Dual-Channel Stopped-Flow Apparatus for Simultaneous Fluorescence, Anisotropy, and FRET Kinetic Data Acquisition for Binary and Ternary Biological Complexes

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Solution Conditions
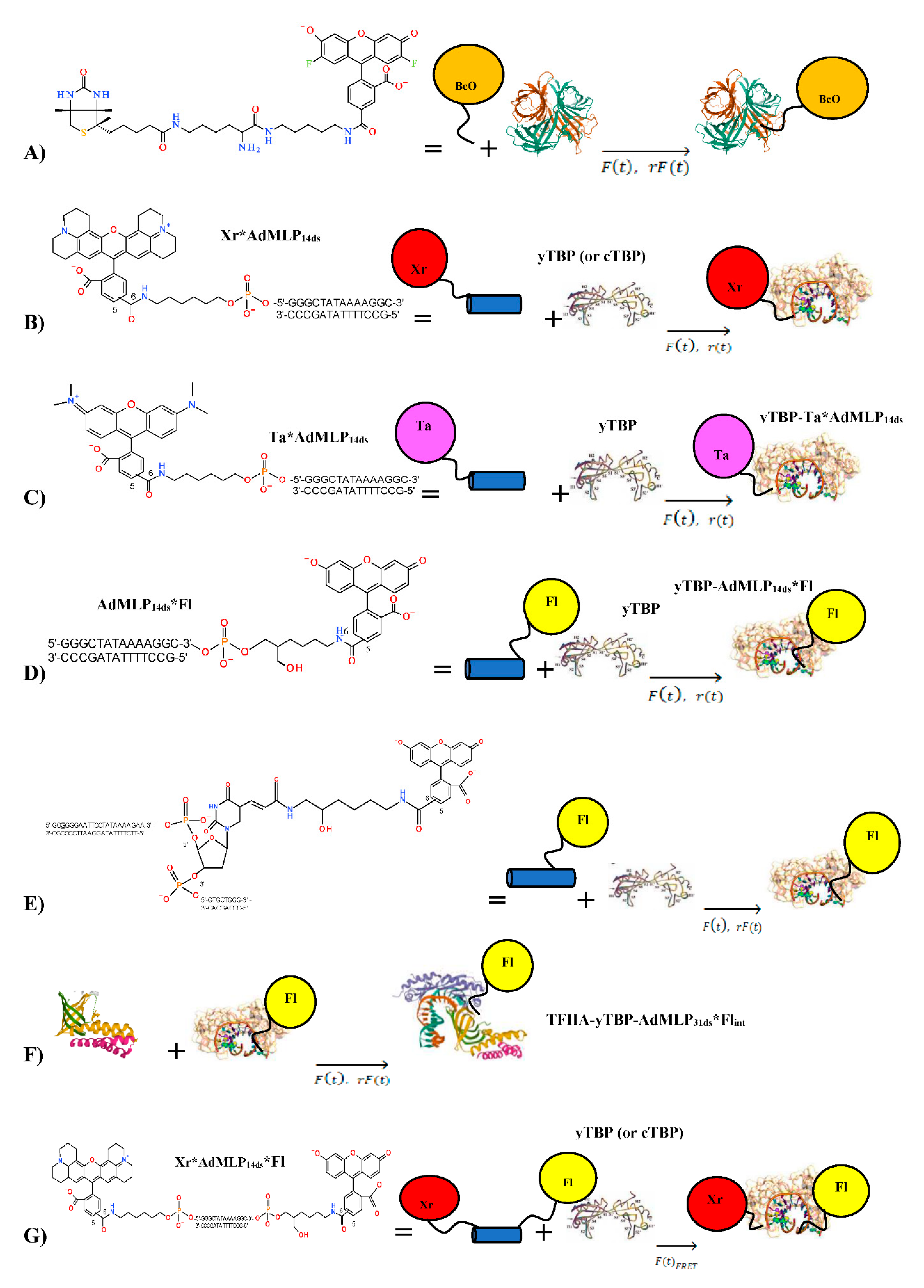
2.2. Biological AV-BcO Materials
2.3. Oligonucleotide Probe Design
2.4. Transcription Factor Proteins
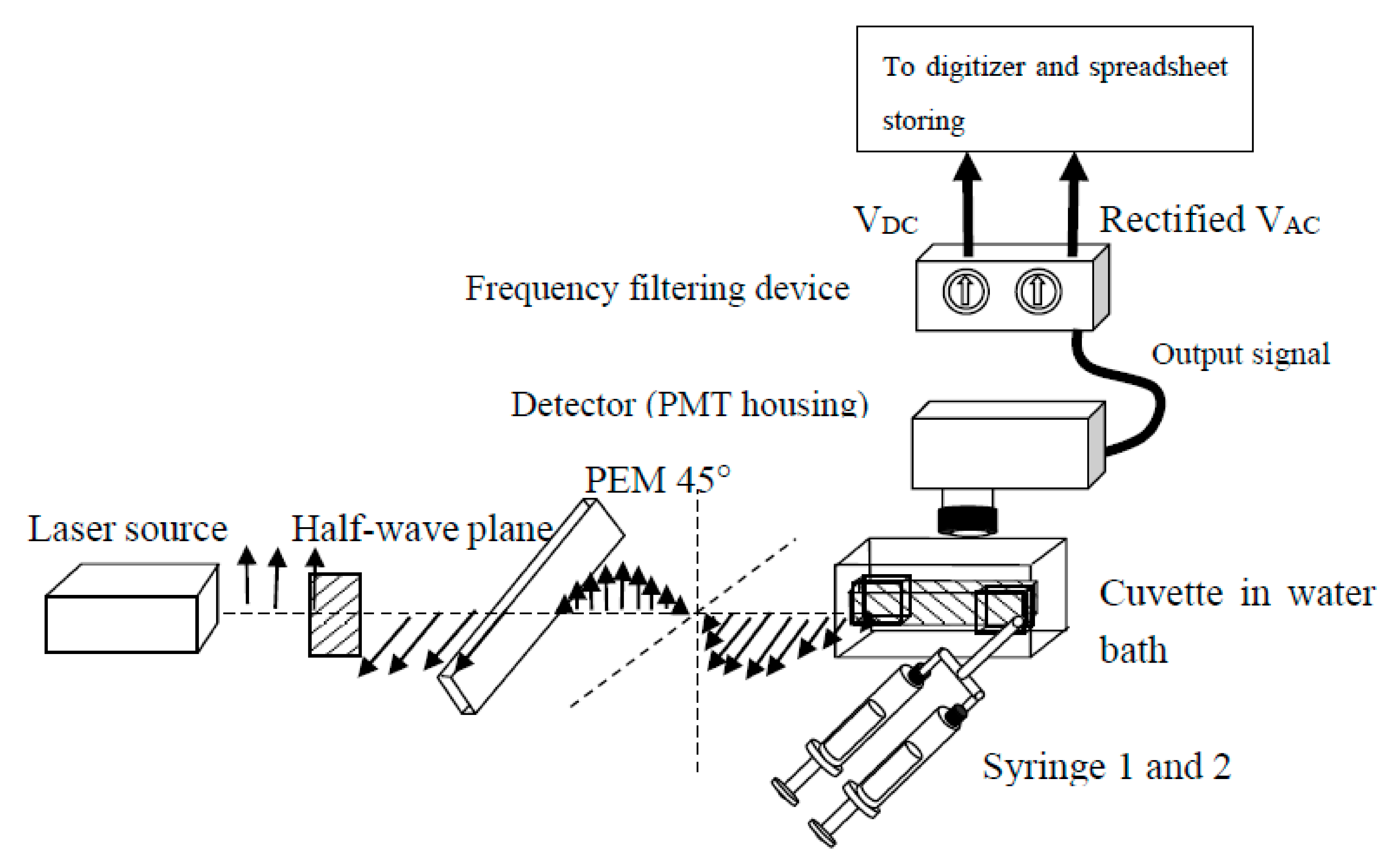
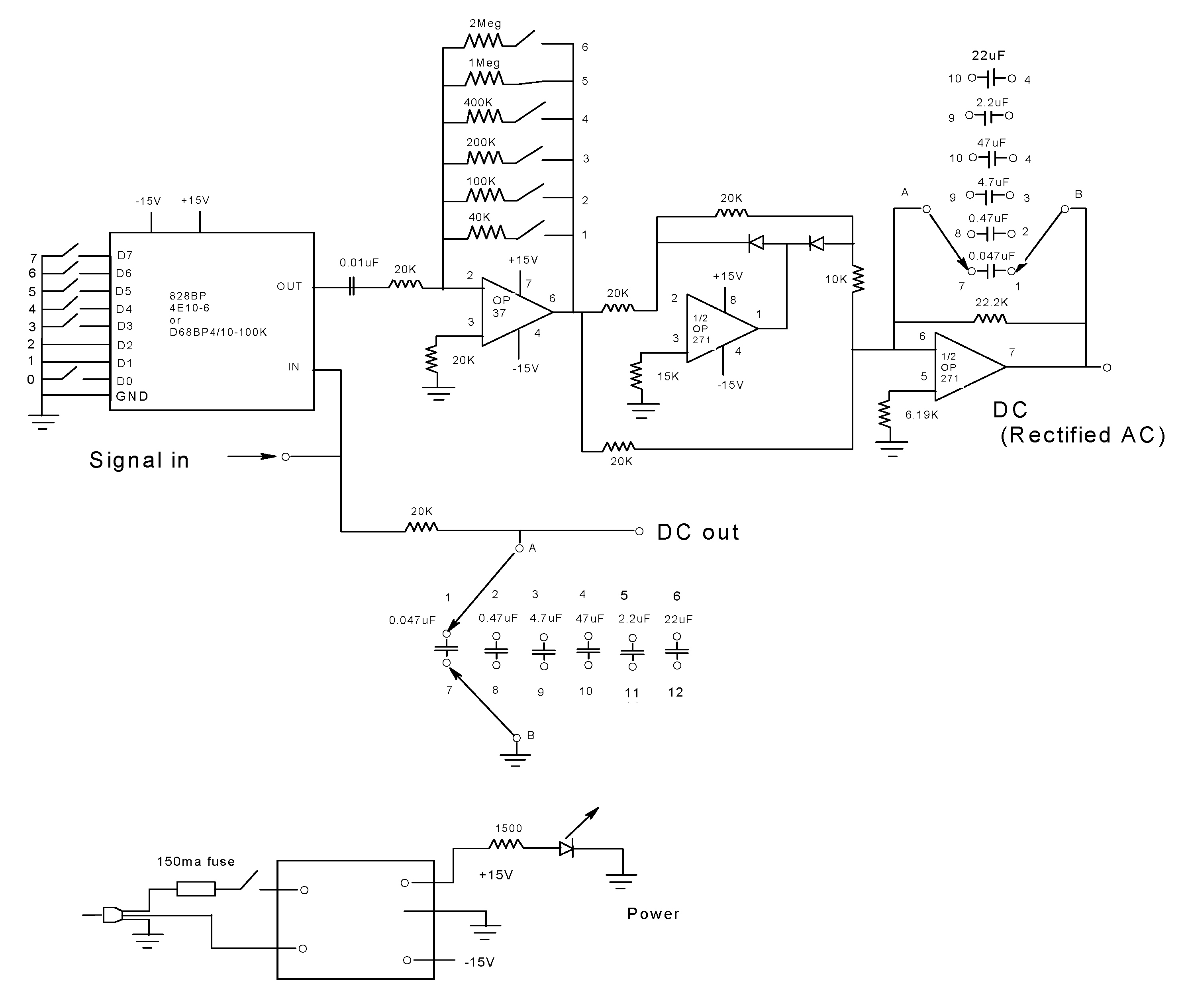
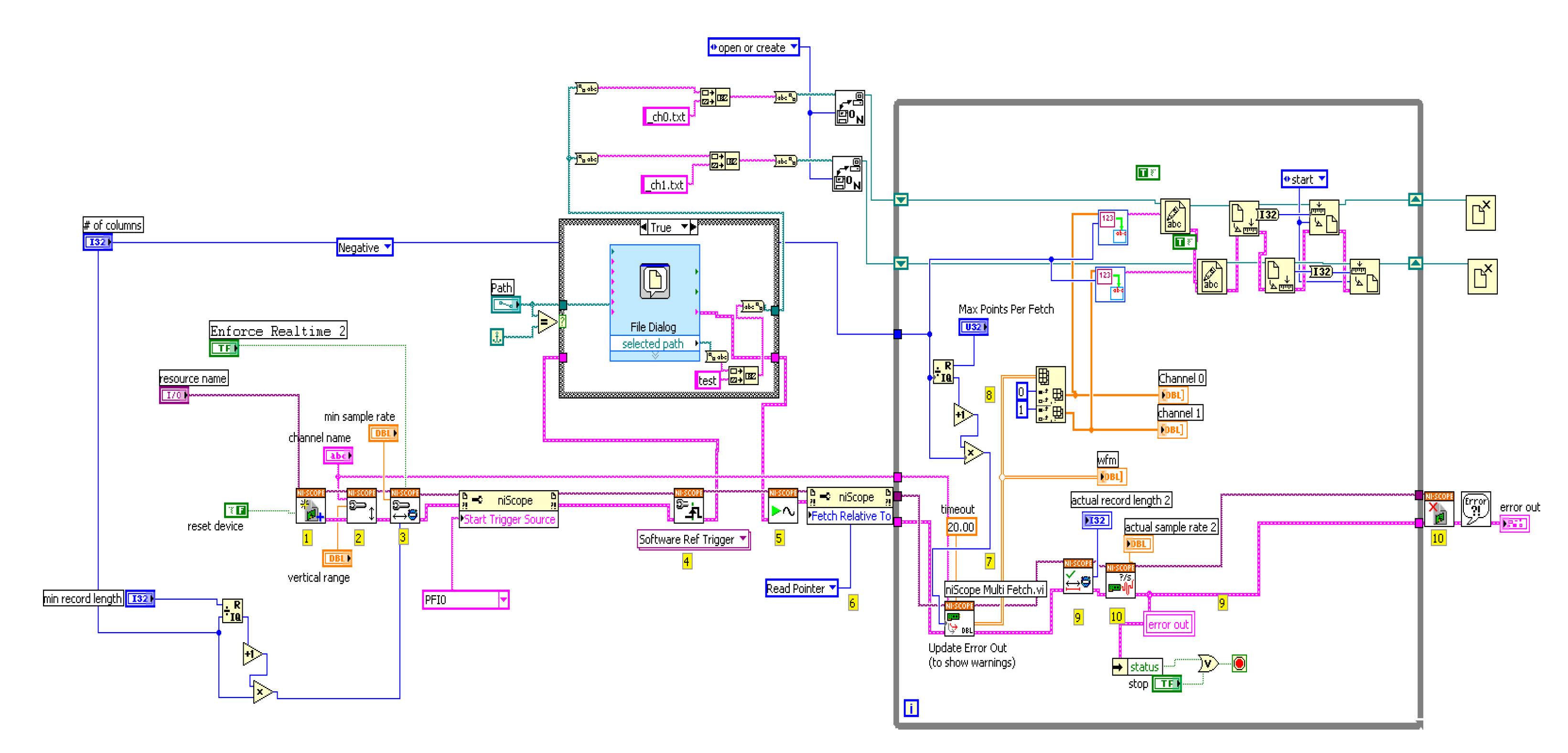
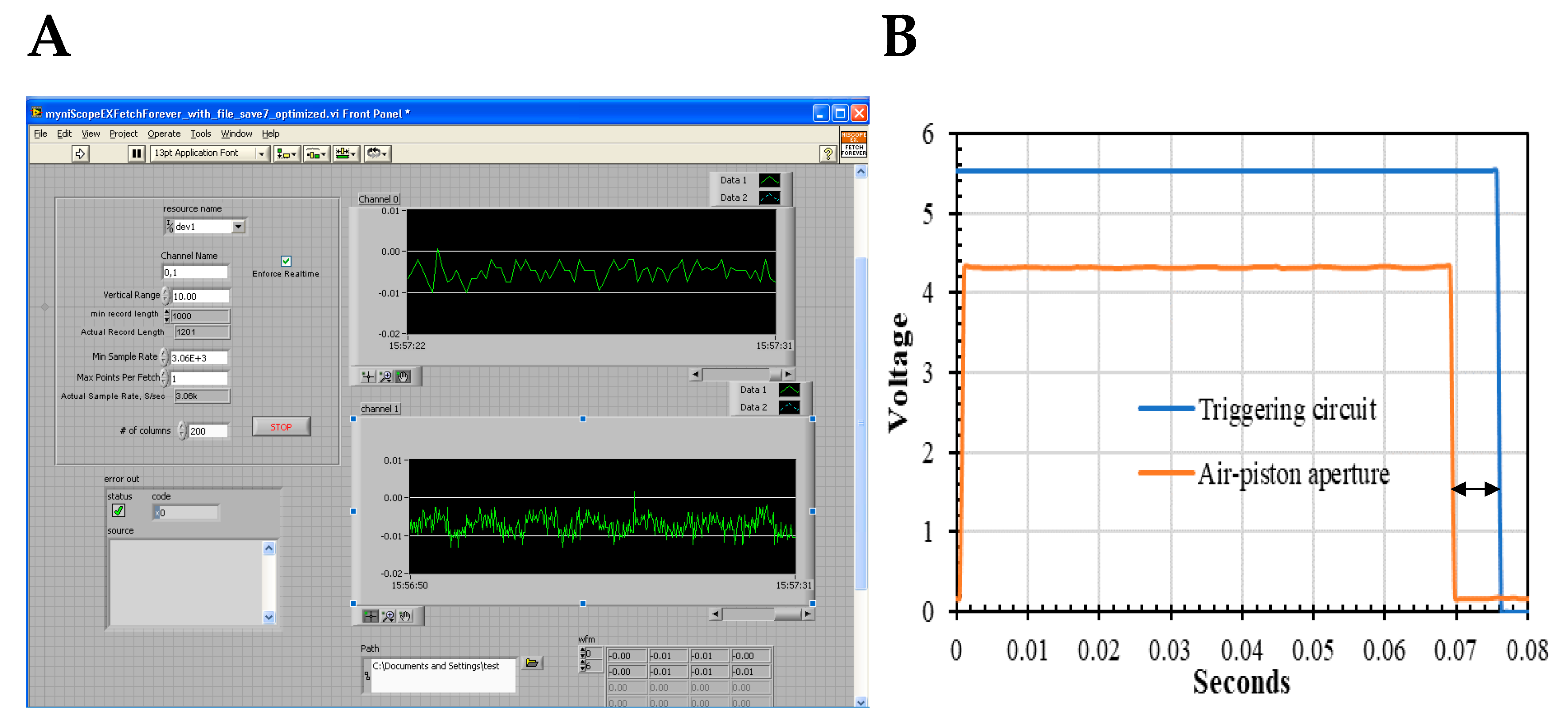
2.5. Dual-Channel SF
2.6. Stopped-Flow Association Kinetics Acquired with Excitation and Emission Polarizers
2.7. Stopped-Flow Association Kinetics Collected at the Magic Angle
2.8. Steady-State Anisotropy, rss
2.9. The F(t), r(t), and rF(t) Sensing Modalities Analysis
2.10. Stopped-Flow Association Kinetics of TBP-AdMLP14ds Collected by Energy Transfer
2.11. Time-Resolved Energy Transfer, trFRET
3. Results and Discussions
3.1. Steady-State Anisotropy
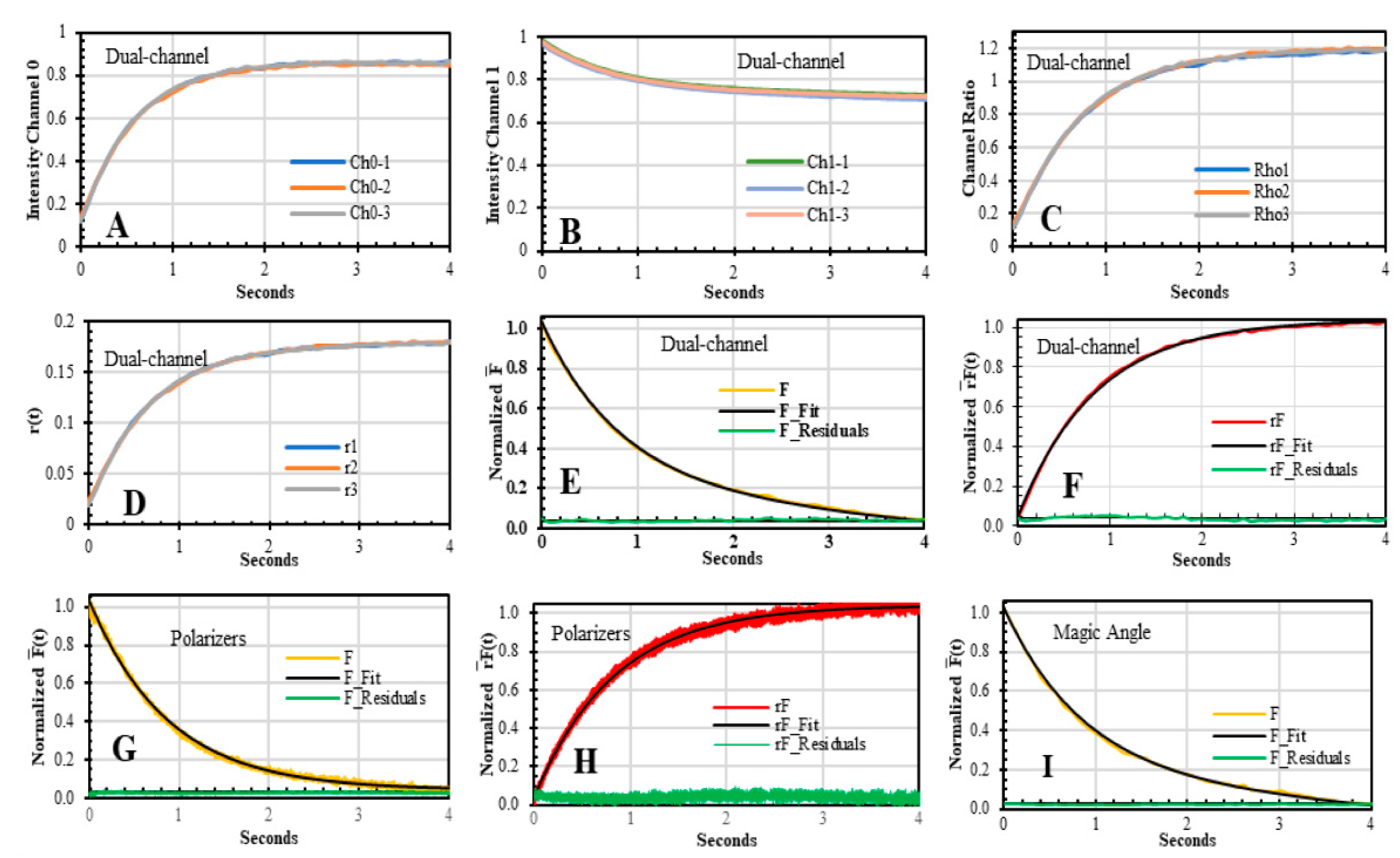
3.2. Dual-Channel SF Validation by Polarizers and Magic Angle Methodologies
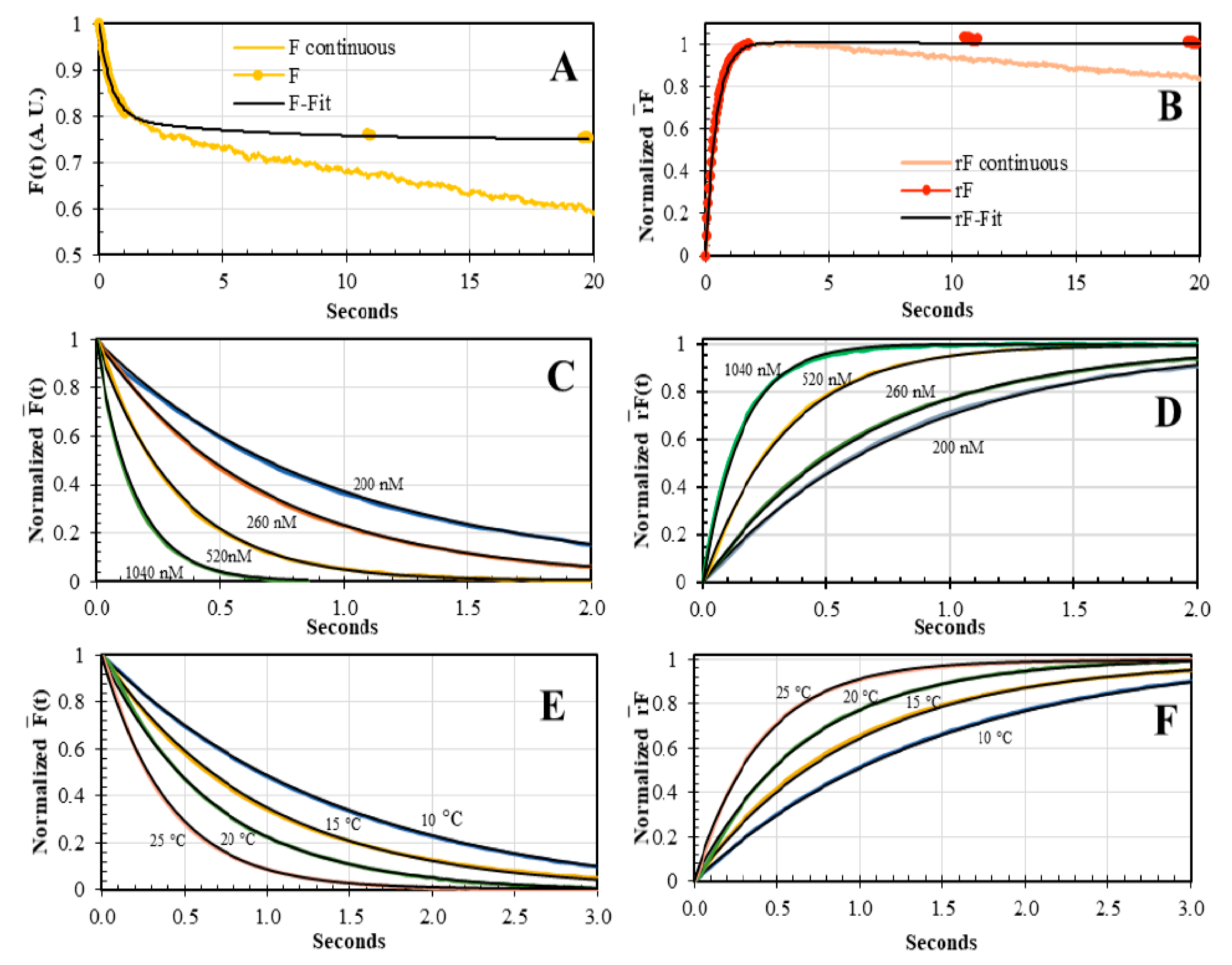
3.3. Concentration and Temperature Dependence of AV-BcO Binding Association Acquired by Dual-Channel SF
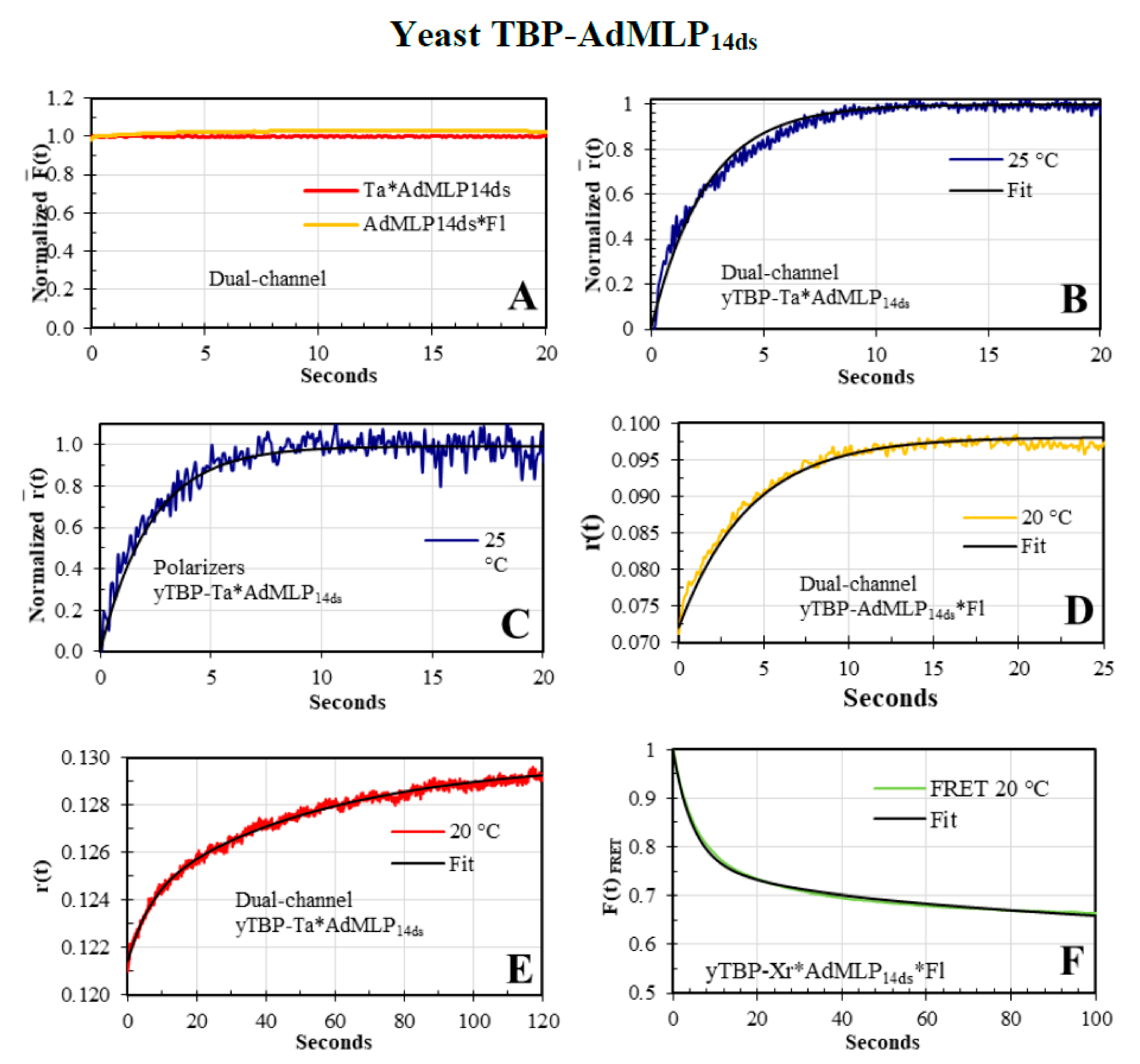
3.4. The yTBP-AdMLP14ds Association Traces Acquired by Dual-Channel and FRET SF Methodologies
3.5. Core TBP-AdMLP14ds Association Kinetics Acquired by Dual-Channel and FRET SF Methodologies
3.6. Ternary Association Kinetics of TFIIA and the Binary yTBP-AdMLP31ds Acquired by Dual-Channel SF
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goss, D.J.; Parkhurst, L.J.; Wahba, A.J. Kinetic studies of the rates and mechanism of assembly of the protein synthesis initiation complex. Biophys. J. 1980, 32, 283–293. [Google Scholar] [CrossRef]
- Delgadillo, R.F.; Whittington, J.D.E.; Parkhurst, L.K.; Parkhurst, L.J. The TATA-Binding Protein Core Domain in Solution Variably Bends TATA Sequences via a Three-Step Binding Mechanism. Biochemistry 2009, 48, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Whittington, J.D.E.; Delgadillo, R.F.; Attebury, T.J.; Parkhurst, L.K.; Daugherty, M.A.; Parkhurst, L.J. TATA-Binding Protein Recognition and Bending of a Consensus Promoter Are Protein Species Dependent. Biochemistry 2008, 47, 7264–7273. [Google Scholar] [CrossRef] [PubMed]
- Galletto, R.; Maillard, R.; Jezewska, M.J.; Bujalowski, W. Global Conformation of the Escherichia coli Replication Factor DnaC Protein in Absence and Presence of Nucleotide Cofactors. Biochemistry 2004, 43, 10988–11001. [Google Scholar] [CrossRef]
