

Vitamin C Affinity to TiO2 Nanotubes: A Computational Study by Hybrid Density Functional Theory Calculations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
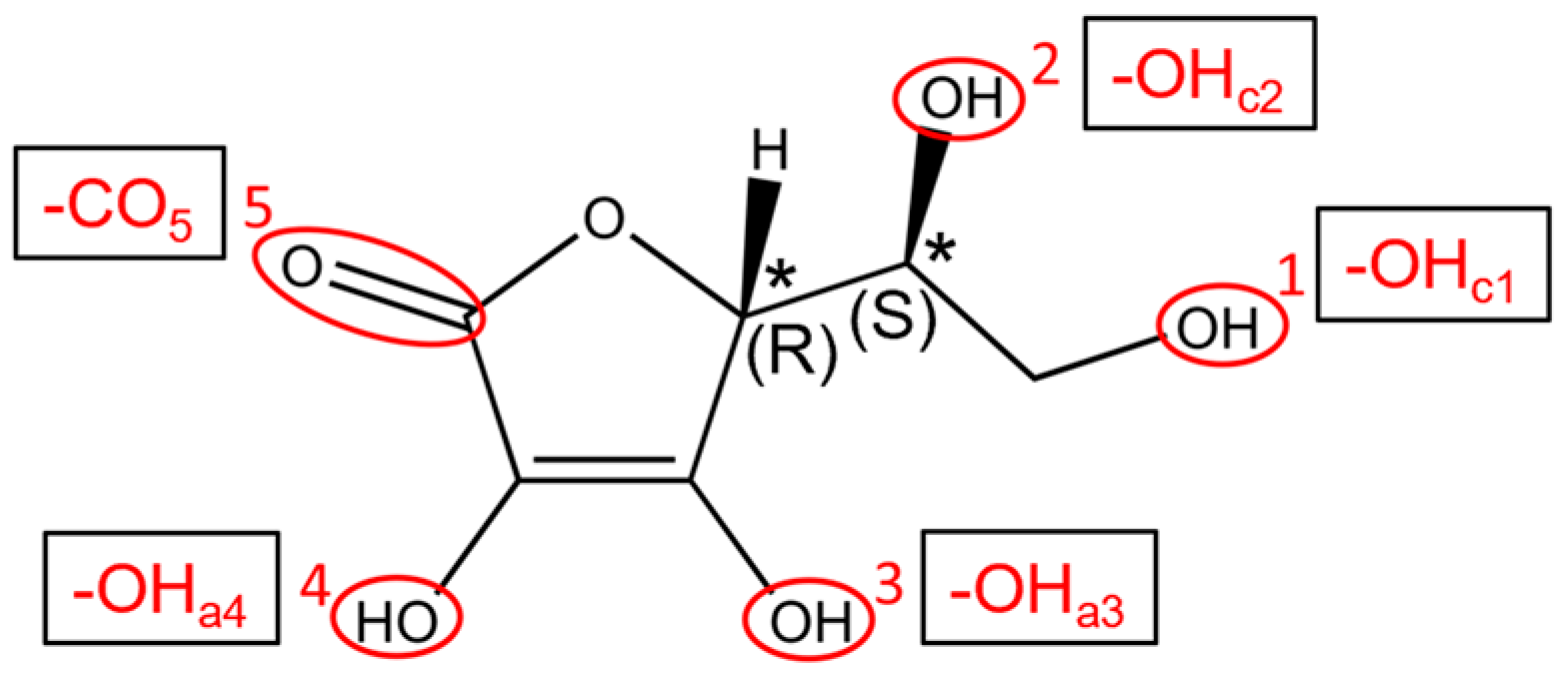
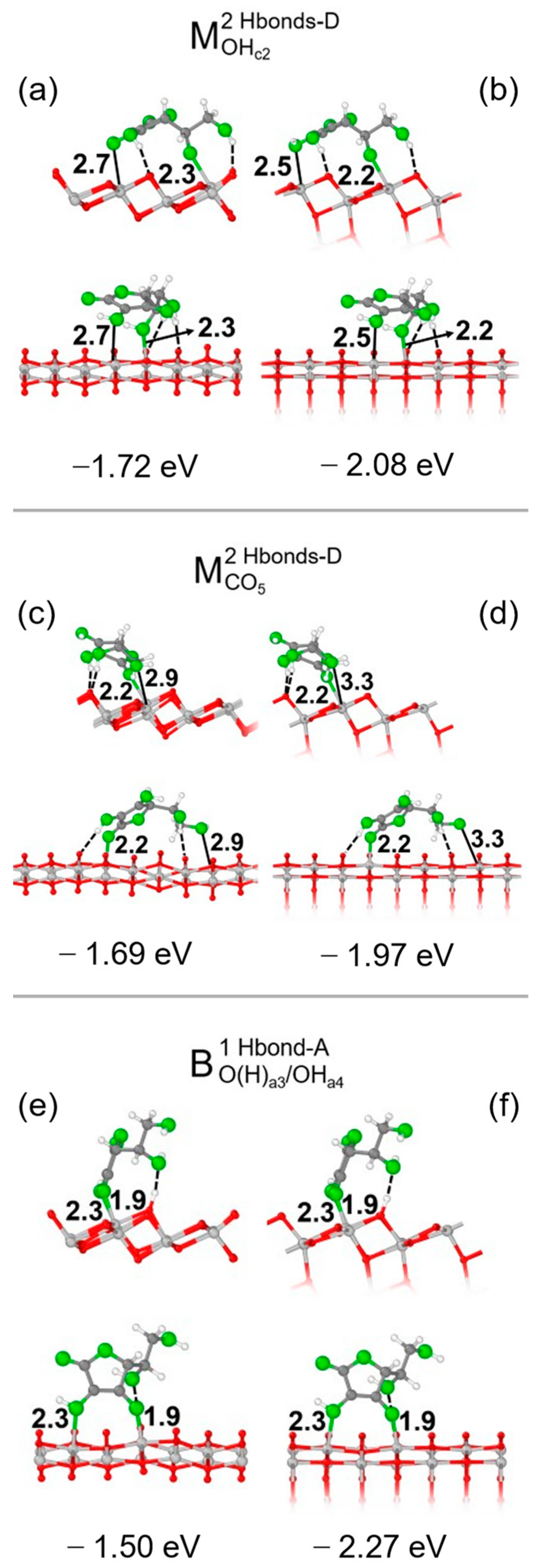
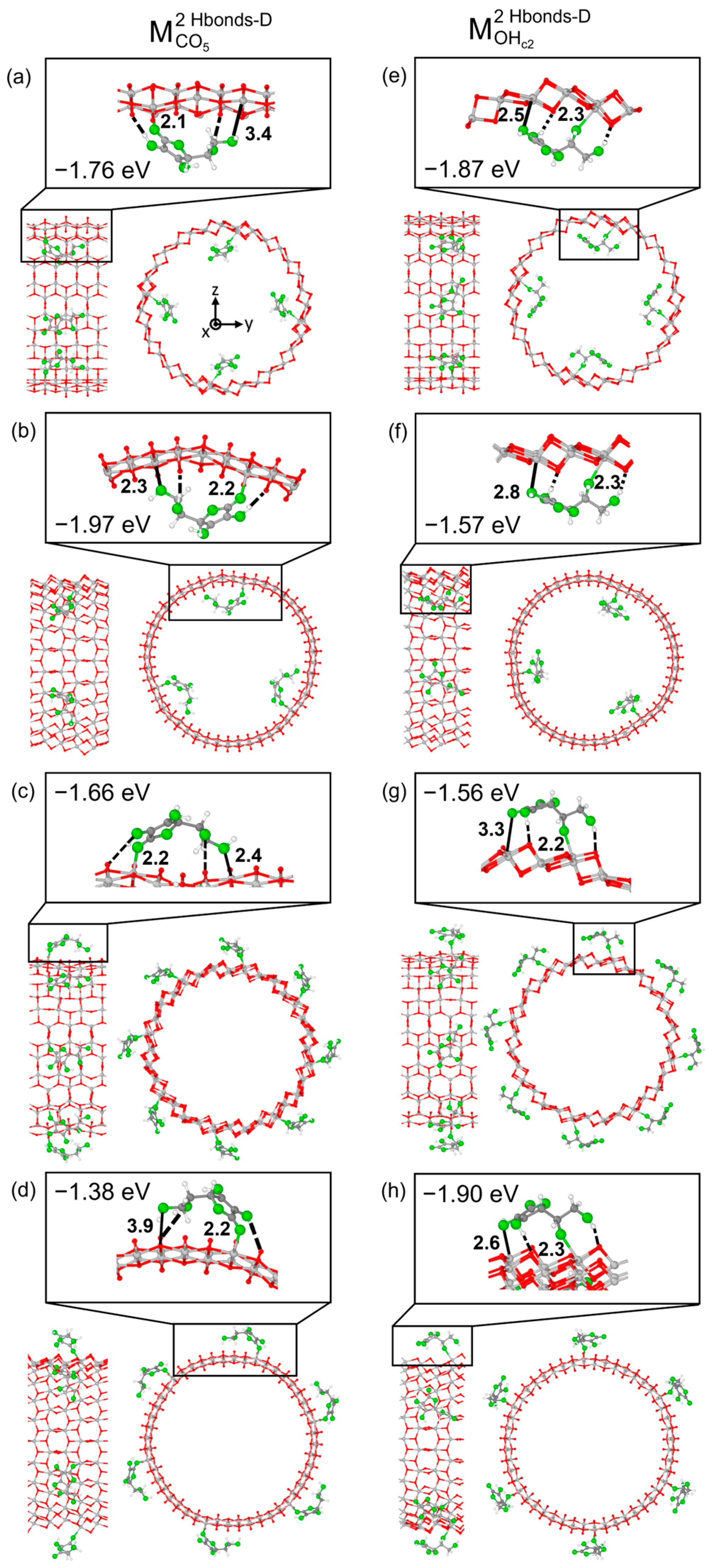
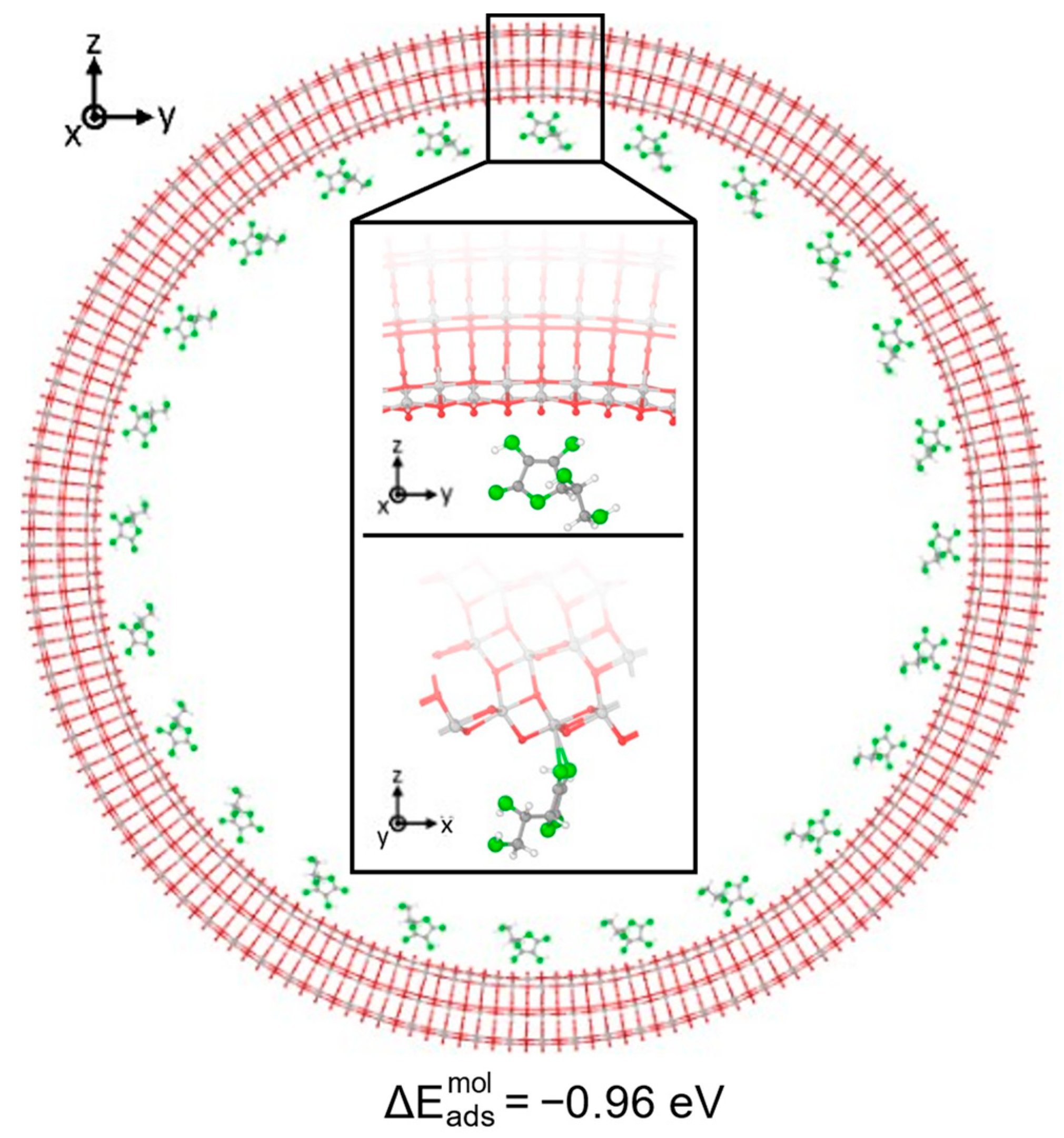
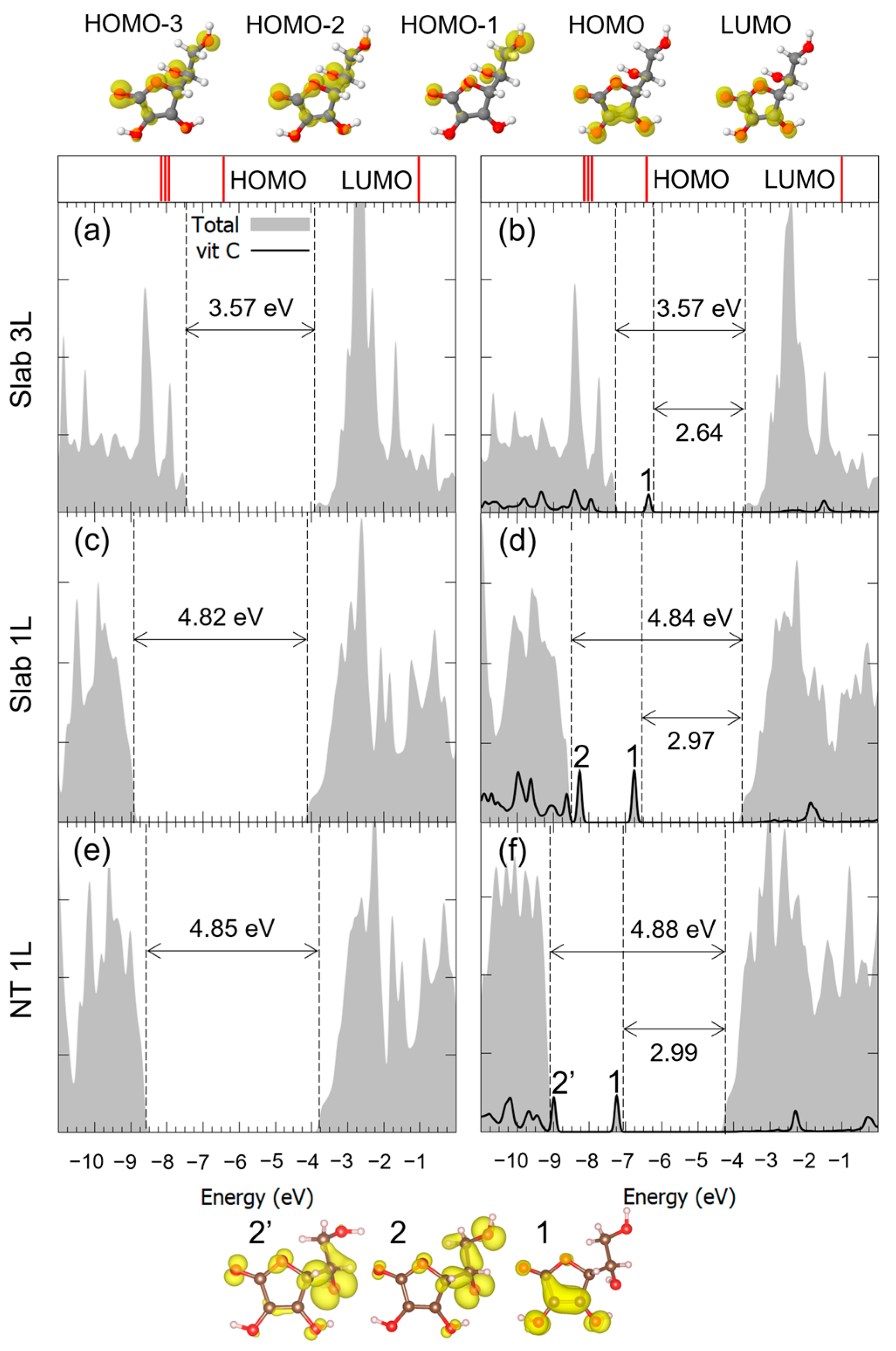
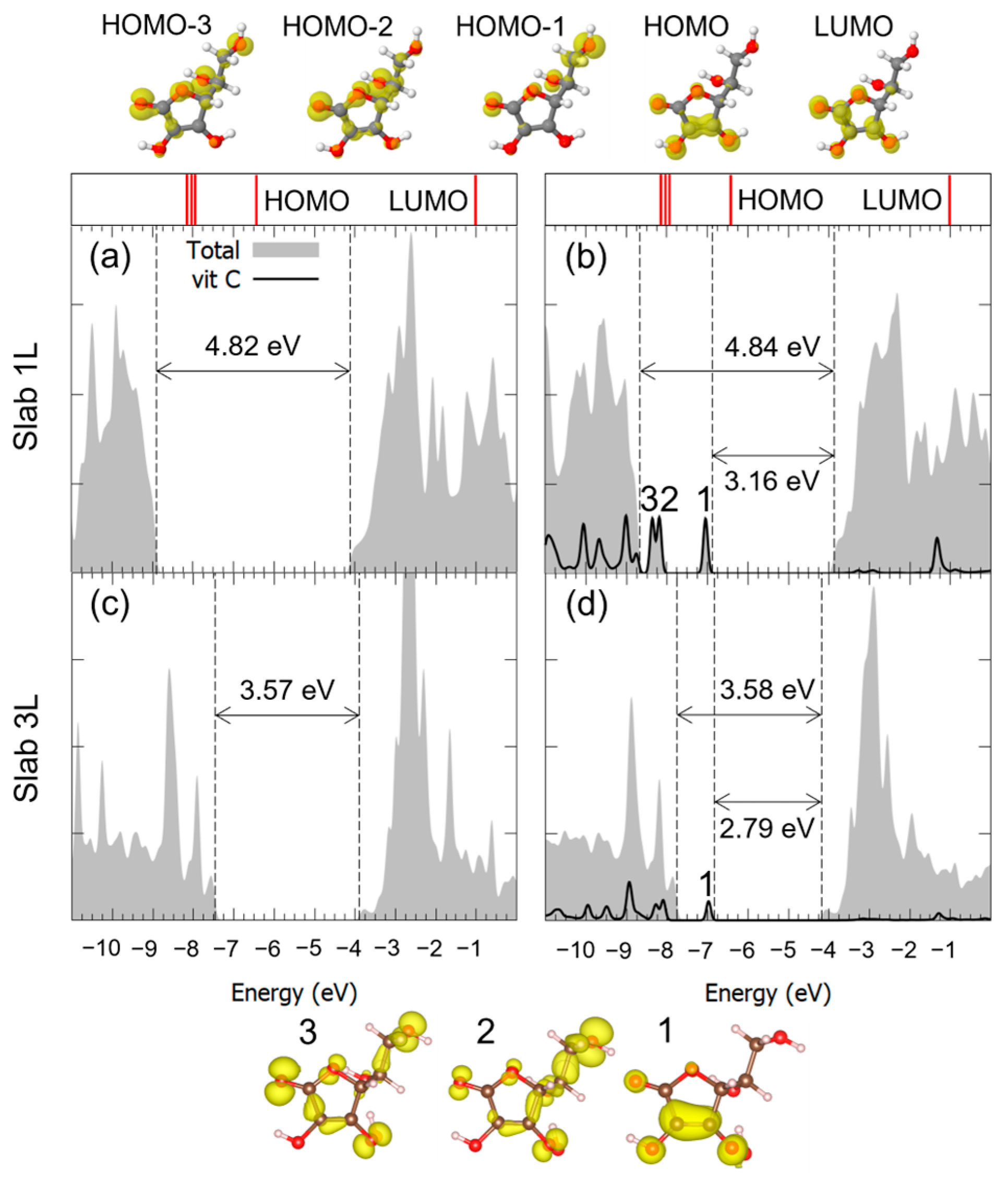
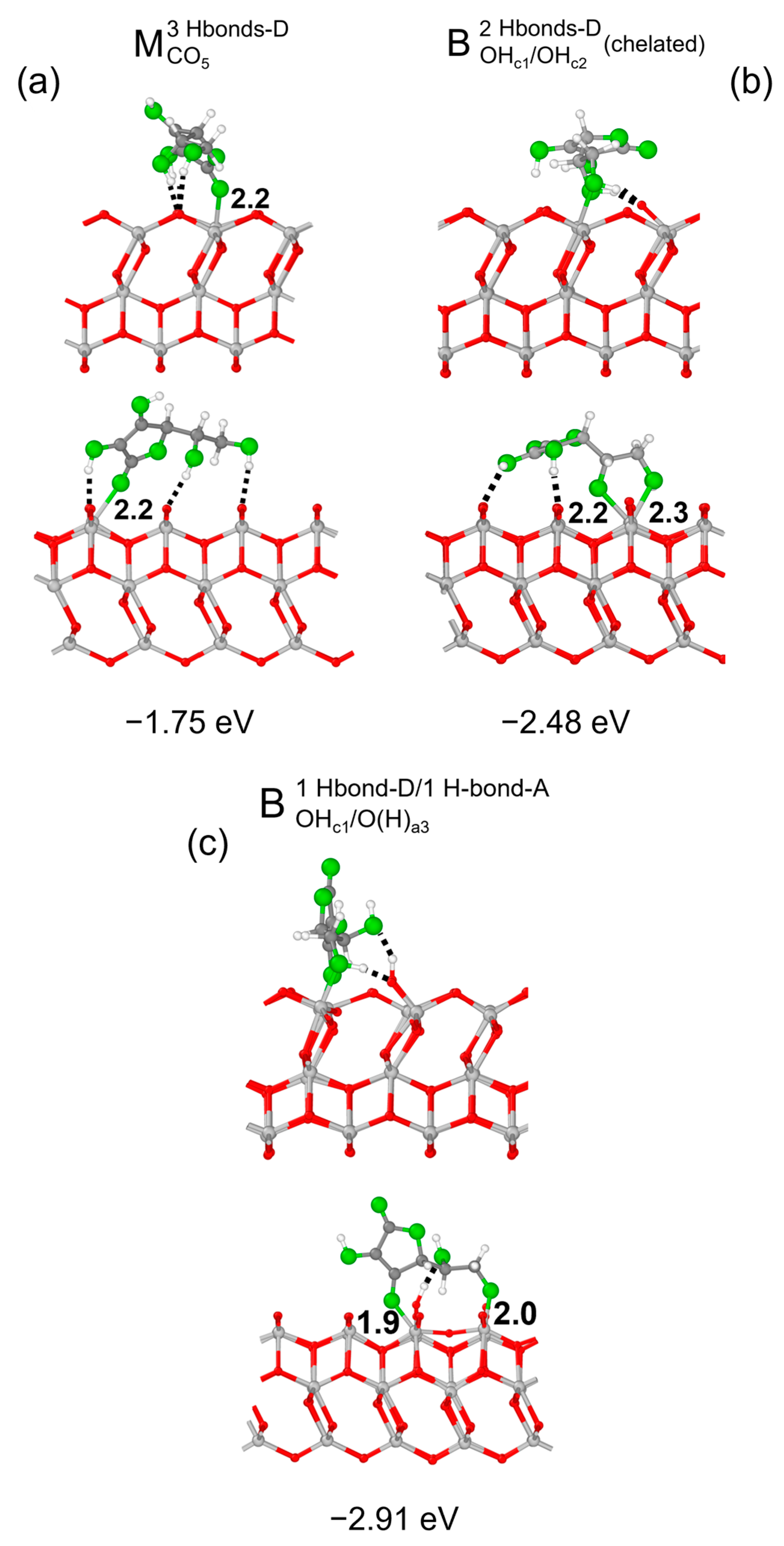
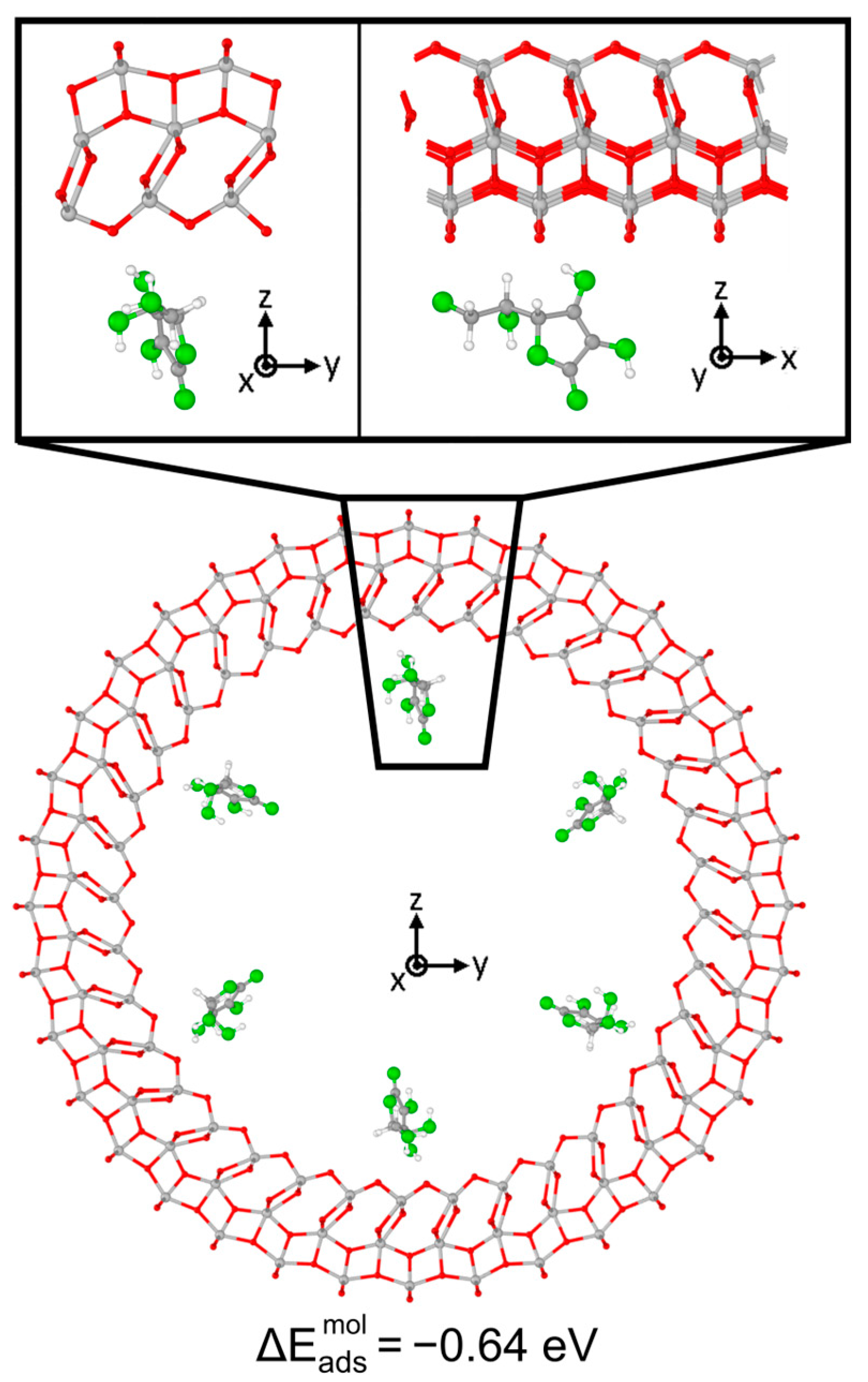
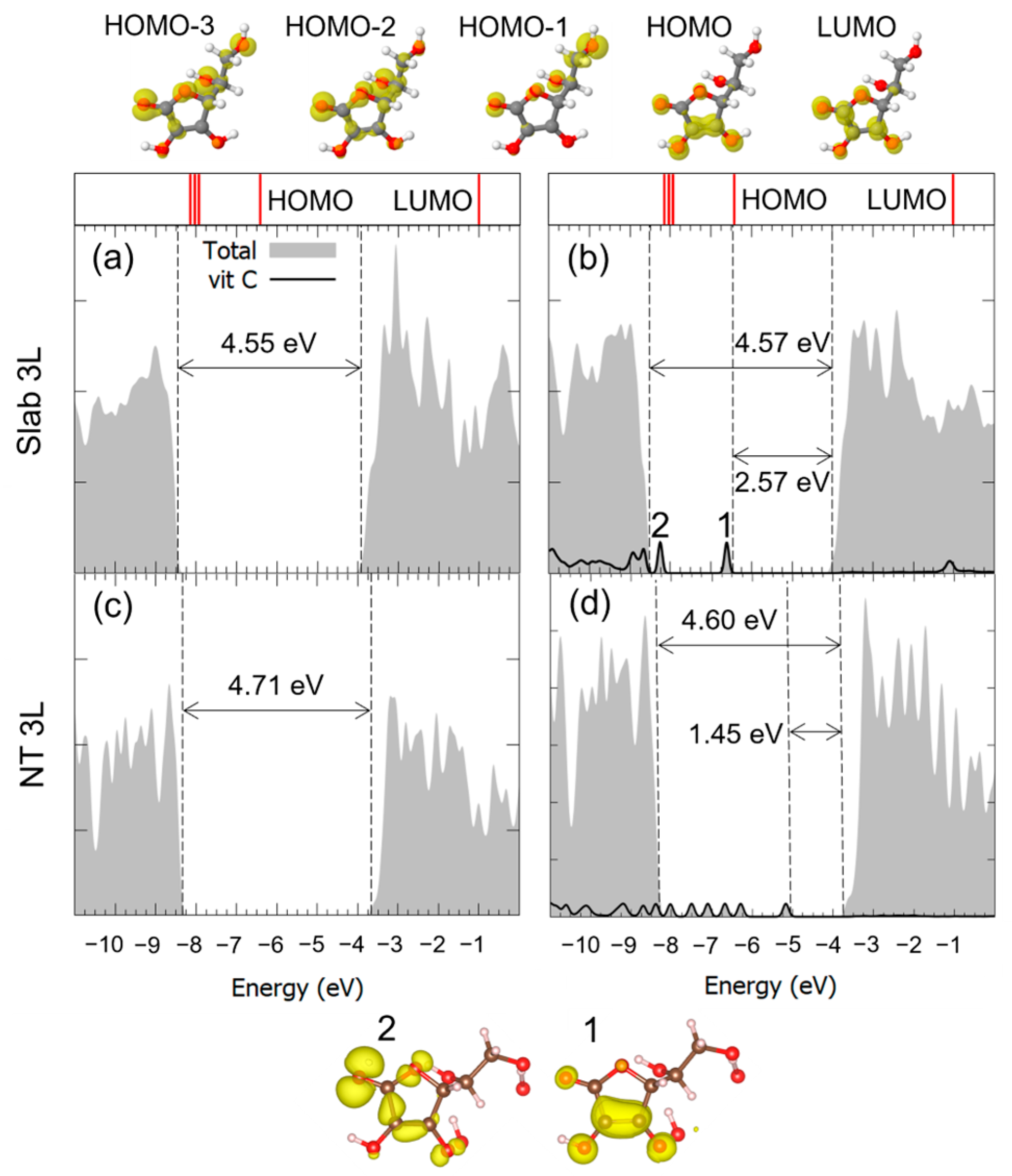
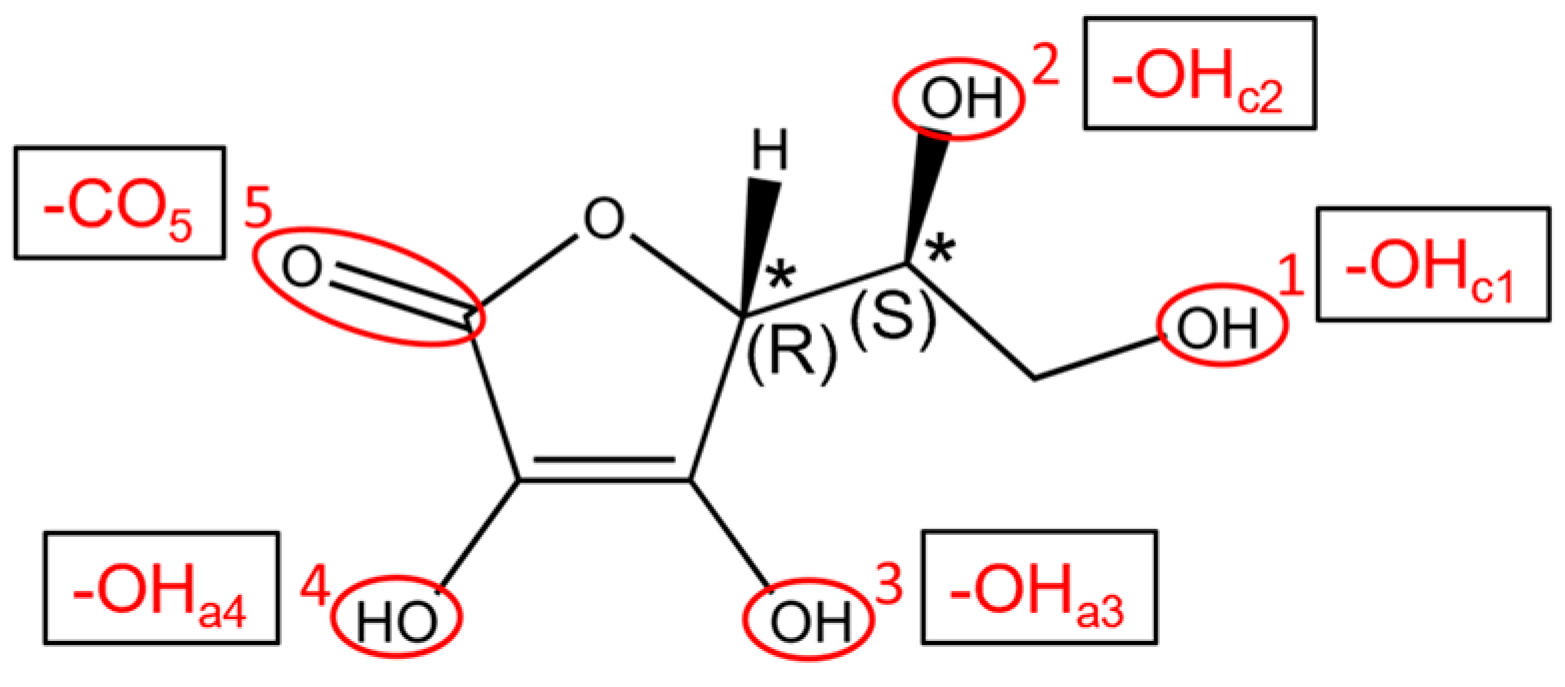
3.1. Vitamin C Adsorbed on the Anatase (101) TiO2 Surface and the Corresponding Nanotubes
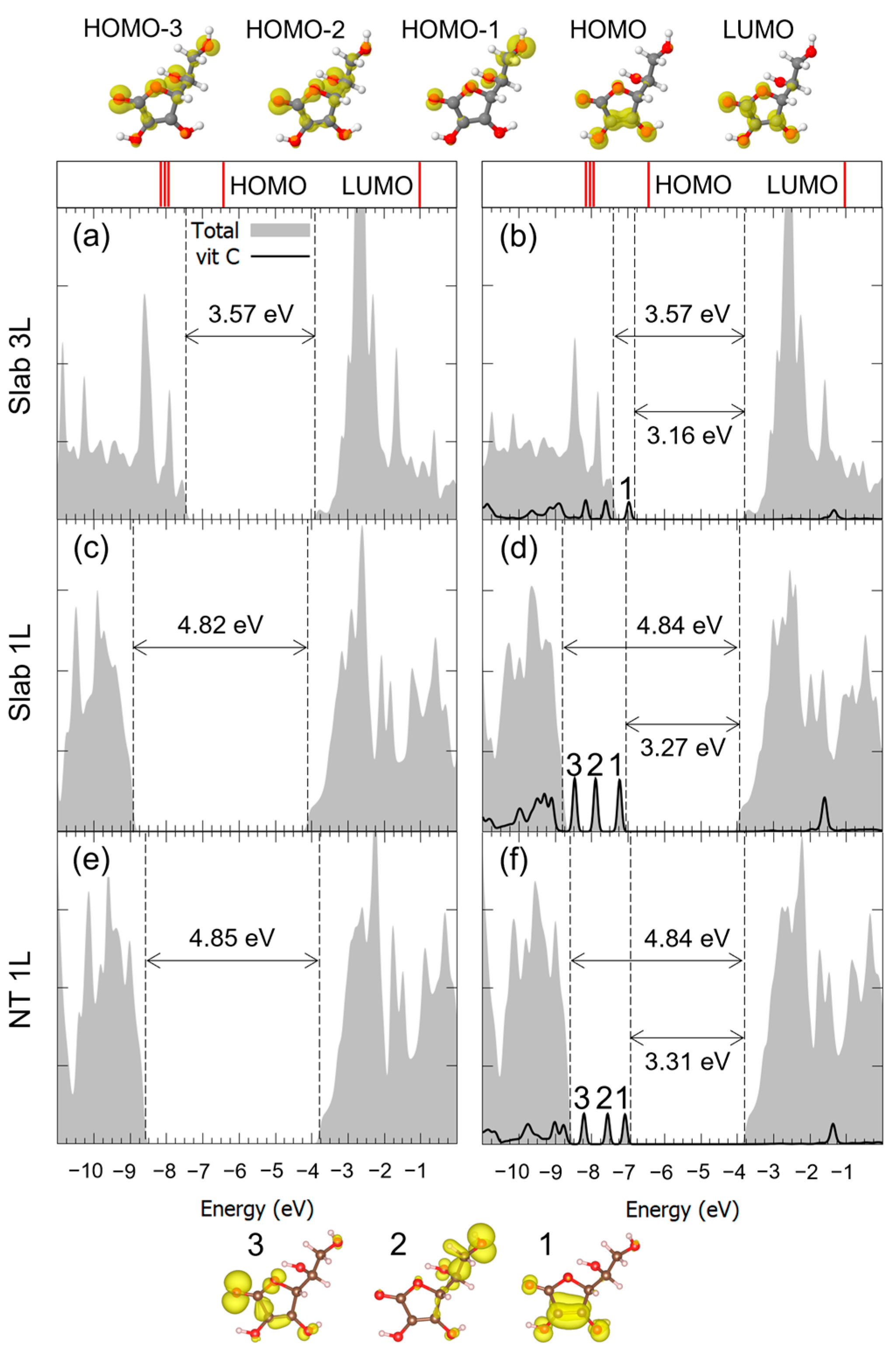
3.2. Vitamin C Adsorbed on the Anatase (001)A TiO2 Surface and the Corresponding Nanotube
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thostenson, E.T.; Ren, Z.; Chou, T.-W. Advances in the science and technology of carbon nanotubes and their composites: A review. Compos. Sci. Technol. 2001, 61, 1899–1912. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G.; Saito, R. Physics of carbon nanotubes. Carbon 1995, 33, 883–891. [Google Scholar] [CrossRef]
- Mintmire, J.W.; White, C.T. Electronic and structural properties of carbon nanotubes. Carbon 1995, 33, 893–902. [Google Scholar] [CrossRef]
