3.2. Electrode Characterization
3.2.1. Electrochemical Characterization
Electrochemical measurements were carried out in order to characterize the modified electrodes at different stages of the fabrication of the MIP-sensor. CV, DPV and EIS techniques were used to assess the response and to characterize the unmodified surfaces and after polymerization, extraction and incubation.
A first step in the electrochemical characterization was taken in the preliminary tests, in which CV tests were conducted in order to choose the most suitable redox probe. In this step, described in more detail, in the optimization section, three different probes were tested, [Fe(CN)6]3−/[Fe(CN)6]4− yielding the best results. During these tests, all the probes tested showed a decrease in the peak current after polymerization, both for the MIP and NIP films, demonstrating the insulating effect of the polymer. Similarly, they all presented a major increase in the peak current after the extraction procedure, only for the MIP film, but due to their different properties (charge, size) and their different interactions with the polymer, the best response were obtained for the [Fe(CN)6]3−/[Fe(CN)6]4− probe. This was once again observed, when after the incubation procedure, the peak current decreases, a decrease which was significantly bigger for the [Fe(CN)6]3−/[Fe(CN)6]4− probe.
The results obtained using the CV technique and [Fe(CN)
6]
3−/[Fe(CN)
6]
4− as redox probe, were confirmed by the DPV and EIS tests, using both BDDE and GCE. Using the DPV technique to measure the signal caused by the reduction of the [Fe(CN)
6]
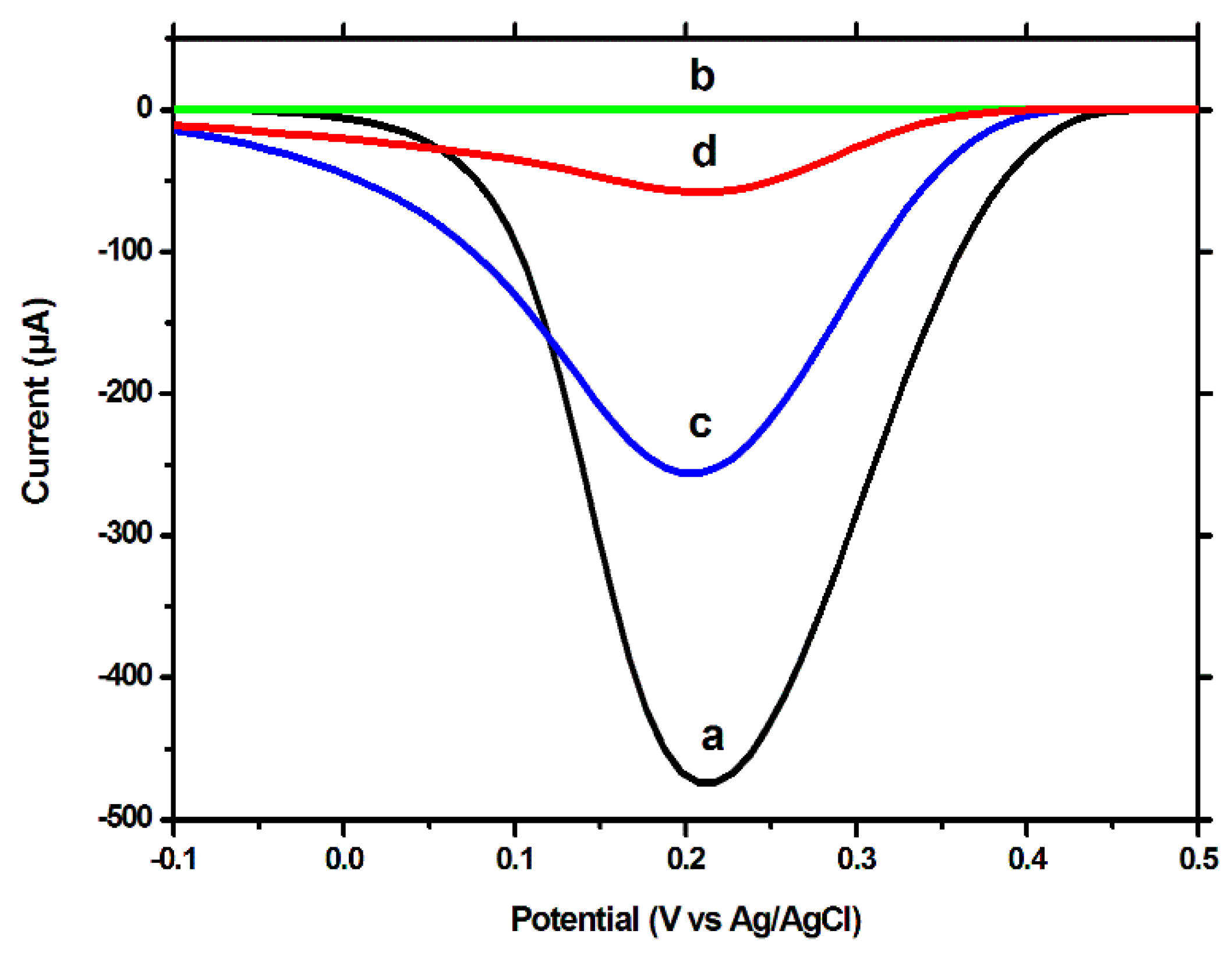
3−, a high peak current was observed for the unmodified electrode (
Figure 3a), which was severely decreased after polymerization (
Figure 3b). After the extraction procedure, the intensity of the peak current increased by a significant degree (
Figure 3c), but not reaching the intensity obtained for the unmodified surface. This difference between the signal for the unmodified electrode and for the MIP-modified electrode, after extraction, a difference which was also visible in the CV tests, indicates that the extraction procedure removed only the template molecules, decreasing the insulating properties of the MIP film, but not removing the film in its entirety, the extracted imprinted film retaining, although to a lesser degree, its insulating properties. After the incubation with CFX, a decrease in the peak current was once again observed; this can be explained by the fact that a large percentage of the imprinted cavities are reoccupied during the incubation process.