- Hartridge, H.H.; Roughton, F.J.W. A method of measuring the velocity of very rapid chemical reactions. Proc. R. Soc. Lond. Ser. A 1923, 104, 376–394. [Google Scholar]
- Chance, B. The accelerated flow method for rapid reactions. J. Franklin Inst. 1940, 229, 737–766. [Google Scholar] [CrossRef]
- Gibson, Q.H. [6] Rapid mixing: Stopped flow. Methods Enzymol. 1969, 187–228. [Google Scholar] [CrossRef]
- Berger, R.L.; Balko, B.; Borcherdt, W.; Friauf, W. High Speed Optical Stopped-Flow Apparatus. Rev. Sci. Instrum. 1968, 39, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Trojanowicz, M. Flow Chemistry in Contemporary Chemical Sciences: A Real Variety of Its Applications. Molecules 2020, 25, 1434. [Google Scholar] [CrossRef]
- Cantor, C.R.; Schimmel, P.R. Techniques for the Study of Biological Structure and Function (Pt. 2); W.H. Freeman and Co.: Oxford, UK, 1980. [Google Scholar]
- Parkhurst, K.M.; Richards, R.M.; Brenowitz, M.; Parkhurst, L.J. Intermediate species possessing bent DNA are present along the pathway to formation of a final TBP-TATA complex. J. Mol. Biol. 1999, 289, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, A.; Gómez-Hens, A.; Perez-Bendito, D. Use of stopped-flow fluorescence polarization immunoassay in drug determinations. Anal. Chim. Acta 1993, 280, 129–135. [Google Scholar] [CrossRef]
- Gaikwad, A.; Gómez-Hens, A.; Perez-Bendito, D. Estimation of opiates in urine by stopped-flow fluorimmunoassay. Fresenius J. Anal. Chem. 1993, 347, 450–453. [Google Scholar] [CrossRef]
- Pérez-Bendito, D.; Gómez-Hens, A.; Gaikwad, A. Direct stopped-flow fluorescence polarization immunoassay of abused drugs and their metabolites in urine. Clin. Chem. 1994, 40, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Sendra, B. Kinetic determination of atrazine in foods based on stopped-flow fluorescence polarization immunoassay. Talanta 1998, 47, 153–160. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Fluorescence Polarization. In Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 1983; pp. 111–112. [Google Scholar]
- Otto, M.R.; Lillo, M.P.; Beechem, J.M. Resolution of multiphasic reactions by the combination of fluorescence total-intensity and anisotropy stopped-flow kinetic experiments. Biophys. J. 1994, 67, 2511–2521. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Fluorescence Anisotropy. In Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 1999; pp. 291–319. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Fluorescence Anisotropy. In Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 2006; pp. 353–382. [Google Scholar] [CrossRef]