- Zhang, M.; Jin, Z.; Zhang, J.; Guo, X.; Yang, J.; Li, W.; Wang, X.; Zhang, Z. Effect of annealing temperature on morphology, structure and photocatalytic behavior of nanotubed H2Ti2O4(OH)2. J. Mol. Catal. A Chem. 2004, 217, 203–210. [Google Scholar] [CrossRef]
- Yu, J.; Yu, H.; Cheng, B.; Zhao, X.; Zhang, Q. Preparation and photocatalytic activity of mesoporous anatase TiO2 nanofibers by a hydrothermal method. J. Photochem. Photobiol. A Chem. 2006, 182, 121–127. [Google Scholar] [CrossRef]
- Losic, A.S.K.G.D.; Bariana, M. Titania nanotube arrays for local drug delivery: Recent advances and perspectives. Expert Opin. Drug Deliv. 2015, 12, 103–127. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, P.; Sun, Y.; Wu, Y.; Mayers, B.; Gates, B.; Yin, Y.; Kim, F.; Yan, H. One-Dimensional Nanostructures: Synthesis, Characterization, and Applications. Adv. Mater. 2003, 15, 353–389. [Google Scholar] [CrossRef]
- Hoyer, P. Formation of a Titanium Dioxide Nanotube Array. Langmuir 1996, 12, 1411–1413. [Google Scholar] [CrossRef]
- Kasuga, T.; Hiramatsu, M.; Hoson, A.; Sekino, T.; Niihara, K. Formation of Titanium Oxide Nanotube. Langmuir 1998, 14, 3160–3163. [Google Scholar] [CrossRef]
- Indira, K.; Mudali, U.K.; Nishimura, T.; Rajendran, N. A Review on TiO2 Nanotubes: Influence of Anodization Parameters, Formation Mechanism, Properties, Corrosion Behavior, and Biomedical Applications. J. Bio-Tribo-Corros. 2015, 1, 28. [Google Scholar] [CrossRef]
