Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory


Abstract
1. Introduction
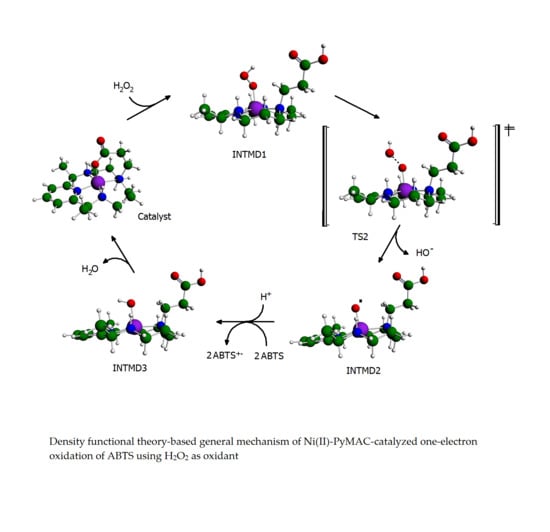
Proposed Reaction Pathways
2. Computational Methods
3. Results and Discussion
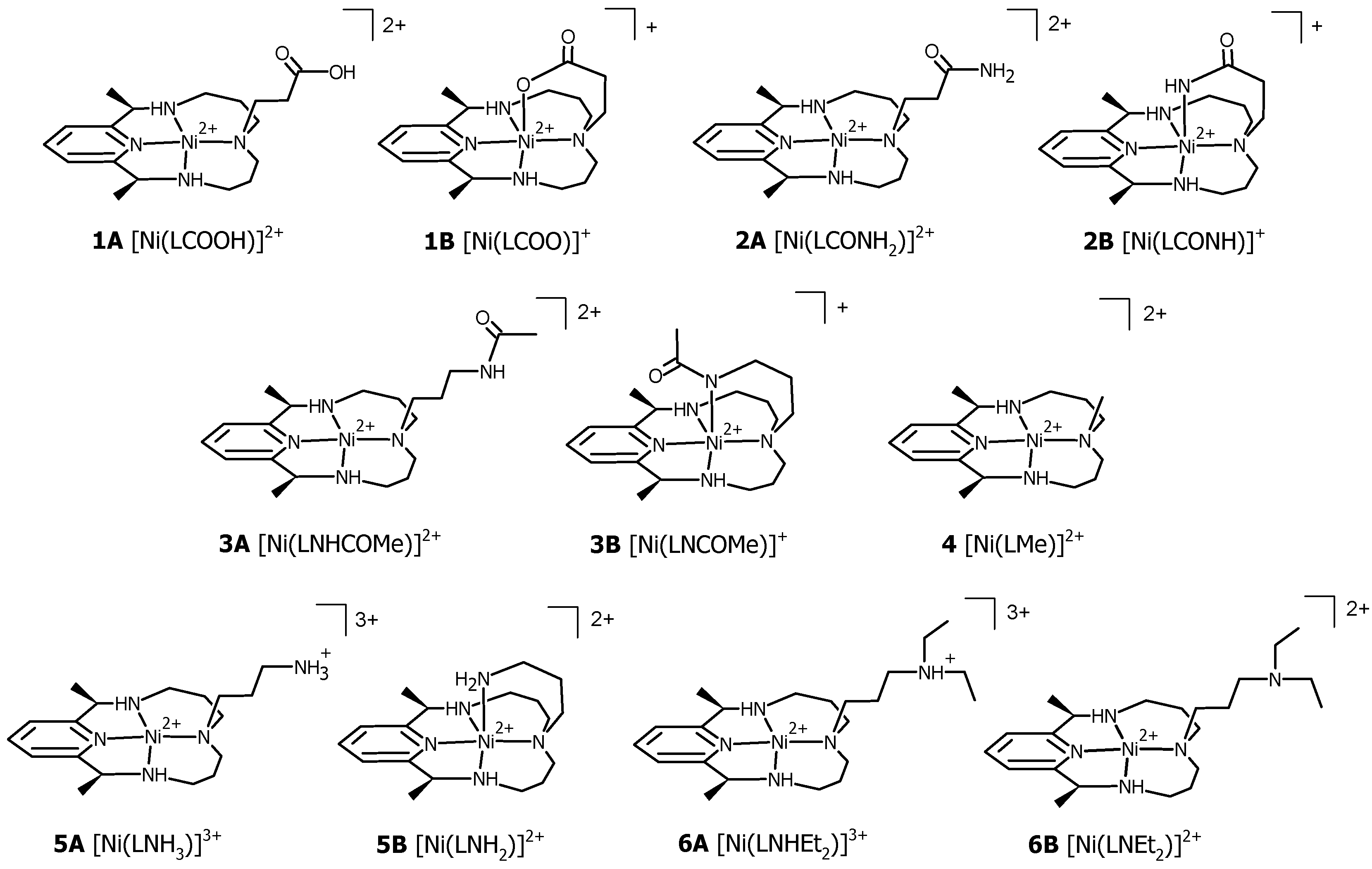
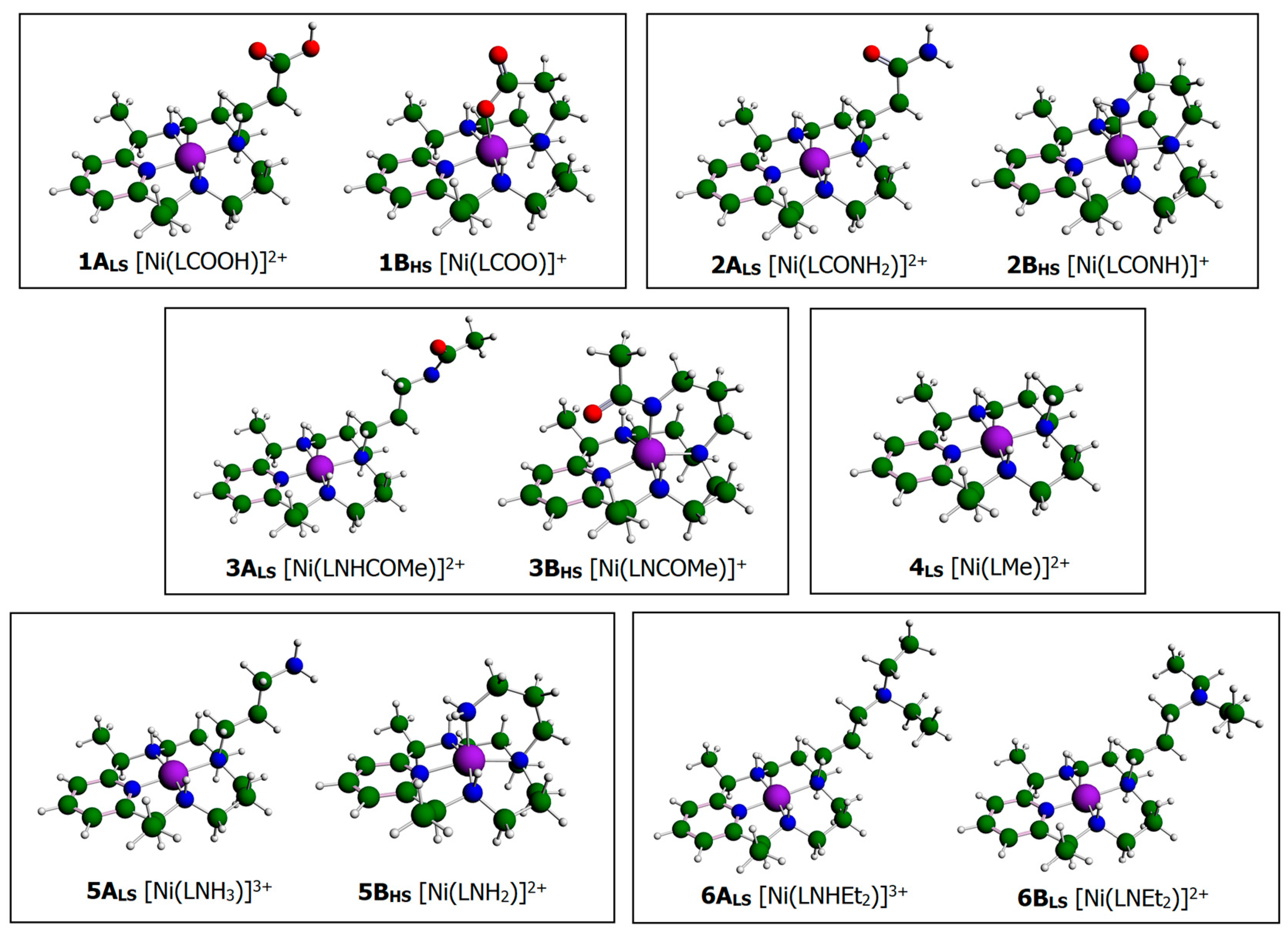