- Canet, D.; Doering, K.; Dobson, C.M.; Dupont, Y. High-Sensitivity Fluorescence Anisotropy Detection of Protein-Folding Events: Application to α-Lactalbumin. Biophys. J. 2001, 80, 1996–2003. [Google Scholar] [CrossRef]
- Beechem, J.M.; Sherman, M.A.; Mas, M.T. Sequential Domain Unfolding in Phosphoglycerate Kinase: Fluorescence Intensity and Anisotropy Stopped-Flow Kinetics of Several Tryptophan Mutants. Biochemistry 1995, 34, 13943–13948. [Google Scholar] [CrossRef]
- Tang, G.-Q.; Patel, S.S. Rapid Binding of T7 RNA Polymerase Is Followed by Simultaneous Bending and Opening of the Promoter DNA. Biochemistry 2006, 45, 4947–4956. [Google Scholar] [CrossRef] [PubMed]
- Galletto, R.; Bujalowski, W. Kinetics of the E. Coli replication Factor DnaC Protein−Nucleotide Interactions. II. Fluorescence Anisotropy and Transient, Dynamic Quenching Stopped-Flow Studies of the Reaction Intermediates. Biochemistry 2002, 41, 8921–8934. [Google Scholar] [CrossRef]
- Rousseau, D.L.; Staros, J.V.; Beechem, J.M. The interaction of epidermal growth factor with its receptor in A431 cell membranes: A stopped-flow fluorescence anisotropy study. Biochemistry 1995, 34, 14508–14518. [Google Scholar] [CrossRef]
- Wilkinson, J.C.; Beechem, J.M.; Staros, J.V. A stopped-flow fluorescence anisotropy method for measuring hormone binding and dissociation kinetics with cell-surface receptors in living cells. J. Recept. Signal Transduct. 2002, 22, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.; Hunter, J.L.; Eccleston, J.F.; Webb, M.R. The Mechanism of Ras GTPase Activation by Neurofibromin. Biochemistry 2003, 42, 3956–3965. [Google Scholar] [CrossRef] [PubMed]
- Bradrick, T.; Philippetis, A.; Georghiou, S. Stopped-flow fluorometric study of the interaction of melittin with phospholipid bilayers: Importance of the physical state of the bilayer and the acyl chain length. Biophys. J. 1995, 69, 1999–2010. [Google Scholar] [CrossRef]
- Badoz, J.; Billardon, M.; Canit, J.C.; Russel, M.F. Sensitive devices to determine the state and degree of polarization of a light beam using a birefringence modulator. J. Opt. 1977, 8, 373–384. [Google Scholar] [CrossRef]
- Wampler, J.E.; DeSa, R.J. Recording Polarization of Fluorescence Spectrometer—A Unique Application of Piezoelectric Birefringence Modulation. Anal. Chem. 1974, 46, 563–567. [Google Scholar] [CrossRef]
- Giblin, D.E. A Modular Instrument for the Measurement of Transient Circular Dichroism, Fluorescence Polarization and Emission Anisotropy; University of Nebraska-Lincoln: Lincoln, NE, USA, 1978. [Google Scholar]