- Lee, H.; Park, T.-H.; Jang, D.-J. Preparation of anatase TiO2 nanotube arrays dominated by highly reactive facets via anodization for high photocatalytic performances. New J. Chem. 2016, 40, 8737–8744. [Google Scholar] [CrossRef]
- Jung, M.-H.; Chu, M.-J.; Kang, M.G. TiO2 nanotube fabrication with highly exposed (001) facets for enhanced conversion efficiency of solar cells. Chem. Commun. 2012, 48, 5016–5018. [Google Scholar] [CrossRef] [PubMed]
- Weon, S.; Choi, E.; Kim, H.; Kim, J.Y.; Park, H.-J.; Kim, S.-M.; Kim, W.; Choi, W. Active 001 Facet Exposed TiO2 Nanotubes Photocatalyst Filter for Volatile Organic Compounds Removal: From Material Development to Commercial Indoor Air Cleaner Application. Environ. Sci. Technol. 2018, 52, 9330–9340. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, Y.; Xiao, P.; Zhang, X.; Yang, Y. Aggregation of TiO2 Nanotubes as Recyclable Catalyst for Photocatalytic Degradation of Methylene Blue. Environ. Eng. Sci. 2010, 27, 281–286. [Google Scholar] [CrossRef]
- Kravanja, K.A.; Suhadolnik, L.; Bele, M.; Maver, U.; Rožanc, J.; Knez, Ž.; Marevci, M.K.; Finšgar, M. The synthesis, surface analysis, and cellular response of titania and titanium oxynitride nanotube arrays prepared on TiAl6V4 for potential biomedical applications. J. Mater. Res. Technol. 2023, 24, 4074–4090. [Google Scholar] [CrossRef]