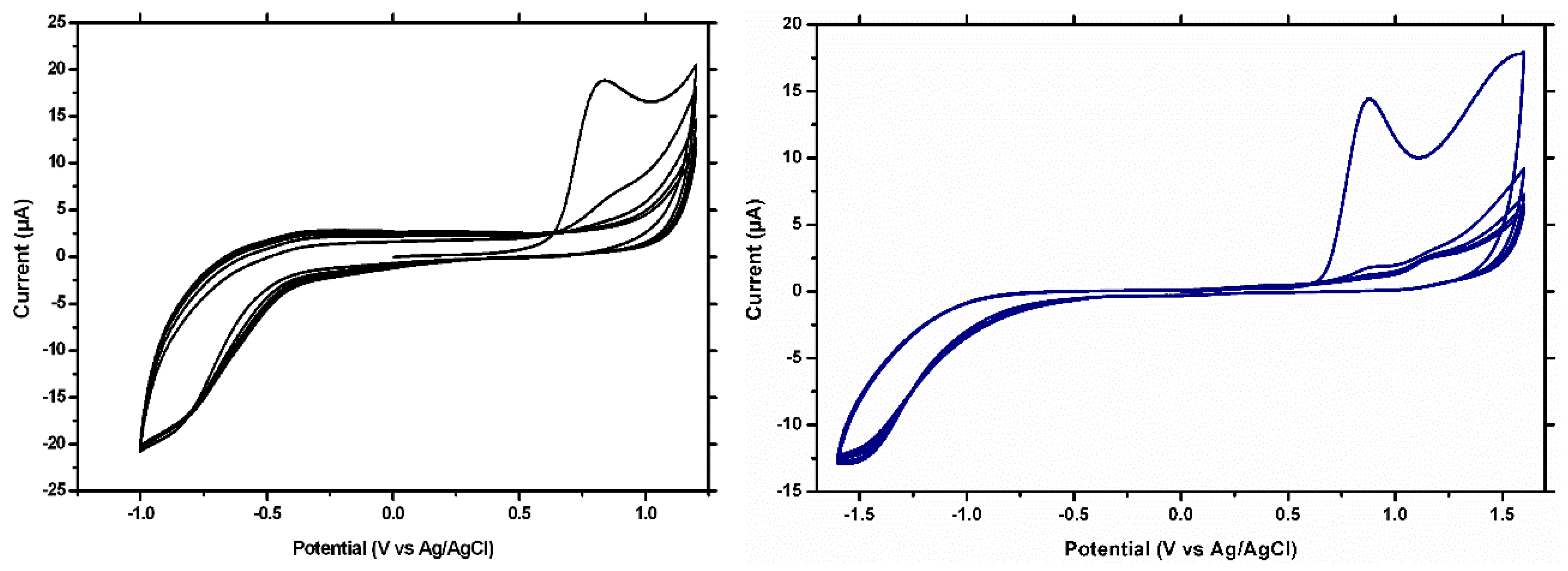
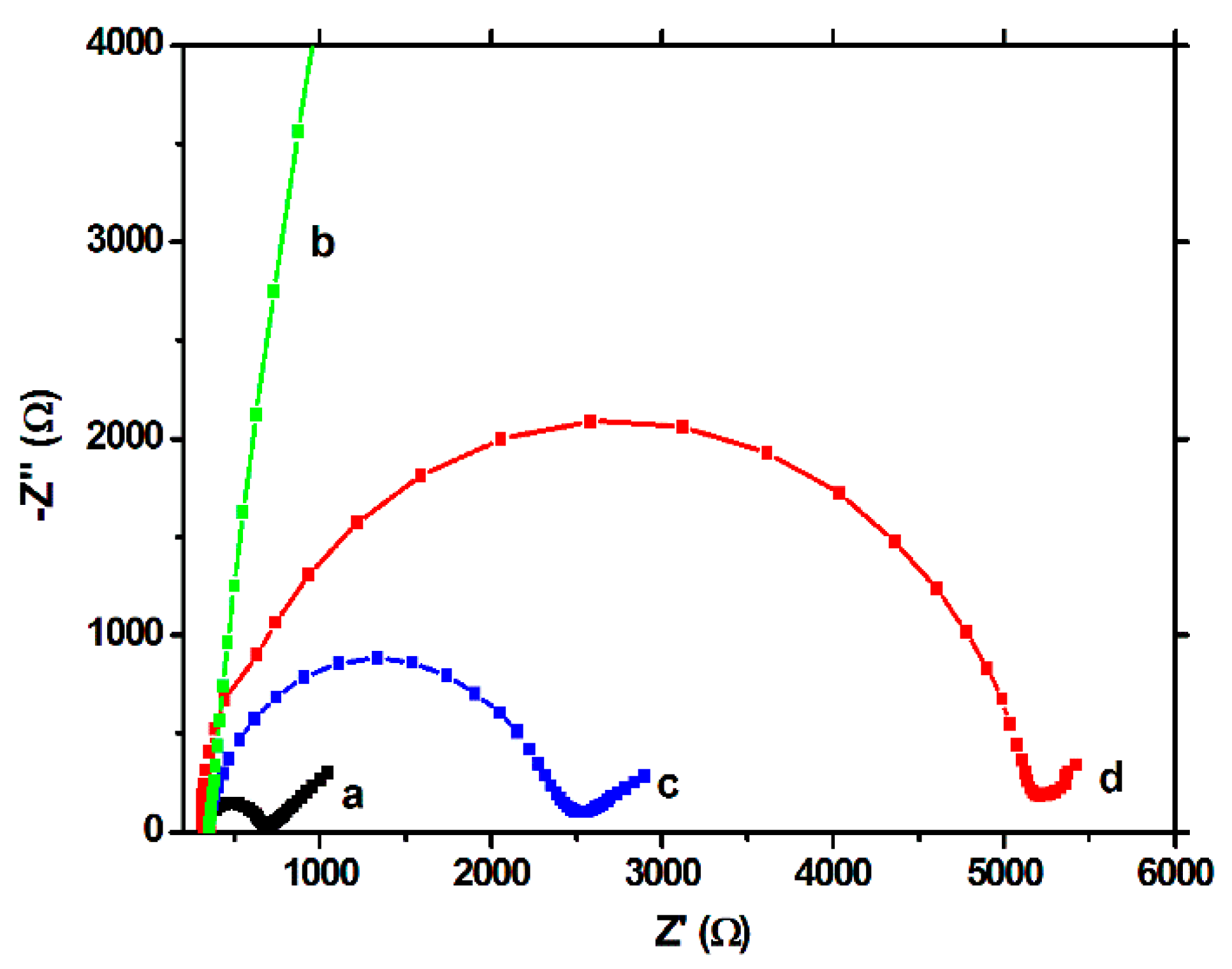
The EIS tests showed a similar behavior. In the Nyquist plots, the semicircle portion, at higher frequencies, corresponds to the electron transfer limited process, and the linear portion, at lower frequencies, may be ascribed to diffusion. Thus, the diameter of the semicircle equals to the electron transfer resistance (Rct), which is correlated with the dielectric and insulating features of the electrode/electrolyte interface, mainly, in our case, of the modifying film. For the unmodified electrode, a low value of Rct was obtained (
Figure 4a), but after polymerization (
Figure 4b), this value increased drastically, confirming once again the strong insulating properties of the polymeric film. After extraction, a decrease of the Rct was observed (
Figure 4c), but the response did not reached the values of the unmodified electrode, showing once again the persistence of the imprinted film on the working surface. The capacity of the MIP to rebind the CFX molecule was proven by the increase of the Rct after the incubation step (
Figure 4d).
All these results demonstrate the modification of the electrode with a MIP film, the successful extraction of the template and the capacity of the MIP to recapture the analyte molecules, CFX. They also show that by removing the template and making available the imprinted cavities, the polymeric film increases its porosity and decreases its insulating properties, the unoccupied cavities allowing an easier electron transfer between the redox probe and the electrode and these changes can be quantified using the signal of the redox probe.
3.2.2. Surface Characterization
SEM images at higher magnification (100 k×) were used to get an insight into the surface morphology of NIP (
Figure 5C) and MIP (
Figure 5B) films and, more interesting, into the surface of the MIP film after the extraction procedure (
Figure 5C).
NIP film surface looks compact and smooth. The MIP film, at a first glance seems similar to the NIP surface, but a more porous morphology and some roughness of the surface can be observed. However, the MIP surface after extraction, definitely presents a higher degree of roughness and a further increase in porosity, indicating that the extraction procedures of the template molecules leads to a change in the morphology of the MIP film.
In contrast to SEM, which provides a two-dimensional projection or a two-dimensional image of a surface, the main advantage of AFM is the fact that it provides a true three-dimensional profile of the surface. Through AFM is it possible to carry out topographic contrast direct height measurements, measure the thickness and roughness of the layer.
In the present work, the formation of both NIP and MIP layers was suggested by the AFM images (
Figure 5D,E), indicating an elevated layer with the surface height of 3.1 nm for NIP and 15 nm for MIP. The root mean square (RMS) roughness obtained was of 0.33 nm for NIP and 0.65 nm for MIP.
