Gemignani, G.; Nielsen, E.I.; Bocci, G.; Karlsson, M.O.; Menichetti, F.; Danesi, R. Population Pharmacokinetics of Daptomycin in Patients Affected by Severe Gram-Positive Infections. Int. J. Antimicrob. Agents 2013, 42, 250–255. [Google Scholar] [CrossRef]

{kind=link}
| β-Lactamases | Substrates | Spectrum of Activity of BLIs | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Active Site | Ambler Class | Representative Enzymes | ||||||||||||
| Pen | Cep | ECep | Cbn | Mb | AVI | REL | VAB | DUR | ZID | NAC | TAN | |||
| Serine | A | PC1 | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||
| TEM-1, TEM-2, SHV-1 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| CTX-M-15, GES-1, VEB-1 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||
| IRT, SHV-10, TEM-30 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||
| CARB-1, PSE-1 | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||
| KPC, SME-1, GES-2 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| C | AmpC, P99, ACT-1, MIR-1 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| GC1, CMY-37 | ✓ | ✓ | ✓ | ✓ | ||||||||||
| D | OXA-1, OXA-10 | ✓ | +/− | +/− | ✓ | ✓ | ✓ | |||||||
| OXA-11, OXA-15 | ✓ | ✓ | ✓ | +/− | +/− | ✓ | ||||||||
| OXA-23, OXA48 | ✓ | ✓ | ✓ | +/− | ✓ | +/− | ✓ | ✓ | ||||||
| MBL | B | IMP, VIM, NDM | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| CphA, Sfh-1 | ✓ | ✓ | ||||||||||||
| Drug | CL 1 (L/h) | Vd (L) | t1/2 (h) | PPB (%) | References |
|---|---|---|---|---|---|
| AVI | 1.59 | 18.0 | 2.0 | 8 | [45] |
| CAZ | 1.54 | 22.0 | 2.0 | 10 | [45] |
| AZT | 140 | 29.4 | 1.7 | 77 | [46,47] |
| CEF | 1.8–3.0 | 28.3 | 2.5 | 20 | [48] |
| VAB | 10.5 | 19.0 | 2.25 | 33 | [34,49,50] |
| MER | 7.7 | 21.0 | 2.30 | 2 | [34,49,50] |
| REL | 8.1 | 21 | 1.7 | 22 | [34,51] |
| IMI | 8.4 | 21.7 | 1.1 | 20 | [35,51,52] |
| DUR | 10.3 | 31.6 | 2.5 | - | [52] |
| SUL | 2.4 | 12.0 | 1.8 | 38 | [52,53] |
| ZID | 7.4 | 17.4 | 1.9 | <15 | [54] |
| NAC | 8.8 | 20.6 | 2.4 | - | [55] |
| TAN | 5.8 | 30–50 | 6.5 | - | [56,57] |
| Drugs and Dosage | Clinical Use | Therapeutic Indications (Duration of Treatment) | Pediatric Use | Ref. |
|---|---|---|---|---|
| CAZ/AVI 1 2/0.5 g q8h 2-h IV infusion | YES | cIAI (5–14 days) cUTI (5–10 days) Pyelonephritis (5–10 days) HAP (7–14 days) VAP (7–14 days) Aerobic G- infections (variable) | YES (age ≥3 months) | [45] |
| MER/VAB 2/2 g q8h 3-h IV infusion | YES | cIAI (5–10 days) cUTI (5–10 days) Pyelonephritis (5–10 days) HAP (7–14 days) VAP (7–14 days) Aerobic G- infections (variable) Bacteremia (variable) | NO | [50] |
| REL/IMI/CIL 0.25/0.5/0.5 g q6h 0.5-h IV infusion | YES | HAP (7–14 days) VAP (7–14 days) Infections from Aerobic G- (variable) Bacteremia (variable) | NO | [51] |
| REL/IMI 0.25/0.5 g q6h 0.5-h IV infusion | NO | HAP (variable) VAP (variable) cUTI (variable) cIAI (variable) | - | [100,101,102] |
| DUR/SUL | NO | Phase III studies: cUTI, HAP, VAP | - | [103,104] |
| DUR/IMI/CIL | NO | Studies in HV | - | [61] |
| ZID/CEF | NO | Phase III studies: cUTI, HAP, VAP | - | [104] |
| NAC/CEF or AZT | NO | Phase I studies: cUTI | - | [104] |
| TAN/CEF | NO | Phase III studies: cUTI | - | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luci, G.; Mattioli, F.; Falcone, M.; Di Paolo, A. Pharmacokinetics of Non-β-Lactam β-Lactamase Inhibitors. Antibiotics 2021, 10, 769. https://doi.org/10.3390/antibiotics10070769
Luci G, Mattioli F, Falcone M, Di Paolo A. Pharmacokinetics of Non-β-Lactam β-Lactamase Inhibitors. Antibiotics. 2021; 10(7):769. https://doi.org/10.3390/antibiotics10070769
Chicago/Turabian StyleLuci, Giacomo, Francesca Mattioli, Marco Falcone, and Antonello Di Paolo. 2021. "Pharmacokinetics of Non-β-Lactam β-Lactamase Inhibitors" Antibiotics 10, no. 7: 769. https://doi.org/10.3390/antibiotics10070769
APA StyleLuci, G., Mattioli, F., Falcone, M., & Di Paolo, A. (2021). Pharmacokinetics of Non-β-Lactam β-Lactamase Inhibitors. Antibiotics, 10(7), 769. https://doi.org/10.3390/antibiotics10070769