- Gusmão, S.B.S.; Ghosh, A.; Marques, T.M.F.; Ferreira, O.P.; Lobo, A.O.; Osajima, J.A.O.; Luz-Lima, C.; Sousa, R.R.M.; Matos, J.M.E.; Viana, B.C. One-Pot Synthesis of Titanate Nanotubes Decorated with Anatase Nanoparticles Using a Microwave-Assisted Hydrothermal Reaction. J. Nanomater. 2019, 2019, 4825432. [Google Scholar] [CrossRef]
- Umek, P.; Cevc, P.; Jesih, A.; Gloter, A.; Ewels, C.P.; Arčon, D. Impact of Structure and Morphology on Gas Adsorption of Titanate-Based Nanotubes and Nanoribbons. Chem. Mater. 2005, 17, 5945–5950. [Google Scholar] [CrossRef]
- Ou, H.-H.; Lo, S.-L. Review of titania nanotubes synthesized via the hydrothermal treatment: Fabrication, modification, and application. Sep. Purif. Technol. 2007, 58, 179–191. [Google Scholar] [CrossRef]
- Jia, H.; Kerr, L.L. Kinetics of drug release from drug carrier of polymer/TiO2 nanotubes composite—pH dependent study. J. Appl. Polym. Sci. 2015, 132, 41570. [Google Scholar] [CrossRef]
- Oh, S.; Moon, K.S.; Lee, S.H. Effect of RGD Peptide-Coated TiO2 Nanotubes on the Attachment, Proliferation, and Functionality of Bone-Related Cells. J. Nanomater. 2013, 2013, 965864. [Google Scholar] [CrossRef]
- Li, L.; Xie, C.; Xiao, X. Polydopamine modified TiO2 nanotube arrays as a local drug delivery system for ibuprofen. J. Drug Deliv. Sci. Technol. 2020, 56, 101537. [Google Scholar] [CrossRef]