- Kornberg, R.D. The molecular basis of eucaryotic transcription. Cell Death Differ. 2007, 14, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef]
- Delgadillo, R.F. Acceptor Detected Fluorescence Resonance Energy Transfer for Measurement up to 250 A and Biophysical Studies on Core TATA Binding Protein-DNA Complex; University of Nebraska-Lincoln: Lincoln, NE, USA, 2009; Available online: http://digitalcommons.unl.edu/dissertations/AAI3388957/ (accessed on 1 October 2020).
- Matsui, T.; Segall, J.; A Weil, P.; Roeder, R.G. Multiple factors required for accurate initiation of transcription by purified RNA polymerase II. J. Biol. Chem. 1980, 255, 11992–11996. [Google Scholar]
- Kramm, K.; Engel, C.; Grohmann, D. Transcription initiation factor TBP: Old friend new questions. Biochem. Soc. Trans. 2019, 47, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, T.W.; Buratowski, S. The basal initiation machinery: Beyond the general transcription factors. Curr. Opin. Cell Biol. 2009, 21, 344–351. [Google Scholar] [CrossRef]
- Liu, Q.; Gabriel, S.E.; Roinick, K.L.; Ward, R.D.; Arndt, K.M. Analysis of TFIIA Function In Vivo: Evidence for a Role in TATA-Binding Protein Recruitment and Gene-Specific Activation. Mol. Cell. Biol. 1999, 19, 8673–8685. [Google Scholar] [CrossRef][Green Version]
- Hieb, A.R.; Halsey, W.A.; Betterton, M.D.; Perkins, T.T.; Kugel, J.F.; Goodrich, J.A. TFIIA Changes the Conformation of the DNA in TBP/TATA Complexes and Increases their Kinetic Stability. J. Mol. Biol. 2007, 372, 619–632. [Google Scholar] [CrossRef]
- Bhuiyan, T.; Timmers, H.M. Promoter Recognition: Putting TFIID on the Spot. Trends Cell Biol. 2019, 29, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Delgadillo, R.F.; Parkhurst, L.J. Spectroscopic Properties of Fluorescein and Rhodamine Dyes Attached to DNA. Photochem. Photobiol. 2010, 86, 261–272. [Google Scholar] [CrossRef]
- Young, C.S.H. The Structure and Function of the Adenovirus Major Late Promoter. Future Hiv-1 Ther. 2003, 213–249. [Google Scholar] [CrossRef]
- Parkhurst, K.M.; Brenowitz, M.; Parkhurst, L.J. Simultaneous Binding and Bending of Promoter DNA by the TATA Binding Protein: Real Time Kinetic Measurements†. Biochemistry 1996, 35, 7459–7465. [Google Scholar] [CrossRef]
- Livnah, O.; Bayer, E.A.; Wilchek, M.; Sussman, J.L. Three-dimensional structures of avidin and the avidin-biotin complex. Proc. Natl. Acad. Sci. USA 1993, 90, 5076–5080. [Google Scholar] [CrossRef]
- Delgadillo, R.F.; Mueser, T.C.; Zaleta-Rivera, K.; Carnes, K.A.; González-Valdez, J.; Parkhurst, L.J. Detailed characterization of the solution kinetics and thermodynamics of biotin, biocytin and HABA binding to avidin and streptavidin. PLoS ONE 2019, 14, e0204194. [Google Scholar] [CrossRef] [PubMed]
