Chitosan-Derived Porous Activated Carbon for the Removal of the Chemical Warfare Agent Simulant Dimethyl Methylphosphonate



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Preparation of Activated Chitosan Carbon
2.2. Adsorption of DMMP on Activated Carbon Derived from Chitosan
2.3. Desorption of DMMP from Activated Carbon Derived from Chitosan
2.4. Instrumentation
3. Results
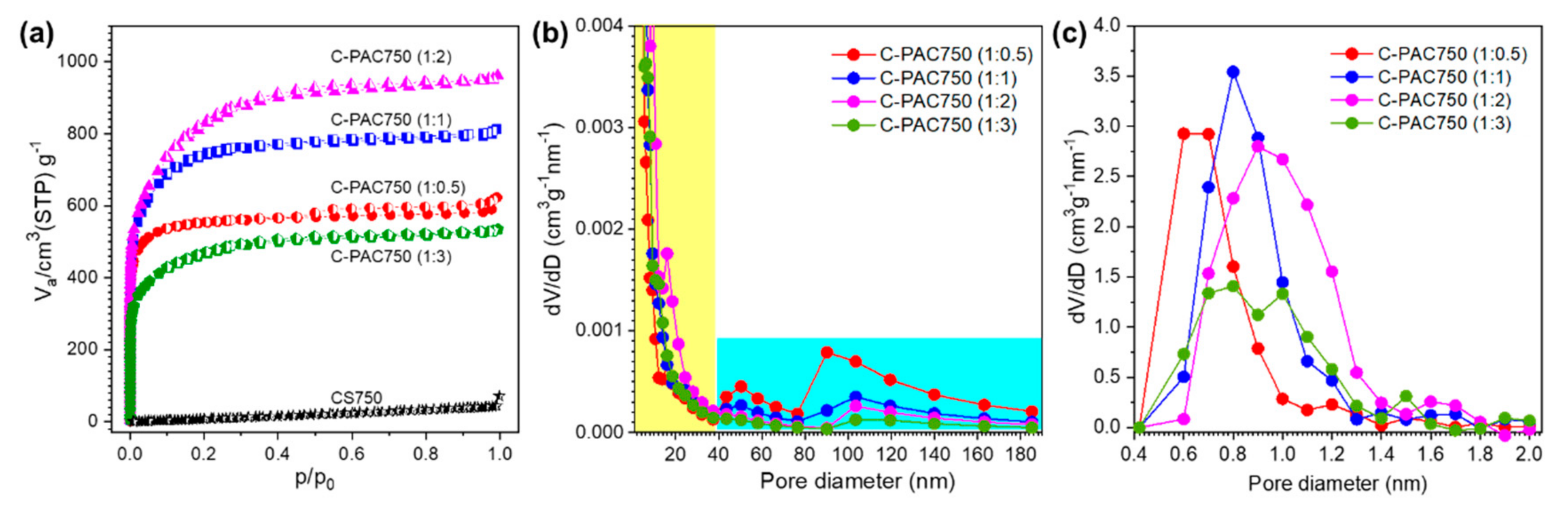
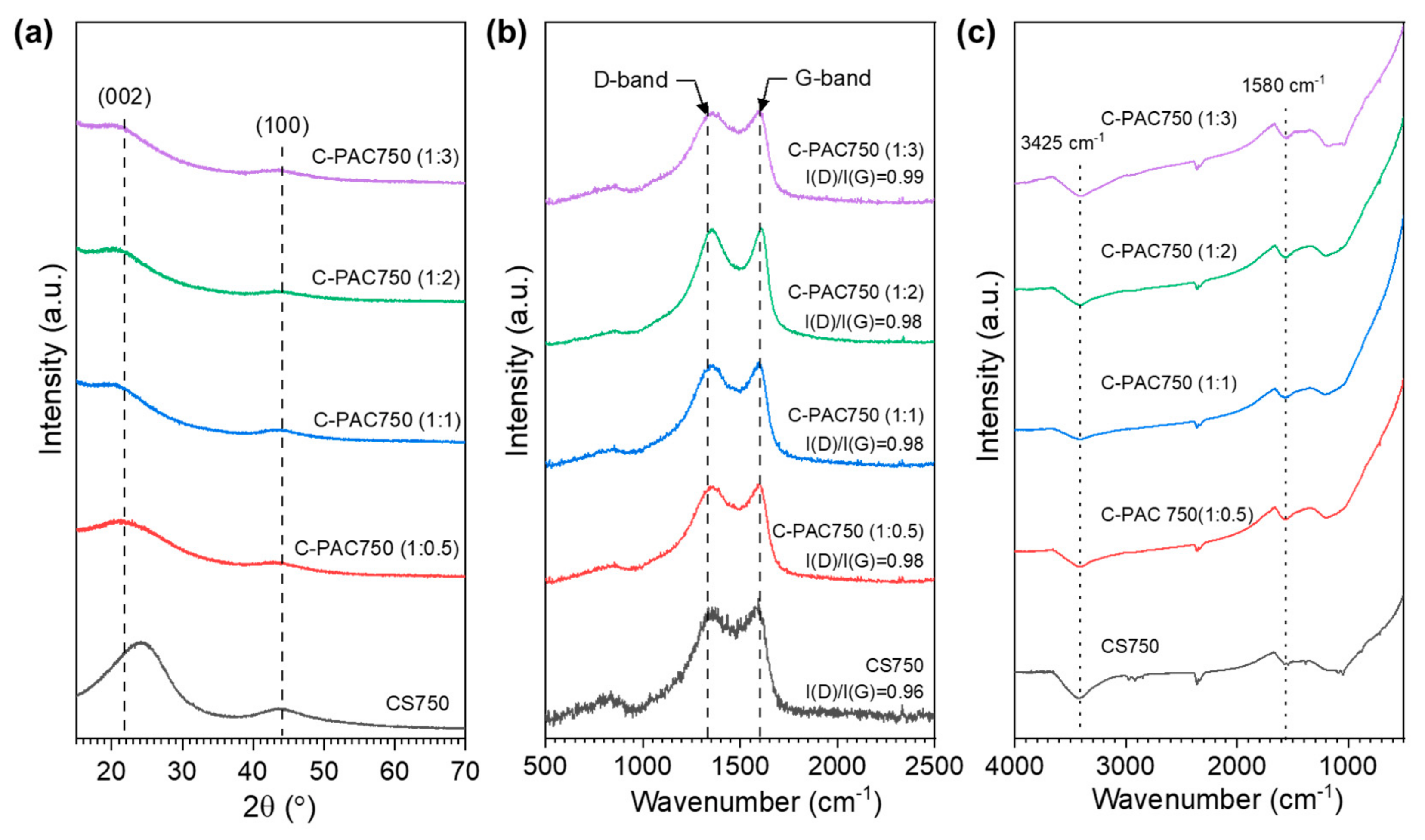
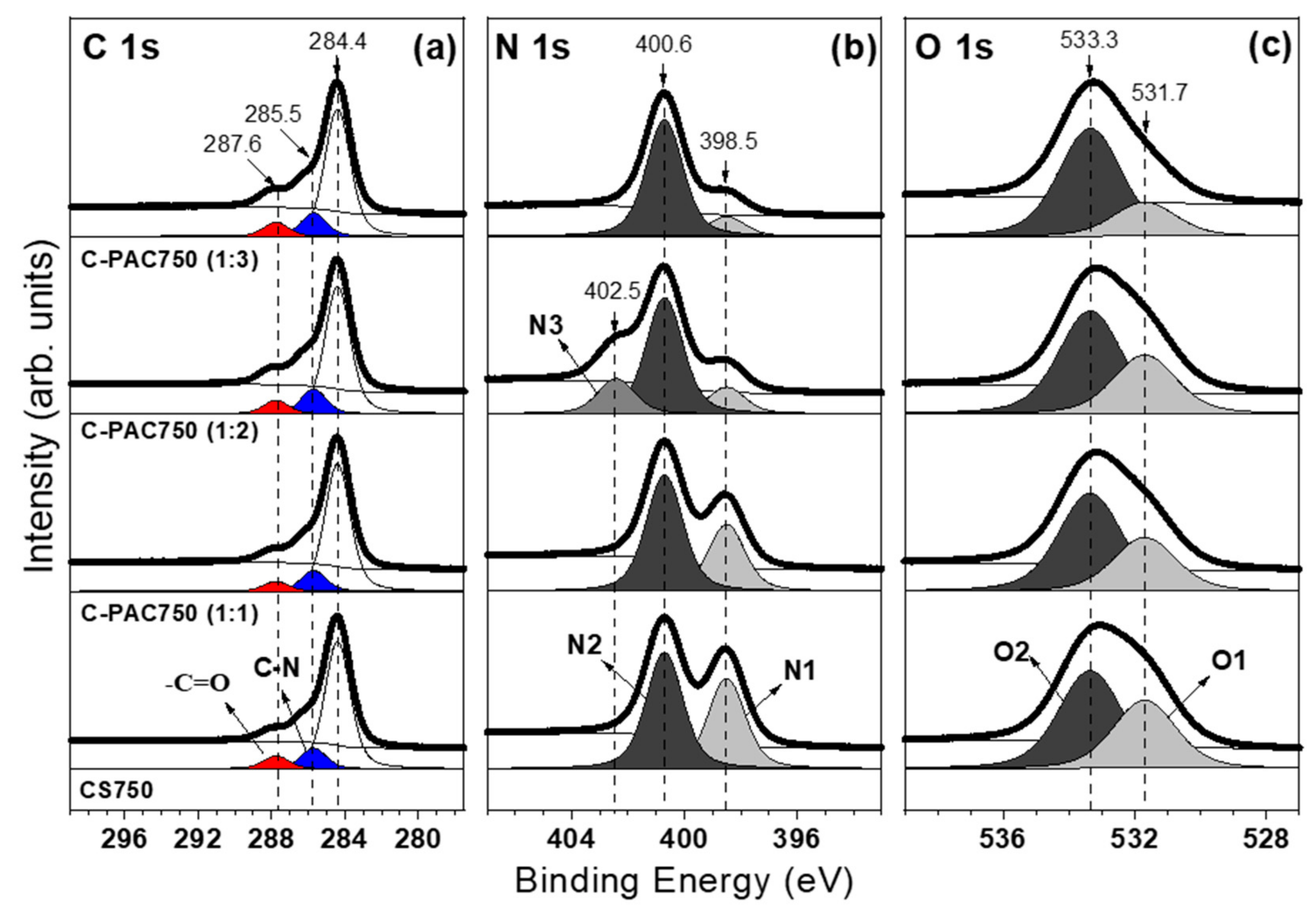
3.1. Chatacterization of C-PAC Samples
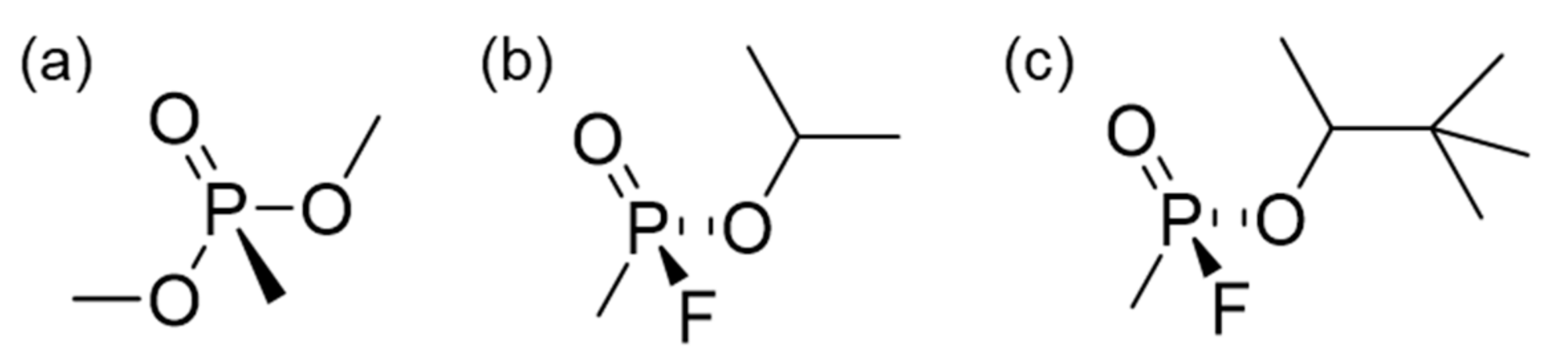
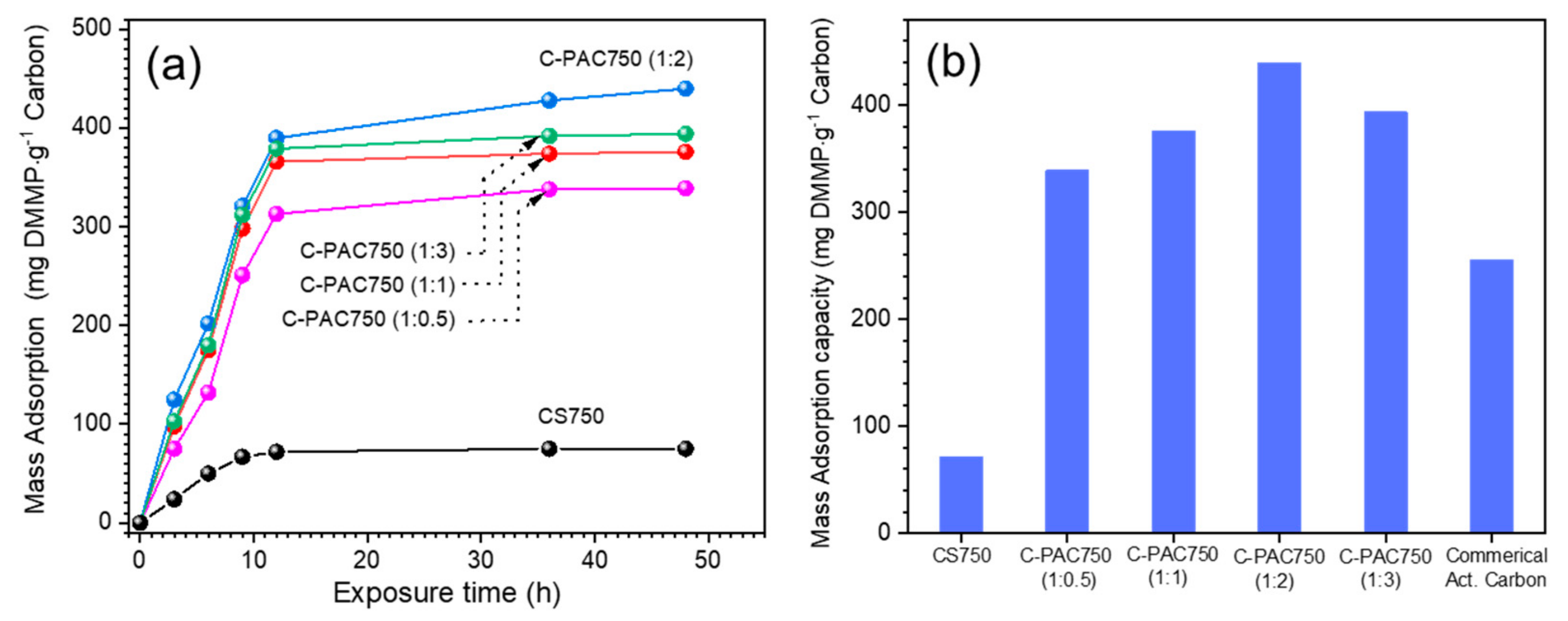
3.2. Evalulation of DMMP Adsorption Properties of C-PAC Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Eubanks, L.M.; Dickerson, T.J.; Janda, K.D. Technological advancements for the detection of and protection against biological and chemical warfare agents. Chem. Soc. Rev. 2007, 36, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Bobbitt, N.S.; Mendonca, M.L.; Howarth, A.J.; Islamoglu, T.; Hupp, J.T.; Farha, O.K.; Snurr, R.Q. Metal-organic frameworks for the removal of toxic industrial chemicals and chemical warfare agents. Chem. Soc. Rev. 2017, 46, 3357–3385. [Google Scholar] [CrossRef] [PubMed]
- Picard, B.; Chataigner, I.; Maddaluno, J.; Legros, J. Introduction to chemical warfare agents, relevant simulants and modern neutralisation methods. Org. Biomol. Chem. 2019, 17, 6528–6537. [Google Scholar] [CrossRef] [PubMed]