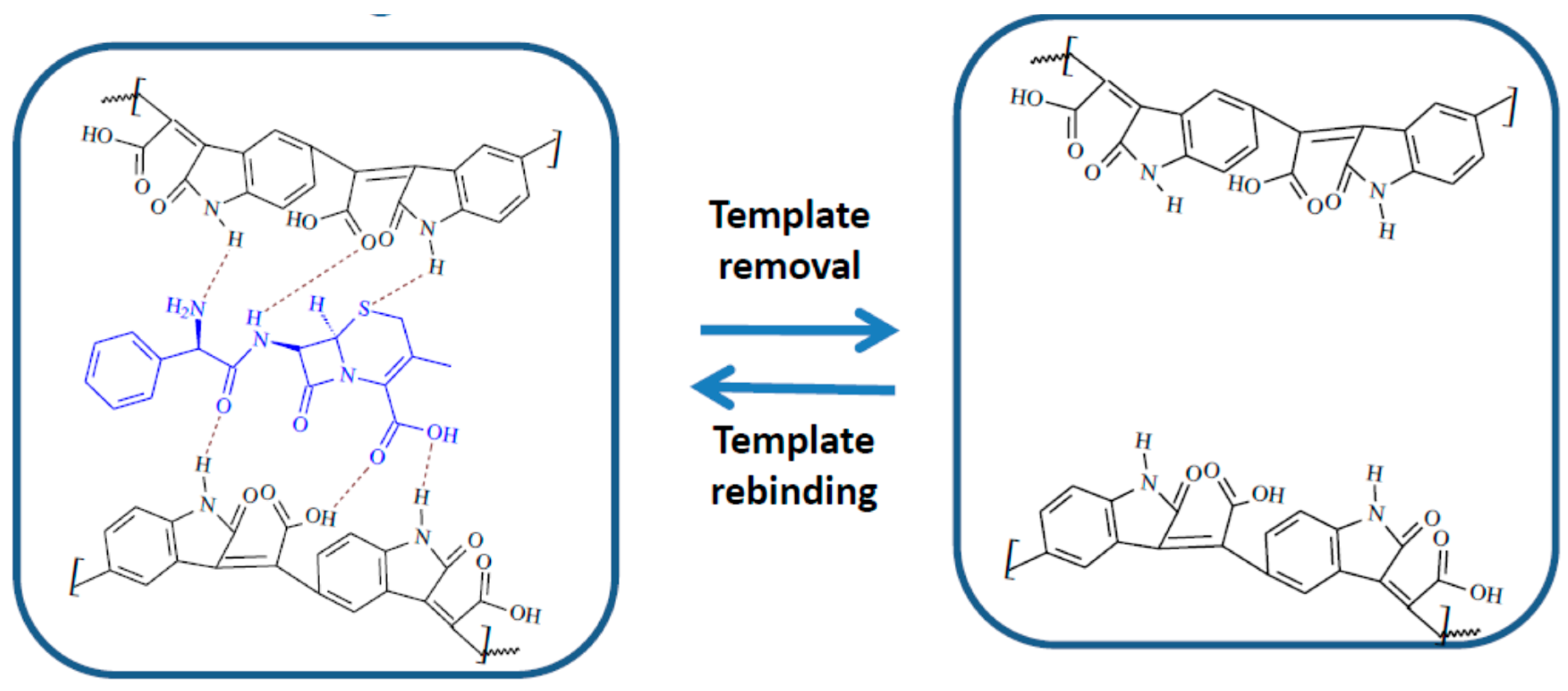
These results can be easily explained by the inclusion of the template molecule in the MIP film, drastically modifying the polymerization process and implicitly the structure of the film. CFX, the template, has a more voluminous molecule than the monomer and also it does not make covalent bonds with the polymer, causing a considerably less structured organization of the polymeric film, causing an increase in both roughness and thickness, for the MIP film.
Similarly to the results obtained through SEM, the thickness of the MIP layer after extraction (
Figure 5F) was 7 nm and the roughness 0.95 nm. These data support the success of the extraction procedure, which causes, by removing the template, the creation of the “imprinted” cavities, which further leads to an increase in roughness. This removal of the template, combined perhaps with the removal of certain residues remnant from the polymerization procedure and with a certain degree of reorganization of the polymeric film, after template removal, is the cause of the decrease in thickness of the MIP layer after extraction.
3.3. Optimization of the MIP Sensor
The development of the MIP-based sensor was obtained in several steps and each one needed to be optimized. The overall development of the sensor can be divided in four main steps: first, the selection of the redox probe for the indirect detection, second, the creation of the initial imprinted polymeric film, containing the template molecule in it, third, the extraction procedure (the removal of the template) and fourth, the incubation with CFX (the recapture or the rebinding of the template). In the optimization of each parameter, the final signal (the signal difference after the extraction and the incubation step) was taken into account, because each one of the four aforementioned consecutive steps influences the final response of the sensor.
The final signal was constructed as a ratio between the difference of the two signals, after extraction and after incubation, and the signal after extraction. Considering that the signal after extraction is almost constant, the denominator part of the ratio remains also constant and the differences in the numerator appear only as a result of the changes in the values of the signal after incubation.
To better characterize the formation of imprinted cavities and to demonstrate that in the incubation process there is truly taking place a specific adsorbtion phenomenon, all the optimization tests carried out for the MIP-electrodes, were also realized using NIP-electrodes. Working in parallel with an MIP and NIP, we could also more easily elucidate the influence of each parameter on the performance of the MIP.
Because in DPV, the technique used for quantification, the signal after extraction is always going to be greater in absolute value and for simplicity we chose the difference to be calculated as the signal after extraction minus the signal after incubation. Because the signal after incubation decreases with increasing concentrations, the difference and also the ratio, increase their values in correlation with the concentration of analyte. In a similar way, the signal was constructed for the CV technique.