- Bleichenbacher, M.; Tan, S.; Richmond, T.J. Novel Interactions Between the Components of Human and Yeast TFIIA/TBP/DNA Complexes. J. Mol. Biol. 2003, 332, 783–793. [Google Scholar] [CrossRef]
- Goerisch, H.; Goss, D.J.; Parkhurst, L.J. Kinetics of ribosome dissociation and subunit association studied in a light-scattering stopped-flow apparatus. Biochemistry 1976, 15, 5743–5753. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Nichols, S.M.; Kahr, B. Mueller matrices in fluorescence scattering. Opt. Lett. 2012, 37, 2835–2837. [Google Scholar] [CrossRef]
- Fisz, J. Another Look at Magic-Angle-Detected Fluorescence and Emission Anisotropy Decays in Fluorescence Microscopy. J. Phys. Chem. A 2007, 111, 12867–12870. [Google Scholar] [CrossRef]
- Parkhurst, L.J.; Parkhurst, K.M.; Powell, R.; Wu, J.; Williams, S. Time-resolved fluorescence resonance energy transfer studies of DNA bending in double-stranded oligonucleotides and in DNA-protein complexes. Biochemistry 2002, 61, 180–200. [Google Scholar] [CrossRef]
- Wu, J.; Parkhurst, K.M.; Powell, R.M.; Parkhurst, L.J. DNA Sequence-dependent Differences in TATA-binding Protein-induced DNA Bending in Solution Are Highly Sensitive to Osmolytes. J. Biol. Chem. 2001, 276, 14623–14627. [Google Scholar] [CrossRef]
- Marquardt, D.W. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Durbin, J.; Watson, G.S. Testing for Serial Correlation in Least Squares Regression. II. Biometrika 1951, 38, 159. [Google Scholar] [CrossRef] [PubMed]
- Swed, F.S.; Eisenhart, C. Tables for Testing Randomness of Grouping in a Sequence of Alternatives. Ann. Math. Stat. 1943, 14, 66–87. [Google Scholar] [CrossRef]
- Cianfrocco, M.A.; Kassavetis, G.A.; Grob, P.; Fang, J.; Juven-Gershon, T.; Kadonaga, J.T.; Nogales, E. Human TFIID Binds to Core Promoter DNA in a Reorganized Structural State. Cell 2013, 152, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Kramm, K.; Schröder, T.; Gouge, J.; Vera, A.M.; Heiss, F.B.; Liedl, T.; Engel, C.; Vannini, A.; Tinnefeld, P.; Grohmann, D.; et al. DNA origami-based single-molecule force spectroscopy unravels the molecular basis of RNA Polymerase III pre-initiation complex stability. bioRxiv 2019, 775528. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Arrhenius, S. Über die Dissociationswärme und den Einfluss der Temperatur auf den Dissociationsgrad der Elektrolyte. Z. Für Phys. Chem. 1889, 4, 96–116. [Google Scholar]