- Holdren, S.; Tsyshevsky, R.; Fears, K.; Owrutsky, J.; Wu, T.; Wang, X.; Eichhorn, B.W.; Kuklja, M.M.; Zachariah, M.R. Adsorption and Destruction of the G-Series Nerve Agent Simulant Dimethyl Methylphosphonate on Zinc Oxide. ACS Catal. 2019, 9, 902–911. [Google Scholar] [CrossRef]
- Plachá, D.; Kovář, P.; Vaněk, J.; Mikeska, M.; Škrlová, K.; Dutko, O.; Řeháčková, L.; Slabotínský, J. Adsorption of nerve agent simulants onto vermiculite structure: Experiments and modelling. J. Hazard. Mater. 2020, 382, 121001. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Gauden, P.A.; Terzyk, A.P.; Neimark, A.V. Screening of carbonaceous nanoporous materials for capture of nerve agents. Phys. Chem. Chem. Phys. 2013, 15, 291–298. [Google Scholar] [CrossRef]
- Osovsky, R.; Kaplan, D.; Nir, I.; Rotter, H.; Elisha, S.; Columbus, I. Decontamination of adsorbed chemical warfare agents on activated carbon using hydrogen peroxide solutions. Environ. Sci. Technol. 2014, 48, 10912–10918. [Google Scholar] [CrossRef]
- Cojocaru, B.; Neaţu, Ş.; Pârvulescu, V.I.; Şomoghi, V.; Petrea, N.; Epure, G.; Alvaro, M.; Garcia, H. Synergism of activated carbon and undoped and nitrogen-doped TiO2 in the photocatalytic degradation of the chemical warfare agents Soman, VX, and Yperite. ChemSusChem 2009, 2, 427–436. [Google Scholar] [CrossRef]