- Jia, H.; Kerr, L.L. Sustained Ibuprofen Release Using Composite Poly(Lactic-co-Glycolic Acid)/Titanium Dioxide Nanotubes from Ti Implant Surface. J. Pharm. Sci. 2013, 102, 2341–2348. [Google Scholar] [CrossRef]
- Pawlik, A.; Jarosz, M.; Syrek, K.; Sulka, G.D. Co-delivery of ibuprofen and gentamicin from nanoporous anodic titanium dioxide layers. Colloids Surf. B Biointerfaces 2017, 152, 95–102. [Google Scholar] [CrossRef]
- Wang, T.; Weng, Z.; Liu, X.; Yeung, K.W.K.; Pan, H.; Wu, S. Controlled release and biocompatibility of polymer/titania nanotube array system on titanium implants. Bioact. Mater. 2017, 2, 44–50. [Google Scholar] [CrossRef]
- Mohan, L.; Anandan, C.; Rajendran, N. Drug release characteristics of quercetin-loaded TiO2 nanotubes coated with chitosan. Int. J. Biol. Macromol. 2016, 93, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Somsanith, N.; Kim, Y.-K.; Jang, Y.-S.; Lee, Y.-H.; Yi, H.-K.; Jang, J.-H.; Kim, K.-A.; Bae, T.-S.; Lee, M.-H. Enhancing of Osseointegration with Propolis-Loaded TiO2 Nanotubes in Rat Mandible for Dental Implants. Materials 2018, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, L.; Yao, C.; Fang, J.; Wu, M. Cytotoxicity Evaluation of pH-Controlled Antitumor Drug Release System of Titanium Dioxide Nanotubes. J. Nanosci. Nanotechnol. 2015, 15, 4143–4148. [Google Scholar] [CrossRef]