In contrast, because in EIS measurements Rct increased in value due to increasing amounts of captured analyte, the difference was reversed in the construction of the signal in the EIS technique. For the optimization part, in the same way, two other signals in each technique were constructed, corresponding to each procedure: a signal after polymerization, using the signal after polymerization and the signal of the unmodified electrode and a signal after extraction, using the signal after extraction and after polymerization. The formulas for the calculation of the signals are summarized below.
For DPV and CV techniques:
Iunmod is the peak height in DPV/CV for the unmodified electrode, Ipolym is the peak height in DPV/CV after polymerization, Iextr is the peak height in DPV after extraction, Iincub is the peak height in DPV after incubation.
Runmodif is the Rct in EIS on the unmodified electrode, Rpolym is the Rct in EIS after polymerization, Rextr is the Rct in EIS obtained after extraction, Rincub is the Rct in EIS obtained after incubation. The response after polymerization is directly correlated to the insulating properties of the imprinted polymer formed after electropolymerization, a higher response value meaning a more insulating film, which is correlated to the thickness, composition and structure of the film. The response after the extraction is directly correlated to the amount of template removed during the extraction step, a higher response value being equal to a higher amount of template removed. In addition, most importantly, the final response is directly correlated to the amount of recaptured analyte, a higher response equals a higher amount of analyte recaptured by the MIP, which can be correlated to a higher concentration in the sample.
3.3.1. The Selection of the Redox Probe
For the quantification and characterization of the sensor, three different redox probes were tested: [Fe(CN)6]3−/4−, an anionic redox probe, 1,1′-ferrocenedimethanol, a neutral redox probe and [Ru(NH3)6]3+, a cationic redox probe, to investigate the most suitable one for the characterization of the modifications suffered by the MIP film and for the CFX quantification. They were chosen based on their particular properties, which could cause different interactions, electrostatic attraction or repulsion, between the probe and the outer layer of the MIP film. Also, another important property of the redox probes is the distance at which their electron transfer is hindered, this property offering a possibility to estimate the thickness of the MIP layer.
As it can be seen from the data presented in
Tables S1 and S2, all three redox probes provided a good signal after polymerization, meaning that regarding the polymerization layer, any of the three probes could be used.
The results for the NIP were similar, to those obtained for MIP, after polymerization. More precisely, a signal of 0.995 was obtained for both MIP and NIP, using [Fe(CN)6]3−/4−, on the GCE. Similar, using the same probe, a signal of 0.986 for the MIP and a signal of 0.993 for the NIP, were obtained on the BDDE. This is in accordance with the fact that both the polymer and the template molecule showed insulating properties, regardless of the probe used.
Nonetheless, after both the extraction and incubation procedures, only the [Fe(CN)
6]
3−/4− probe can still detect the film and more importantly, can detect the rebinding of the template. This can be easily explained by the differences in the distances at which each redox probe can realize its electron transfer, Ru
3+ and ferocene being able to conduct the direct electron transfer at a greater distance than the [Fe(CN)
6]
3−/4− [
19,
20]. This finding is in accordance with the data from SEM and AFM, which estimates the thickness of the MIP film, before extraction at approximately 15 nm and under 10 nm after extraction. Another possible factor is the influence of the charge of the redox probe. The MIP film, due to its acidic moieties, causes electrostatic repulsion with the anionic probe ([Fe(CN)
6]
3−/4−), but does not interact electrostatically with the neutral probe (1,1′-ferrocenedimethanol) or will even attract the cationic probe (Ru
3+). All these results led us to choose [Fe(CN)6]
3−/4− as redox probe for the next studies.
For the NIP modified electrodes, the behavior of the three probes tested was similar to that of the MIP, only the [Fe(CN)6]3−/4− probe being able to detect the film, after the extraction step. However, very important, there were clear differences regarding the signal after extraction and after incubation, using the [Fe(CN)6]3−/4− probe, for the NIP, comparatively to the MIP. First, a much smaller signal, was obtained after extraction for NIP, comparatively to the one obtained for the MIP, For the GCE, 6.320 for MIP and 1.138 for NIP were obtained and, similarly, on the BDDE, 8.348 for MIP and 0.735 for NIP. These results clearly show, that even though the signal of the NIP after extraction is changed, its modification is much smaller than that of the MIP, demonstrating the resulting imprinting process due to the removal of the template. Also, these results predicted a higher sensitivity, caused by a more pronounced imprinting process on the BDDE, in comparison to the GCE.
Second, after incubation, there were also clear differences, even though modifications in the signal were taking place on both of the electrodes. The values obtained, after incubation, for MIP and NIP, were 0.436 and 0.176, on the GCE, respectively, 0.532 and 0.123, on the BDDE. For this step, the modification can be explained by the changes of the polymer and its surface, caused by the incubation procedure, in a different solvent, but also by the phenomenon of nonspecific adsorption of the analyte, on the outer surface of the polymeric film. These two processes take place on both types of surfaces, MIP and NIP, the process that distinguishes them being the specific absorption, the template recapture inside the polymeric film. This way, the final signal for the NIP is caused by the nonspecific adsorption of the template and by the influence of the incubation, whereas the signal of MIP is caused mainly by the specific recapture of the template inside the polymeric film.