- Jin, Z.; Yan, X.; Yu, Y.; Zhao, G. Sustainable activated carbon fibers from liquefied wood with controllable porosity for high-performance supercapacitors. J. Mater. Chem. A 2014, 2, 11706–11715. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, E.; Zhao, G. Preparation of liquefied wood-based activated carbon fibers by different activation methods for methylene blue adsorption. RSC Adv. 2015, 5, 70287–70296. [Google Scholar] [CrossRef]
- Tsubouchi, N.; Nishio, M.; Shinohara, Y.; Bud, J.; Mochizuki, Y. Production of activated carbon from peat by with natural soda ash and effect of nitrogen addition on the development of surface area. Fuel Process. Technol. 2018, 176, 76–84. [Google Scholar] [CrossRef]
- Yang, K.; Peng, J.; Srinivasakannan, C.; Zhang, L.; Xia, H.; Duan, X. Preparation of high surface area activated carbon from coconut shells using microwave heating. Bioresour. Technol. 2010, 101, 6163–6169. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Correa, C.; Stollovsky, M.; Hehr, T.; Rauscher, Y.; Rolli, B.; Kruse, A. Influence of the Carbonization Process on Activated Carbon Properties from Lignin and Lignin-Rich Biomasses. ACS Sustain. Chem. Eng. 2017, 5, 8222–8233. [Google Scholar] [CrossRef]
- Saleem, J.; Shahid, U.B.; Hijab, M.; Mackey, H.; McKay, G. Production and applications of activated carbons as adsorbents from olive stones. Biomass Convers. Biorefin. 2019, 9, 775–802. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, L.; Zhang, G.; Shu, Z.; Shi, J. Chitosan derived nitrogen-doped microporous carbons for high performance CO2 capture. Carbon N. Y. 2013, 61, 423–430. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Li, K.; Wang, Z.; Tian, P.; Liu, D.; Yang, T.; Wang, J. Activated carbon derived from chitosan as air cathode catalyst for high performance in microbial fuel cells. J. Power Sources 2018, 378, 1–9. [Google Scholar] [CrossRef]
- Ilnicka, A.; Lukaszewicz, J.P. Synthesis of N-rich microporous carbon materials from chitosan by alkali activation using Na2CO3. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2015, 201, 66–71. [Google Scholar] [CrossRef]
- Hao, P.; Zhao, Z.; Leng, Y.; Tian, J.; Sang, Y.; Boughton, R.I.; Wong, C.P.; Liu, H.; Yang, B. Graphene-based nitrogen self-doped hierarchical porous carbon aerogels derived from chitosan for high performance supercapacitors. Nano Energy 2015, 15, 9–23. [Google Scholar] [CrossRef]
- Tong, X.; Chen, Z.; Zhuo, H.; Hu, Y.; Jing, S.; Liu, J.; Zhong, L. Tailoring the physicochemical properties of chitosan-derived N-doped carbon by controlling hydrothermal carbonization time for high-performance supercapacitor application. Carbohydr. Polym. 2019, 207, 764–774. [Google Scholar] [CrossRef]
- Huang, J.; Liang, Y.; Hu, H.; Liu, S.; Cai, Y.; Dong, H.; Zheng, M.; Xiao, Y.; Liu, Y. Ultrahigh-surface-area hierarchical porous carbon from chitosan: Acetic acid mediated efficient synthesis and its application in superior supercapacitors. J. Mater. Chem. A 2017, 5, 24775–24781. [Google Scholar] [CrossRef]