- Arrhenius, S. Über die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren. Z. Für Phys. Chem. 1889, 4, 226–248. [Google Scholar]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. Trans. Faraday Soc. 1935, 31, 875–894. [Google Scholar] [CrossRef]
- Pihan, E.; Delgadillo, R.F.; Tonkin, M.L.; Pugnière, M.; Lebrun, M.; Boulanger, M.J.; Douguet, D. Computational and biophysical approaches to protein-protein interaction inhibition of Plasmodium falciparum AMA1/RON2 complex. J. Comput Aided Mol. Des. 2015, 29, 525–539. [Google Scholar] [CrossRef]
- Delgadillo, R.F.; Parker, M.L.; Lebrun, M.; Boulanger, M.J.; Douguet, D. Stability of the Plasmodium falciparum AMA1-RON2 Complex Is Governed by the Domain II (DII) Loop. PLoS ONE 2016, 11, e0144764. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
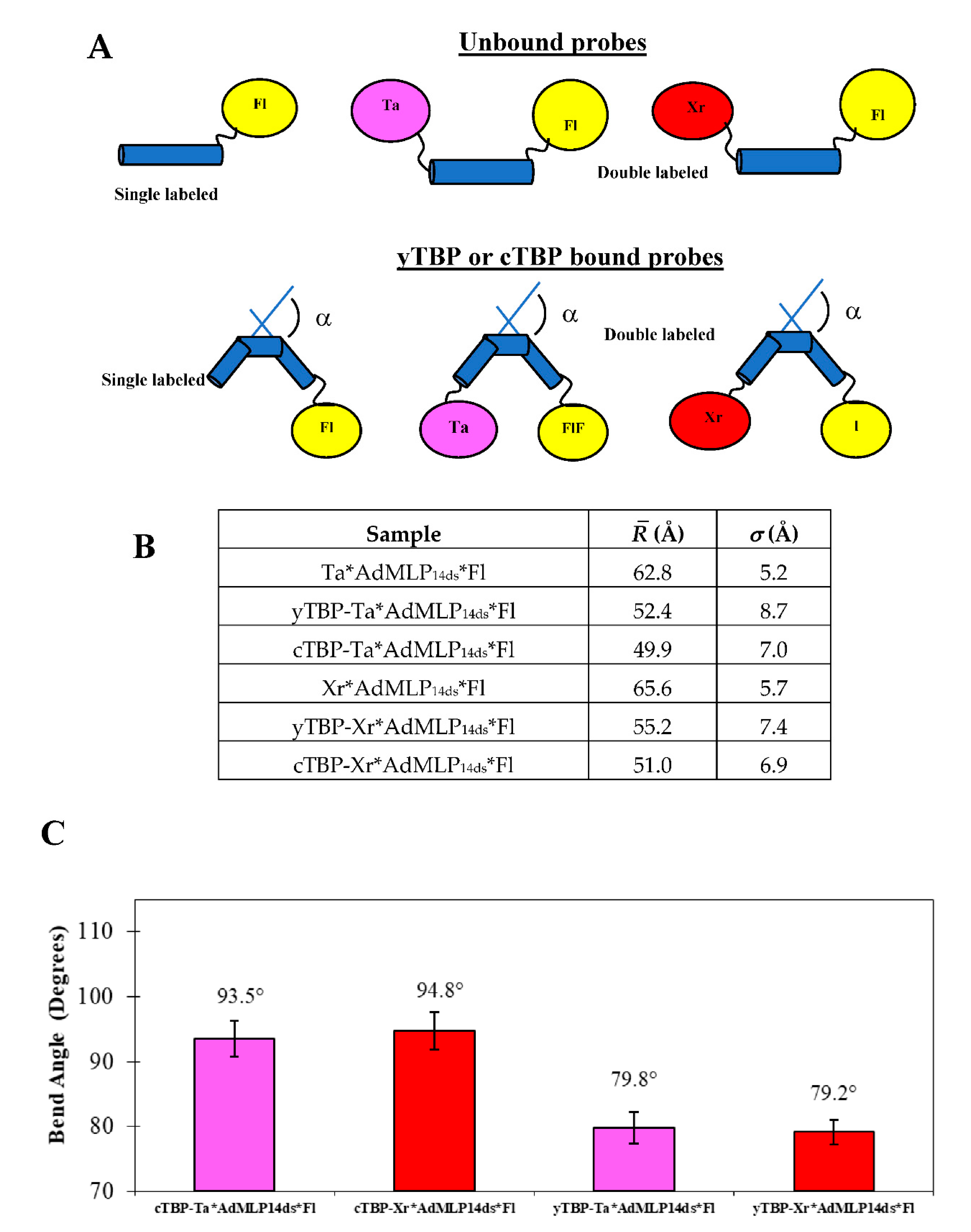{kind=link}
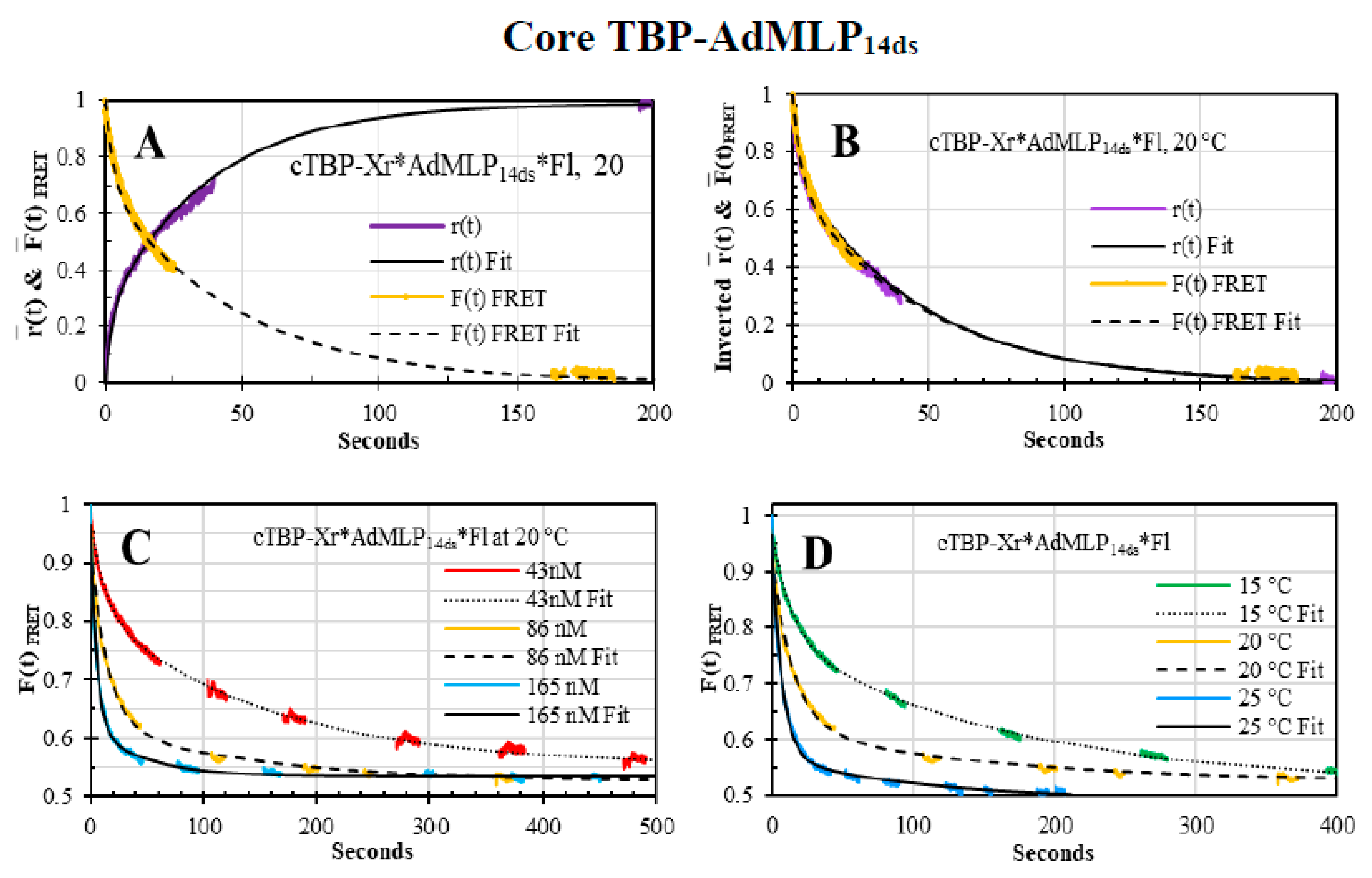{kind=link}
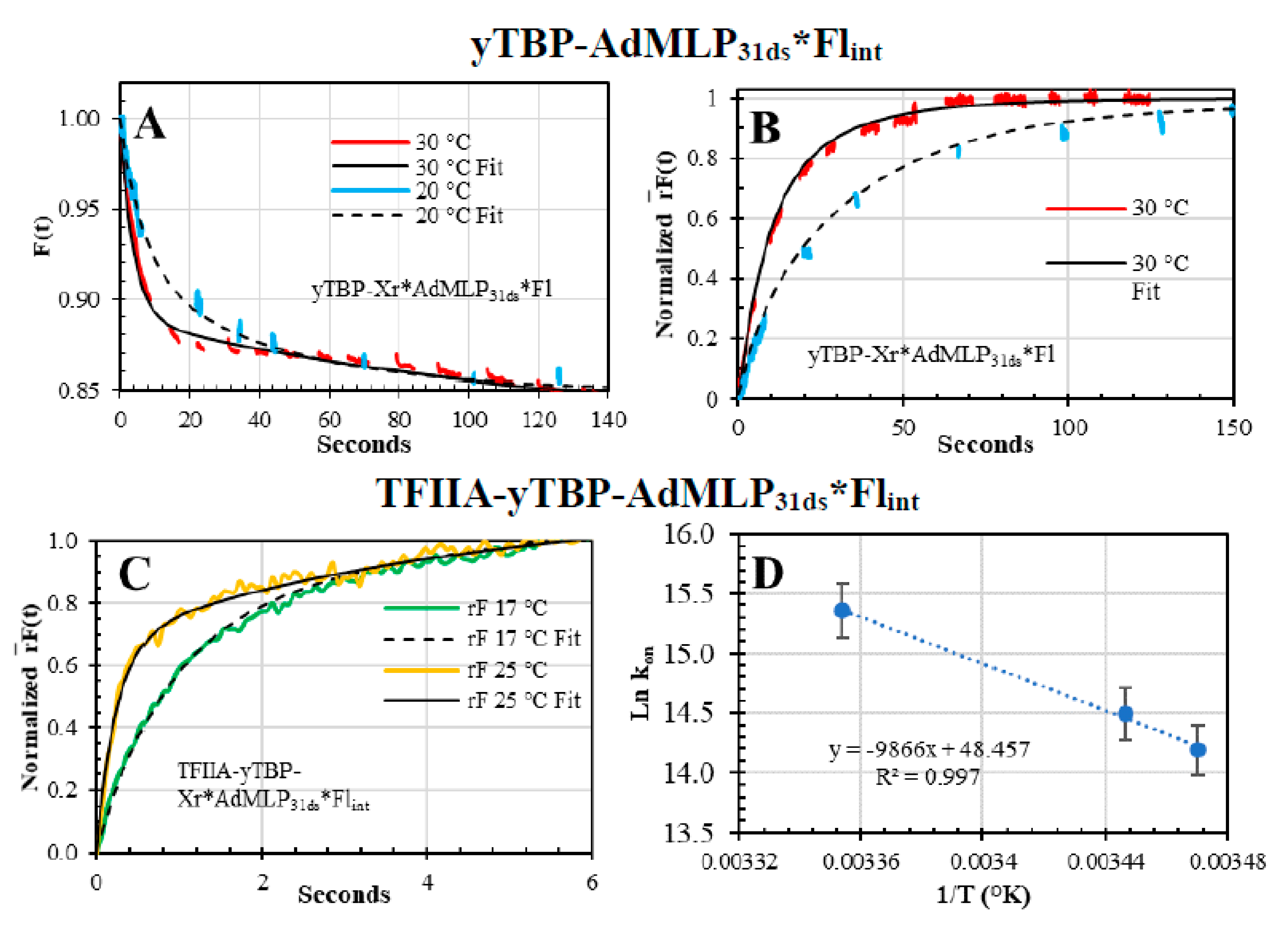{kind=link}
| SF Methodology | Signal | Reaction | Syringe 1 a | Syringe 2 |
|---|---|---|---|---|
| Magic angle | F(t) | AV-BcO | BcO | AV |
| Polarizers | r(t), F(t),& rF(t) | AV-BcO | BcO | AV |
| yTBP-Ta*AdMLP14ds*Fl | 5′-Xr*GGGCTATAAAAGGC*Fl-3′ 3′-CCCGATATTTTCCG-5′ | yTBP | ||
| cTBP-Xr*AdMLP14ds | 5′-Xr*GGGCTATAAAAGGC*Fl-3′ 3′-CCCGATATTTTCCG-5′ | cTBP | ||
| Dual-Channel | r(t), F(t),& rF(t) | AV-BcO | BcO | AV |
| yTBP-Ta*AdMLP14ds | 5′-Ta*GGGCTATAAAAGGC-3′ 3′-CCCGATATTTTCCG-5′ | yTBP | ||
| yTBP-AdMLP14ds*Fl | 5′-GGGCTATAAAAGGC*Fl-3′ 3′-CCCGATATTTTCCG-5′ | yTBP | ||
| yTBP-AdMLP31ds*Flint | 5′-GCGGGGAATTCCTATAAAAGAA(T-Fl)GTGCTGGG-3′ 3′-CGCCCCTTAAGGATATTTTCTTACACGACCC-5′ | yTBP | ||
| yTFIIA-yTBP-AdMLP31ds*Flint b | yTBP + 5′-GCGGGGAATTCCTATAAAAGAA(T-Fl)GTGCTGGG-3′ 3′-CGCCCCTTAAGGATATTTTCTTACACGACCC-5′ | yTFIIA | ||
| FRET | F(t)FRET | yTBP-Xr*AdMLP14ds*Fl | 5′-Xr*GGGCTATAAAAGGC*Fl-3′ 3′-CCCGATATTTTCCG-5′ | yTBP |
| cTBP-Xr*AdMLP14ds*Fl | 5′-Xr*GGGCTATAAAAGGC*Fl-3′ 3′-CCCGATATTTTCCG-5′ | cTBP |
| Sample | rss (Free Probe) a | rss (Complex) b | QY (Free Probe) | QY (Complex) |
|---|---|---|---|---|
| BcO (25 °C) | 0.018 ± 0.001 c 0.025 ± 0.001 d | +AV: 0.180 ± 0.003 c +AV: 0.177 ± 0.004 d | 0.91 ± 0.01 | 0.68 ± 0.02 |
| BcO (20 °C) | 0.025 ± 0.001 d | +AV: 0.185 ± 0.004 d | ||
| BcO (15 °C) | 0.054 ± 0.001 d | +AV: 0.176 ± 0.004 d | ||
| BcO (10 °C) | 0.055 ± 0.001 d | +AV: 0.202 ± 0.004 d | ||
| Ta*AdMLP14ds (25 °C) | 0.164 ± 0.002 d | +yTBP: 0.192 ± 0.010 d | 0.20 ± 0.01 | 0.20 ± 0.01 |
| Ta*AdMLP14ds (20 °C) | 0.122 ± 0.002 d | +yTBP: 0.131 ± 0.005 d | 0.20 ± 0.01 | 0.20 ± 0.01 |
| AdMLP14ds*Fl (20 °C) | 0.071 ± 0.013 d 0.068 ± 0.008 c | +yTBP: 0.097 ± 0.002 d | 0.22 ± 0.01 | 0.22 ± 0.01 |