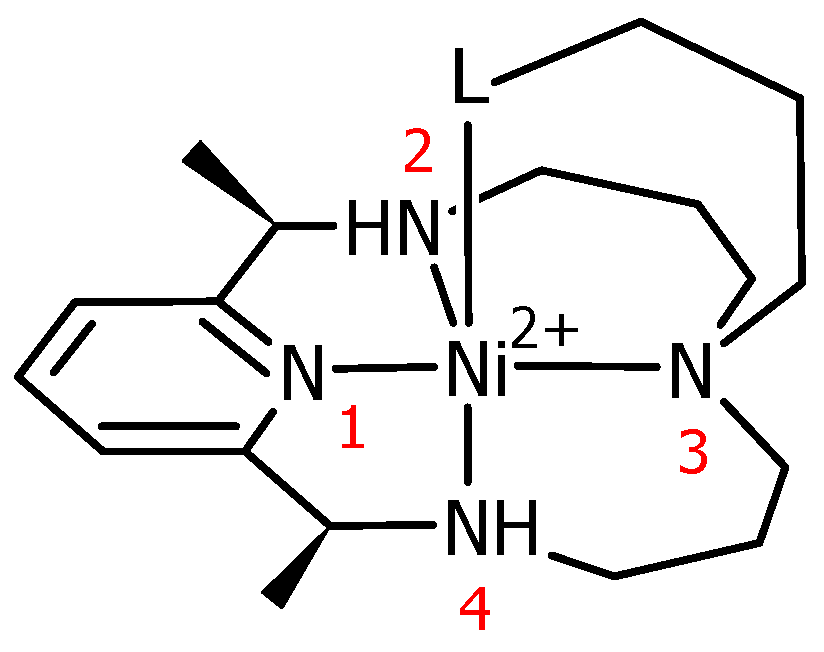
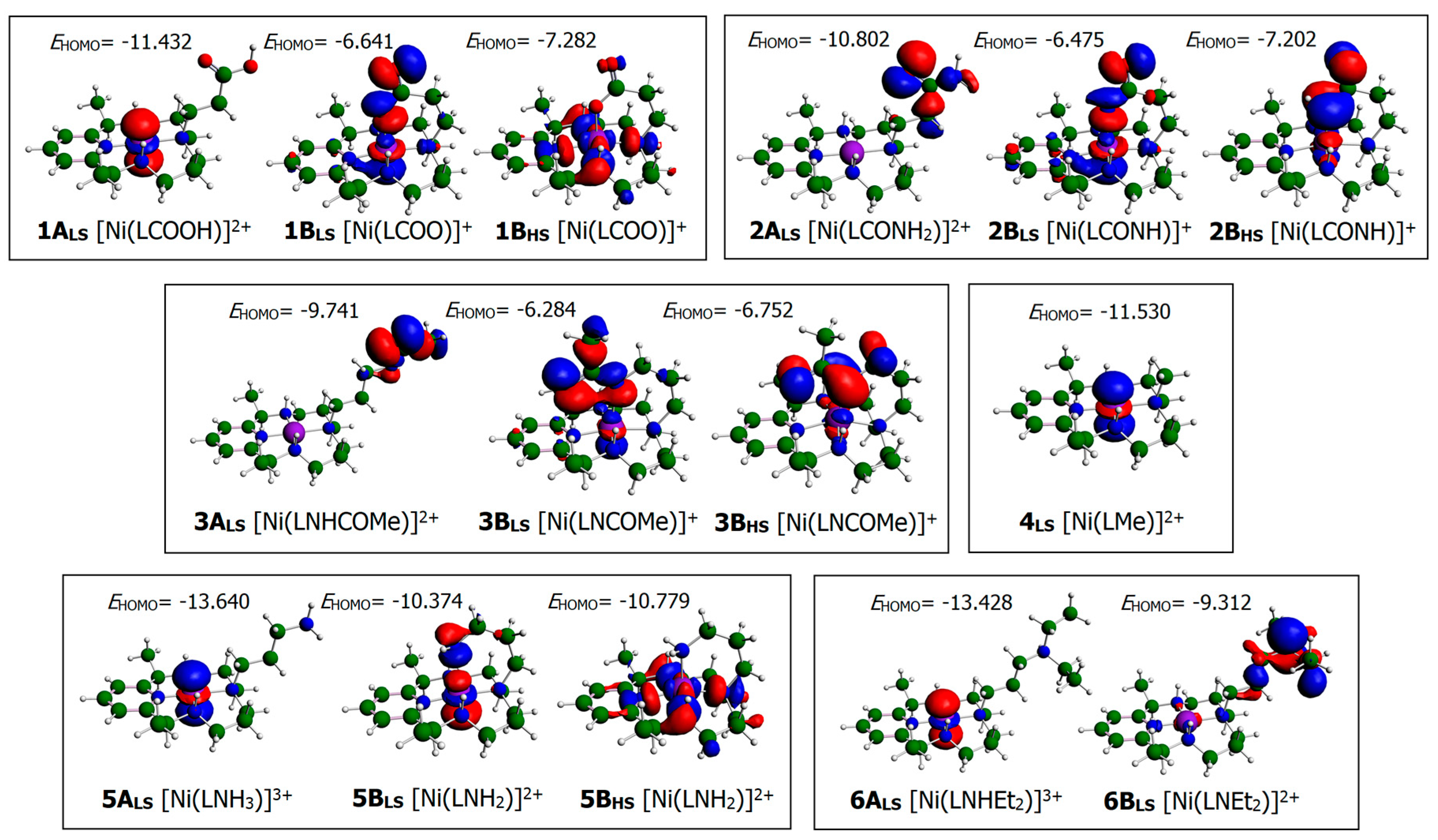
3.1. Structural Characterization of Ni(II)–PyMAC Complexes