- Jin, Q.; Li, Y.; Yang, D.; Cui, J. Chitosan-derived three-dimensional porous carbon for fast removal of methylene blue from wastewater. RSC Adv. 2018, 8, 1255–1264. [Google Scholar] [CrossRef]
- Kucinska, A.; Cyganiuk, A.; Lukaszewicz, J.P. A microporous and high surface area active carbon obtained by the heat-treatment of chitosan. Carbon 2012, 50, 3098–3101. [Google Scholar] [CrossRef]
- Cheng, J.; Xu, Q.; Wang, X.; Li, Z.; Wu, F.; Shao, J.; Xie, H. Ultrahigh-surface-area nitrogen-doped hierarchically porous carbon materials derived from chitosan and betaine hydrochloride sustainable precursors for high-performance supercapacitors. Sustain. Energy Fuels 2019, 3, 1215–1224. [Google Scholar] [CrossRef]
- Bokobza, L.; Bruneel, J.-L.; Couzi, M. Raman Spectra of Carbon-Based Materials (from Graphite to Carbon Black) and of Some Silicone Composites. C 2015, 1, 77–94. [Google Scholar] [CrossRef]
- Girgis, B.S.; Temerk, Y.M.; Gadelrab, M.M.; Abdullah, I.D. X-ray Diffraction Patterns of Activated Carbons Prepared under Various Conditions. Carbon Lett. 2007, 8, 95–100. [Google Scholar] [CrossRef]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cançado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1291. [Google Scholar] [CrossRef]
- Becerril, H.A.; Mao, J.; Liu, Z.; Stoltenberg, R.M.; Bao, Z.; Chen, Y. Evaluation of solution-processed reduced graphene oxide films as transparent conductors. ACS Nano 2008, 2, 463–470. [Google Scholar] [CrossRef]