- Khodabandeh, R.; Mohammadpour, F.; Zolghadr, A.R.; Klein, A. Zn capped Al2O3 and TiO2 nanoporous arrays as pH sensitive drug delivery systems: A combined experimental and simulation study. New J. Chem. 2020, 44, 16602–16612. [Google Scholar] [CrossRef]
- Yang, W.; Deng, C.; Liu, P.; Hu, Y.; Luo, Z.; Cai, K. Sustained release of aspirin and vitamin C from titanium nanotubes: An experimental and stimulation study. Mater. Sci. Eng. C 2016, 64, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Lazzeri, M.; Vittadini, A.; Selloni, A. Structure and energetics of stoichiometric TiO2 anatase surfaces. Phys. Rev. B 2001, 63, 155409. [Google Scholar] [CrossRef]
- De Angelis, F.; Di Valentin, C.; Fantacci, S.; Vittadini, A.; Selloni, A. Theoretical Studies on Anatase and Less Common TiO2 Phases: Bulk, Surfaces, and Nanomaterials. Chem. Rev. 2014, 114, 9708–9753. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.M.; Szieberth, D.; Zicovich-Wilson, C.M.; Demichelis, R. Anatase(001) 3 ML Nanotubes, The First TiO2 Nanotube With Negative Strain Energies: A DFT Prediction. J. Phys. Chem. Lett. 2010, 1, 2854–2857. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Szieberth, D.; Noel, Y. DFT modeling of anatase nanotubes. J. Mater. Chem. 2011, 21, 4568–4580. [Google Scholar] [CrossRef]