| Xr*AdMLP4ds (20 °C) | 0.122 ± 0.003 d 0.122 ± 0.001 c | +cTBP: 0.130 ± 0.002 d | 0.10 ± 0.01 | 0.10 ± 0.01 |
| AdMLP31ds*Flint (20 °C) | 0.043 ± 0.004 c 0.040 ± 0.004 d | +yTBP: 0.201 ± 0.005 d | 0.83 ± 0.03 | 0.71 ± 0.03 |
| yTBP-AdMLP31ds*Flint (17 °C) | 0.201 ± 0.005 d | +yTFIIA: 0.260 ± 0.002 d | 0.71 ± 0.03 | 0.82 ± 0.03 |
| yTBP-AdMLP31ds*Flint (25 °C) | 0.198 ± 0.005 d | +yTFIIA: 0.217 ± 0.002 d | 0.71 ± 0.03 | 0.72 ± 0.03 |
| Fluorescence, a | λ (s−1) | kon (×10−6 M−1s−1) | Error (%) c |
|---|---|---|---|
| Dual channel | 1.186 ± 0.043 | 5.93 ± 0.22 | 3.6 |
| Polarizers | 1.187 ± 0.083 | 5.94 ± 0.42 | 7.0 |
| Magic angle | 1.199 ± 0.099 | 5.99 ± 0.60 | 8.3 |
| rF(t)b | λ (s−1) | kon(×10−6M−1s−1) | Error (%)d |
| Dual channel | 1.183 ± 0.023 | 5.92 ± 0.12 | 2.0 |
| Polarizers | 1.198 ± 0.039 | 5.99 ± 0.20 | 3.3 |
| Magic angle | NA | NA | NA |
| 200 nM | 260 nM | 520 nM | 1040 nM | ||||||
|---|---|---|---|---|---|---|---|---|---|
| λ(s−1) | |||||||||
| 10 °C | NA | NA | 0.629 ± 0.042 | 0.690 ± 0.035 | 1.355 ± 0.047 | 1.372 ± 0.003 | 2.825 ± 0.107 | 2.620 ± 0.012 | |
| 15 °C | NA | NA | 1.058 ± 0.016 | 1.035 ± 0.005 | 1.840 ± 0.055 | 1.840 ± 0.009 | 4.000 ± 0.108 | 4.200 ± 0.365 | |
| 20 °C | 1.186 ± 0.043 | 1.183 ± 0.023 | 1.491 ± 0.027 | 1.543 ± 0.014 | 3.045 ± 0.085 | 3.024 ± 0.030 | 6.209 ± 0.571 | 6.357 ± 0.058 | |
| 25 °C | NA | NA | 2.465 ± 0.014 | 2.473 ± 0.018 | 4.920 ± 0.093 | 4.920 ± 0.034 | 10.031 ± 0.401 | 9.810 ± 0.091 | |
| kon ×10−6 M−1s−1 | kon(Average) | ||||||||
| 10 °C | NA | NA | 2.419 ± 0.162 | 2.655 ± 0.133 | 2.606 ± 0.091 | 2.638 ± 0.006 | 2.520 ± 0.010 | 2.716 ± 0.012 | 2.592 ± 0.107 |
| 15 °C | NA | NA | 4.069 ± 0.061 | 3.980 ± 0.021 | 3.539 ± 0.106 | 3.539 ± 0.018 | 3.846 ± 0.104 | 4.038 ± 0.351 | 3.835 ± 0.242 |
| 20 °C | 5.931 ± 0.216 | 5.931 ± 0.117 | 5.733 ± 0.103 | 5.935 ± 0.054 | 5.855 ± 0.164 | 5.815 ± 0.058 | 5.970 ± 0.549 | 6.113 ± 0.056 | 5.904 ± 0.133 |
| 25 °C | NA | NA | 9.479 ± 0.052 | 9.513 ± 0.068 | 9.461 ± 0.180 | 9.461 ± 0.066 | 9.646 ± 0.386 | 9.433 ± 0.088 | 9.499 ± 0.077 |
| Sample | χ | DW | Z | α | τ(ns) | α | τ(ns) | φ(ns) a | φ(ns) | αiτi (ns) b |
|---|---|---|---|---|---|---|---|---|---|---|
| AdMLP14ds*Fl | 0.985 ± 0.030 | 1.965 ± 0.135 | 0.069 ± 0.400 | 0.340 ± 0.033 | 0.852 ± 0.098 | 0.660 ± 0.033 | 3.729 ± 0.043 | 0.105 ± 0.012 | 0.895 ± 0.012 | 2.749 ± 0.132 |
| Xr*AdMLP14ds*Fl | 1.008 ± 0.030 | 2.027 ± 0.115 | −0.075 ± 0.352 | 0.494 ± 0.033 | 0.645 ± 0.097 | 0.506 ± 0.033 | 2.493 ± 0.119 | 0.201 ± 0.022 | 0.799 ± 0.022 | 1.575 ± 0.066 |