- Holzinger, M.; Abraham, J.; Whelan, P.; Graupner, R.; Ley, L.; Hennrich, F.; Kappes, M.; Hirsch, A. Functionalization of single-walled carbon nanotubes with (R-)oxycarbonyl nitrenes. J. Am. Chem. Soc. 2003, 125, 8566–8580. [Google Scholar] [CrossRef]
- Al-Gaashani, R.; Najjar, A.; Zakaria, Y.; Mansour, S.; Atieh, M.A. XPS and structural studies of high quality graphene oxide and reduced graphene oxide prepared by different chemical oxidation methods. Ceram. Int. 2019, 45, 14439–14448. [Google Scholar] [CrossRef]
- Lee, H.; Kim, Y.; Kim, M.J.; Kim, K.J.; Kim, B.K. Comparative Study of the Catalytic Activities of Three Distinct Carbonaceous Materials through Photocatalytic Oxidation, CO Conversion, Dye Degradation, and Electrochemical Measurements. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Huynh, K.; Holdren, S.; Hu, J.; Wang, L.; Zachariah, M.R.; Eichhorn, B.W. Dimethyl Methylphosphonate Adsorption Capacities and Desorption Energies on Ordered Mesoporous Carbons. ACS Appl. Mater. Interfaces 2017, 9, 40638–40644. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Hicks, Z.; Ganteför, G.; Eichhorn, B.W.; Bowen, K.H. Adsorption and Decomposition of DMMP on Size-Selected (WO3)3 Clusters. ChemistrySelect 2018, 3, 3718–3721. [Google Scholar] [CrossRef]
- Son, Y.R.; Kim, M.K.; Ryu, S.G.; Kim, H.S. Rapid Capture and Hydrolysis of a Sulfur Mustard Gas in Silver-Ion-Exchanged Zeolite Y. ACS Appl. Mater. Interfaces 2018, 10, 40651–40660. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET a (m2·g−1) | Vtot b (cm3·g−1) | Vmicro c (cm3·g1) | d d (nm) |