The behavior of the response of the NIP, in relation to the response of the MIP, was similar to that obtained in the first step, the one regarding the redox probe, in all the tests carried out in the other three steps of optimization.
3.3.2. The Optimization of the Formation of the Imprinted Polymeric Film
The second optimized step in the development of the MIP sensor was the electropolymerization procedure. This is probably the most important step, because during this step a strong interaction between the template molecule, the monomer and the electrogenerated polymer must be assured. The most important parameters for this step are those regarding the composition of the polymerization mixture. The concentration of the monomer, the concentration of the template and implicit the ratio between these two components have been optimized in order to assure a good monomer-template interaction. Also, these parameters influence the thickness and the structure of the imprinted polymeric film resulting after polymerization. A thicker layer makes possible the creation of more imprinted “pockets”, but also can affect the accessibility to this “binding pockets” in the extraction and rebinding steps.
Another parameter that can be easily manipulated, in the case of electropolymerization through the use of cyclic voltammetry is the number of cycles of the procedure, which influences mainly the thickness of the layer and also its stability. The optimization of the main parameters regarding the polymerization procedure is presented in
Tables S3 and S4.
As it can be seen from the data presented in
Tables S3 and S4, the first optimized parameter was the concentration of the monomer, with three concentrations being tested (0.1, 1 and 5 mM), while the concentration of CFX was kept constant at 0.05 mM, varying in this way also the ratio monomer:template. Using the same concentrations of the monomer but no template, three NIP-sensors were fabricated, tested and compared. For comparison all three signals – after polymerization, after extraction and after incubation were taken into account. As it can be seen the value of S
polym was very high in all cases, especially due to the highly insulating nature of the polymer. Also, as it can be seen from the similar S
polym, for different number of cycles, the insulating properties of our polymer were less influenced by the thickness of the layer, a high response being obtained even only after 2 cycles.
Considering Sextr and Sincub, a number of 5 cycles proved to give better results compared to 2 or 10 cycles. This can be explained by the equilibrium that occurs between the number of cycles and the amount of template that can be removed and thus, also rebounded. A lower number of cycles lead to a less structured polymer, which means not enough cavities formed or a large number of not fully formed cavities, which are not stable enough during the extraction step or simply are not able to rebind the template, due to a smaller number of binding sites. On the other hand, a higher number of cycles can lead to a higher value of thickness and to an excessively structured polymer, which due to its high thickness and high rigidity, hinders the proper extraction and recapture of the template molecule.
Also, regarding the Sincub, which varies the most and is the most important response, being the final criteria in choosing the optimal parameters, good polymerization and extraction do not necessarily assure a good rebinding of the analyte. From the data obtained, a concentration of 1 mM for the monomer and of 0.5 mM for the template, with a procedure using five cycles, provided the best results in terms of the amount of recaptured analyte.
3.3.3. The Optimization of the Extraction Procedure
The solvent and the duration of the incubation were the main parameters of the incubation procedure that were optimized (
Tables S5 and S6). Three options were tested as solvents for extraction, based on the solubility of CFX, CFX being highly soluble in MeOH and alkaline aqueous solutions. The best results were obtained using an aqueous solution of NaOH 0.1 M. This can be explained by the fact that the use of a pure non-aqueous solvent such as MeOH affects severely the integrity of the imprinted film, which even though it can cause a better response after extraction, it also affects the binding capacities of the imprinted cavities. Instead, the use of an alkaline aqueous solution provided the balance between stability, being a similar medium with the one in which the polymerization was realized, and efficiency, the change in pH, towards more basic, offering a strong enough change in the solubility of the template in order to determine the break of the bonds between the polymer and template.
Regarding the duration of the procedure, 30 min offered enough time for a satisfactory extraction, without any significant damage to the film. This can be seen by the fact that up to 30 min the amount of rebound template increases, but for 60 min, the amount is lower, which may indicate that the prolonged period of immersion in the strong alkaline solution may have affected the integrity of the film.
3.3.4. The Optimization of the Incubation Procedure
For the incubation procedure, similar parameters as for the extraction were optimized. The solvent tested was PBS, the same as the one used for polymerization and ultrapure water. Even though PBS yielded slightly better results, water offered the advantage of bearing more resemblance to the matrix of the targeted samples. That is why, taking into account the applicability of the method, water was chosen as incubation medium. Regarding the duration of the incubation, the captured amount of analyte seemed to reach a plateau at 30 min, there being no large differences between the values at 30 and 60 min. Therefore, the final parameters chosen for the incubation procedure were water as the medium and 30 min of incubation (
Table S7).
3.4. Analytical Performance of the MIP-Based Sensor
The analytical performance of the developed sensor was tested under the optimized conditions.
3.4.1. Calibration Curve and Limit of Detection
Because the process of rebinding the template can be simulated as an adsorption process, for the MIP surfaces it is required to establish the adsorption isotherm governing the rebinding of the template. The data obtained for our work presented a logarithmic growth, the Freundlich adsorption isotherm, being the one that fitted the best our results. The fact that this empirical equation fitted our data can be explained by the heterogeneity of our imprinted surface, in which characteristics of the Langmuir model, such as a monolayer of adsorbate and equivalency between adsorption sites cannot be fulfilled, the Freundlich model being developed for more irregular and complex surfaces. The unfitted data presented a logarithmic growth of the signal in relation to the concentration of the template, reaching a plateau at higher concentrations, a sign of the fact that all active imprinted cavities have been filled. This type of behavior is characteristic to MIP-based sensors [
21,
22,
23].