| Ta*AdMLP14ds*Fl | 0.984 ± 0.040 | 1.942 ± 0.067 | −0.021 ± 0.170 | 0.520 ± 0.014 | 0.607 ± 0.034 | 0.480 ± 0.014 | 2.230 ± 0.037 | 0.279 ± 0.049 | 0.949 ± 0.173 | 1.385 ± 0.042 |
| cTBP-AdMLP14ds*Fl | 1.009 ± 0.022 | 1.987 ± 0.132 | −0.006 ± 0.404 | 0.315 ± 0.013 | 0.826 ± 0.123 | 0.685 ± 0.013 | 3.823 ± 0.084 | 0.090 ± 0.012 | 0.910 ± 0.012 | 2.878 ± 0.096 |
| yTBP-AdMLP14ds*Fl | 1.016 ± 0.029 | 1.916 ± 0.086 | −0.050 ± 0.349 | 0.330 ± 0.018 | 0.852 ± 0.060 | 0.670 ± 0.018 | 3.998 ± 0.127 | 0.095 ± 0.005 | 0.905 ± 0.005 | 2.959 ± 0.149 |
| cTBP-Xr*AdMLP14ds*Fl | 1.018 ± 0.026 | 2.017 ± 0.157 | 0.160 ± 0.313 | 0.715 ± 0.036 | 0.428 ± 0.043 | 0.285 ± 0.036 | 1.763 ± 0.112 | 0.381 ± 0.048 | 0.619 ± 0.048 | 0.805 ± 0.034 |
| yTBP-Xr*AdMLP14ds*Fl | 1.016 ± 0.010 | 1.916 ± 0.045 | −0.185 ± 0.114 | 0.668 ± 0.013 | 0.529 ± 0.035 | 0.332 ± 0.013 | 2.077 ± 0.047 | 0.339 ± 0.010 | 0.661 ± 0.010 | 1.042 ± 0.040 |
| cTBP-Ta*AdMLP14ds*Fl | 1.011 ± 0.024 | 1.972 ± 0.142 | 0.388 ± 0.234 | 0.772 ± 0.012 | 0.434 ± 0.010 | 0.228 ± 0.012 | 1.601 ± 0.022 | 0.335 ± 0.010 | 0.364 ± 0.014 | 0.699 ± 0.010 |
| yTBP-Ta*AdMLP14ds*Fl | 0.974 ± 0.058 | 1.902 ± 0.106 | 0.299 ± 0.344 | 0.806 ± 0.008 | 0.564 ± 0.032 | 0.194 ± 0.008 | 2.264 ± 0.068 | 0.509 ± 0.007 | 0.491 ± 0.007 | 0.894 ± 0.046 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgadillo, R.F.; Carnes, K.A.; Valles-Villarreal, N.; Olmos, O.; Zaleta-Rivera, K.; Parkhurst, L.J. Dual-Channel Stopped-Flow Apparatus for Simultaneous Fluorescence, Anisotropy, and FRET Kinetic Data Acquisition for Binary and Ternary Biological Complexes. Biosensors 2020, 10, 180. https://doi.org/10.3390/bios10110180
Delgadillo RF, Carnes KA, Valles-Villarreal N, Olmos O, Zaleta-Rivera K, Parkhurst LJ. Dual-Channel Stopped-Flow Apparatus for Simultaneous Fluorescence, Anisotropy, and FRET Kinetic Data Acquisition for Binary and Ternary Biological Complexes. Biosensors. 2020; 10(11):180. https://doi.org/10.3390/bios10110180
Chicago/Turabian StyleDelgadillo, Roberto F., Katie A. Carnes, Nestor Valles-Villarreal, Omar Olmos, Kathia Zaleta-Rivera, and Lawrence J. Parkhurst. 2020. "Dual-Channel Stopped-Flow Apparatus for Simultaneous Fluorescence, Anisotropy, and FRET Kinetic Data Acquisition for Binary and Ternary Biological Complexes" Biosensors 10, no. 11: 180. https://doi.org/10.3390/bios10110180
APA StyleDelgadillo, R. F., Carnes, K. A., Valles-Villarreal, N., Olmos, O., Zaleta-Rivera, K., & Parkhurst, L. J. (2020). Dual-Channel Stopped-Flow Apparatus for Simultaneous Fluorescence, Anisotropy, and FRET Kinetic Data Acquisition for Binary and Ternary Biological Complexes. Biosensors, 10(11), 180. https://doi.org/10.3390/bios10110180