|---|---|---|---|---|
| CS750 | 57 | 0.09 | 0.03 | 0.64 |
| C-PAC750 (1:0.5) | 2084 | 0.96 | 0.88 | 1.84 |
| C-PAC750 (1:1) | 2643 | 1.26 | 1.20 | 1.90 |
| C-PAC750 (1:2) | 2850 | 1.48 | 1.41 | 2.08 |
| C-PAC750 (1:3) | 1639 | 0.82 | 0.79 | 2.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Son, Y.R.; Yoo, H.; Cha, H.G.; Lee, H.; Kim, H.S. Chitosan-Derived Porous Activated Carbon for the Removal of the Chemical Warfare Agent Simulant Dimethyl Methylphosphonate. Nanomaterials 2019, 9, 1703. https://doi.org/10.3390/nano9121703
Yu H, Son YR, Yoo H, Cha HG, Lee H, Kim HS. Chitosan-Derived Porous Activated Carbon for the Removal of the Chemical Warfare Agent Simulant Dimethyl Methylphosphonate. Nanomaterials. 2019; 9(12):1703. https://doi.org/10.3390/nano9121703
Chicago/Turabian StyleYu, Hyejin, Ye Rim Son, Hyeonji Yoo, Hyun Gil Cha, Hangil Lee, and Hyun Sung Kim. 2019. "Chitosan-Derived Porous Activated Carbon for the Removal of the Chemical Warfare Agent Simulant Dimethyl Methylphosphonate" Nanomaterials 9, no. 12: 1703. https://doi.org/10.3390/nano9121703
APA StyleYu, H., Son, Y. R., Yoo, H., Cha, H. G., Lee, H., & Kim, H. S. (2019). Chitosan-Derived Porous Activated Carbon for the Removal of the Chemical Warfare Agent Simulant Dimethyl Methylphosphonate. Nanomaterials, 9(12), 1703. https://doi.org/10.3390/nano9121703