- Szieberth, D.; Ferrari, A.M.; D’Arco, P.; Orlando, R. Ab initio modeling of trititanate nanotubes. Nanoscale 2011, 3, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.M.; Lessio, M.; Rérat, M. Prediction of electronic (hyper)polarizabilities of titania nanotubes: A DFT periodic study. Comput. Mater. Sci. 2013, 68, 280–286. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual; University of Torino: Torino, Italy, 2018; p. 461. [Google Scholar]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Ronchi, C.; Selli, D.; Pipornpong, W.; Di Valentin, C. Proton Transfers at a Dopamine-Functionalized TiO2 Interface. J. Phys. Chem. C 2019, 123, 7682–7695. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10, 405–410. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Burdett, J.K.; Hughbanks, T.; Miller, G.J.; Richardson, J.W.J.; Smith, J.V. Structural-electronic relationships in inorganic solids: Powder neutron diffraction studies of the rutile and anatase polymorphs of titanium dioxide at 15 and 295 K. J. Am. Chem. Soc. 1987, 109, 3639–3646. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Noël, Y.; Ferrari, A.M.; Orlando, R.; De La Pierre, M.; Dovesi, R. On the use of symmetry in SCF calculations. The case of fullerenes and nanotubes. AIP Conf. Proc. 2012, 1456, 248–255. [Google Scholar]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Alessio, M.; Civalleri, B.; Peintinger, M.F.; Bredow, T.; Grimme, S. Geometrical Correction for the Inter- and Intramolecular Basis Set Superposition Error in Periodic Density Functional Theory Calculations. J. Phys. Chem. A 2013, 117, 9282–9292. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, M. The Merck Index: An Encyclopedia of Chemical Drugs and Biologicals, 13th ed.; Merck: Whitehouse Station, NJ, USA, 2001. [Google Scholar]
- Nunzi, F.; De Angelis, F. DFT Investigations of Formic Acid Adsorption on Single-Wall TiO2 Nanotubes: Effect of the Surface Curvature. J. Phys. Chem. C 2011, 115, 2179–2186. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugolotti, A.; Dolce, M.; Di Valentin, C. Vitamin C Affinity to TiO2 Nanotubes: A Computational Study by Hybrid Density Functional Theory Calculations. Nanomaterials 2024, 14, 261. https://doi.org/10.3390/nano14030261
Ugolotti A, Dolce M, Di Valentin C. Vitamin C Affinity to TiO2 Nanotubes: A Computational Study by Hybrid Density Functional Theory Calculations. Nanomaterials. 2024; 14(3):261. https://doi.org/10.3390/nano14030261
Chicago/Turabian StyleUgolotti, Aldo, Mirko Dolce, and Cristiana Di Valentin. 2024. "Vitamin C Affinity to TiO2 Nanotubes: A Computational Study by Hybrid Density Functional Theory Calculations" Nanomaterials 14, no. 3: 261. https://doi.org/10.3390/nano14030261
APA StyleUgolotti, A., Dolce, M., & Di Valentin, C. (2024). Vitamin C Affinity to TiO2 Nanotubes: A Computational Study by Hybrid Density Functional Theory Calculations. Nanomaterials, 14(3), 261. https://doi.org/10.3390/nano14030261