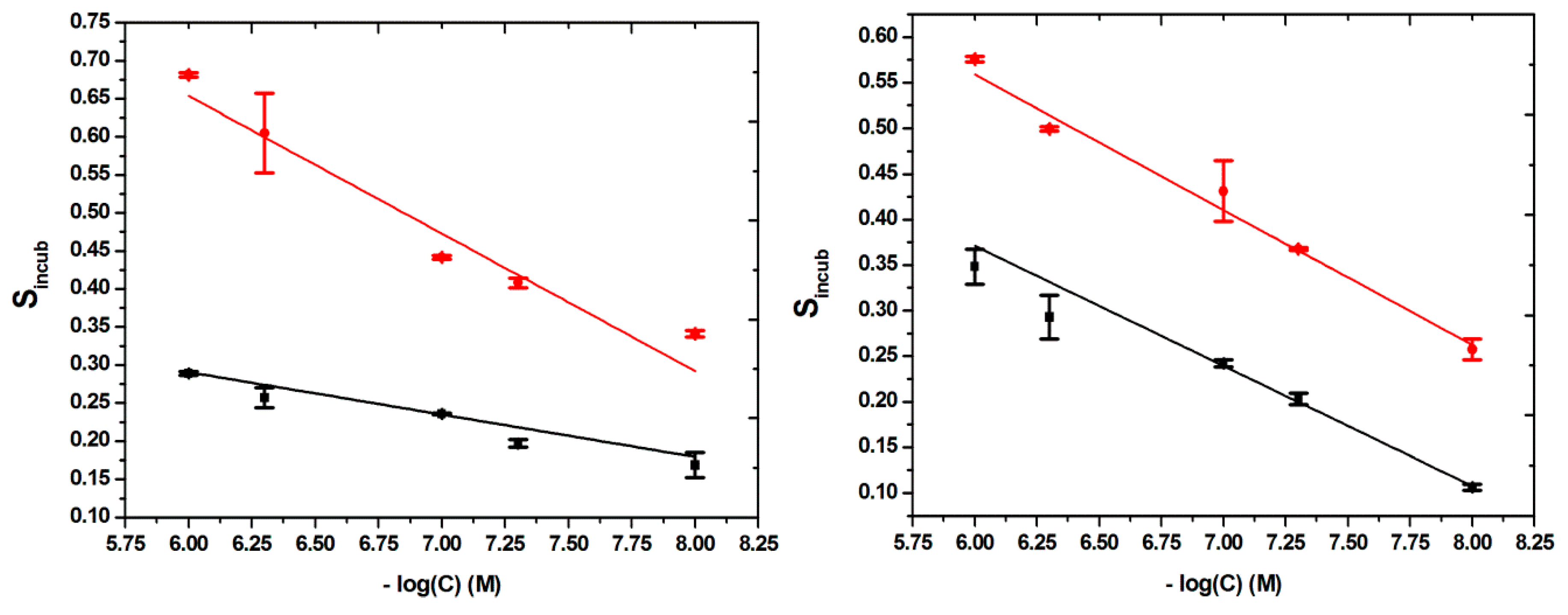
That is why, for the calibrations curves, in order to obtain a linear correlation, the signal was plotted against the negative value of the decimal logarithm of the concentrations, four different calibration curves being constructed, one for each type of modified surface, in the same range of concentrations: 10–1000 nM.
The linear relationships found, with their correlated equations and their correlation coefficients (R2), were as it follows: for MIP-BDDE, S
incub = −0.1809 × (−log(C) (M)) + 1.7392; with R
2 = 0.90669, for NIP-BDDE, S
incub = −0.05566 × (−log(C) (M)) + 0.62457; with R
2 = 0.93433, for MIP-GCE, S
incub =−0.1483 × (−log(C) (M)) + 1.44859; with R
2 = 0.97922 and for NIP-GCE, S
incub = −0.13175 × (−log(C) (M)) + 1.16155; with R
2 = 0.99273 (
Figure 6).
For the determination of the limit of detection (LOD) the following equation was used: LOD = 3S
b/m, in which
Sb is the standard deviation of the response of the blank solution and
m is the slope of the calibration curve. An LOD of 3.2 nM and 4.9 nM was obtained for the BDDE and the GCE, respectively. These results are better than the one obtained by other electrochemical methods and even instrumental methods, as seen in
Table S8.
From the results presented it can be deduced that the BDDE-modified surface presented better sensitivity, even if you take into account only the MIP signal or the difference in response for the MIP and NIP surfaces. This can be attributed to larger number of imprinted cavities, for the film on the BDDE surface, caused by several factors, such as the larger potential window, the better conducting properties of the BDDE and also to the possibility of a higher incorporation of the template, in the process of its electroxidation. However, the GCE offered a greater reproducibility and a better correlation for the results, which can also be easily explained by the more homogeneous composition of the GCE surface, compared to the BDDE surface, known for its more heterogeneous nature, which causes some researchers to avoid, it when wanting to develop a biomimetic sensor. Considering, all this, it can be said that even though the BDDE offered a slightly greater sensitivity, one should choose the GCE, when wanting to develop a biomimetic platform, because of the very heterogeneous composition of the BDDE surface and slightly less reproducible response, which could affect the performance of the developed analytical platform.
3.4.2. Precision and Reusability of the Imprinted Sensor
In order to check the reproducibility of the developed method, 5 different modified BDDEs and 5 different modified GCEs were fabricated and applied to the analysis of a 100 nM CFX solution. The relative standard deviations for the two electrodes were 4.92% and 2.66%, respectively. This shows that even though the LOD obtained with the BDDE is lower, its precision is worse compared to the GCE.
In order to test the reusability of a MIP-based sensor, after a first CFX analysis, an extraction procedure was applied to the sensor and a second CFX analysis was performed. The results presented a great variability, with no reliable results for the second analysis using the same sensor.
3.4.3. Selectivity Studies
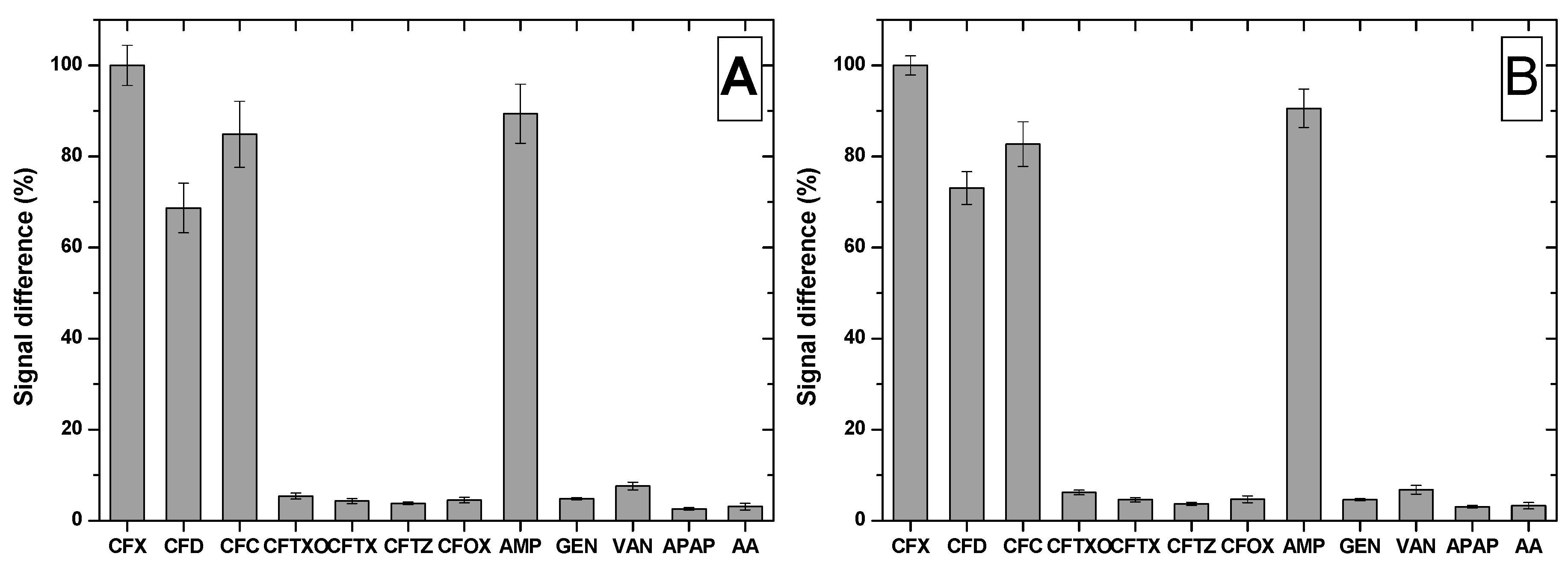
The selectivity of the MIP-based sensor was tested by performing the incubation step in solutions containing other cephalosporins antibiotics (cefadroxil (CFD), cefaclor (CFC), ceftriaxone (CFTXO), cefotaxime (CFTX), ceftazidime (CFTZ), cefuroxime (CFOX)), ampicillin (AMP) that belongs to the penicillin class, two other antibiotics from a different classes (GEN, VAN) and two pharmaceutical compounds intensively used (ascorbic acid (AA) and acetaminophen (APAP)).
The incubation with CFTXO, CFTX, CFTZ or CFOX led to no significant signal modification, because even though these molecules present the common cephalosporin nucleus, they have different substituents, impairing them to be bound in the cavities of the MIP film (
Figure 7). In contrast, CFD and CFC, two cephalosporins with a similar and respective identical one side chain to the one of CFX, were bound in the cavities of the MIP, suggesting the importance of the side chain in the fabrication of the MIP film. This is supported by the results obtained after incubation with AMP, a penicillin with the side chain identical to the one of CFX and different core nucleus, AMP being bound also by the MIP film.
In the case of the compounds that are not structurally related to CFX, they presented low affinity for the MIP film fabricated on both electrodes, the sensor proving to be selective for CFX.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}