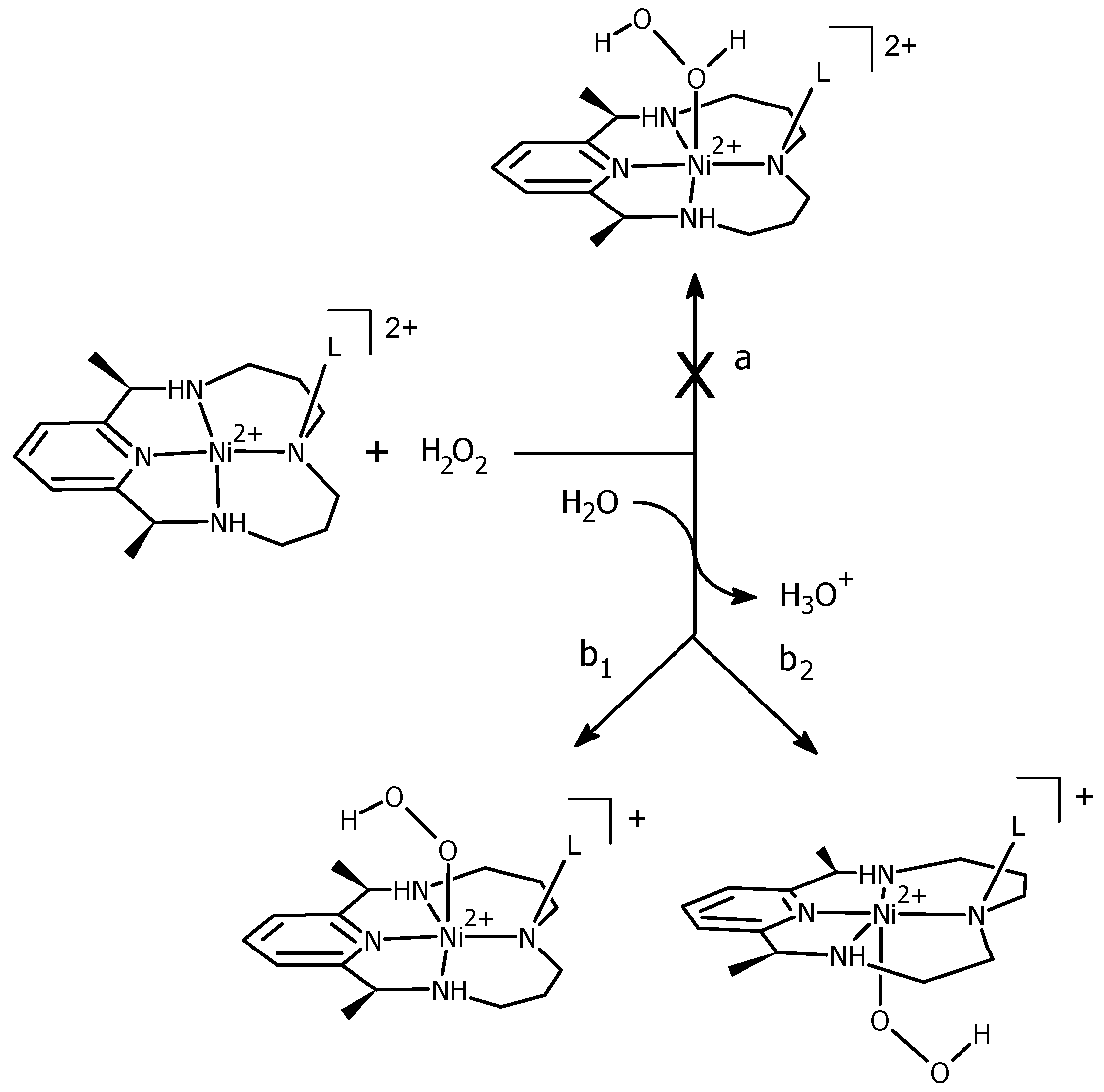
3.2. H2O2 Activation and Reactivity with Ni(II)–PyMACs
3.3. Structure and Energetics of Nickel-Hydroperoxo Species
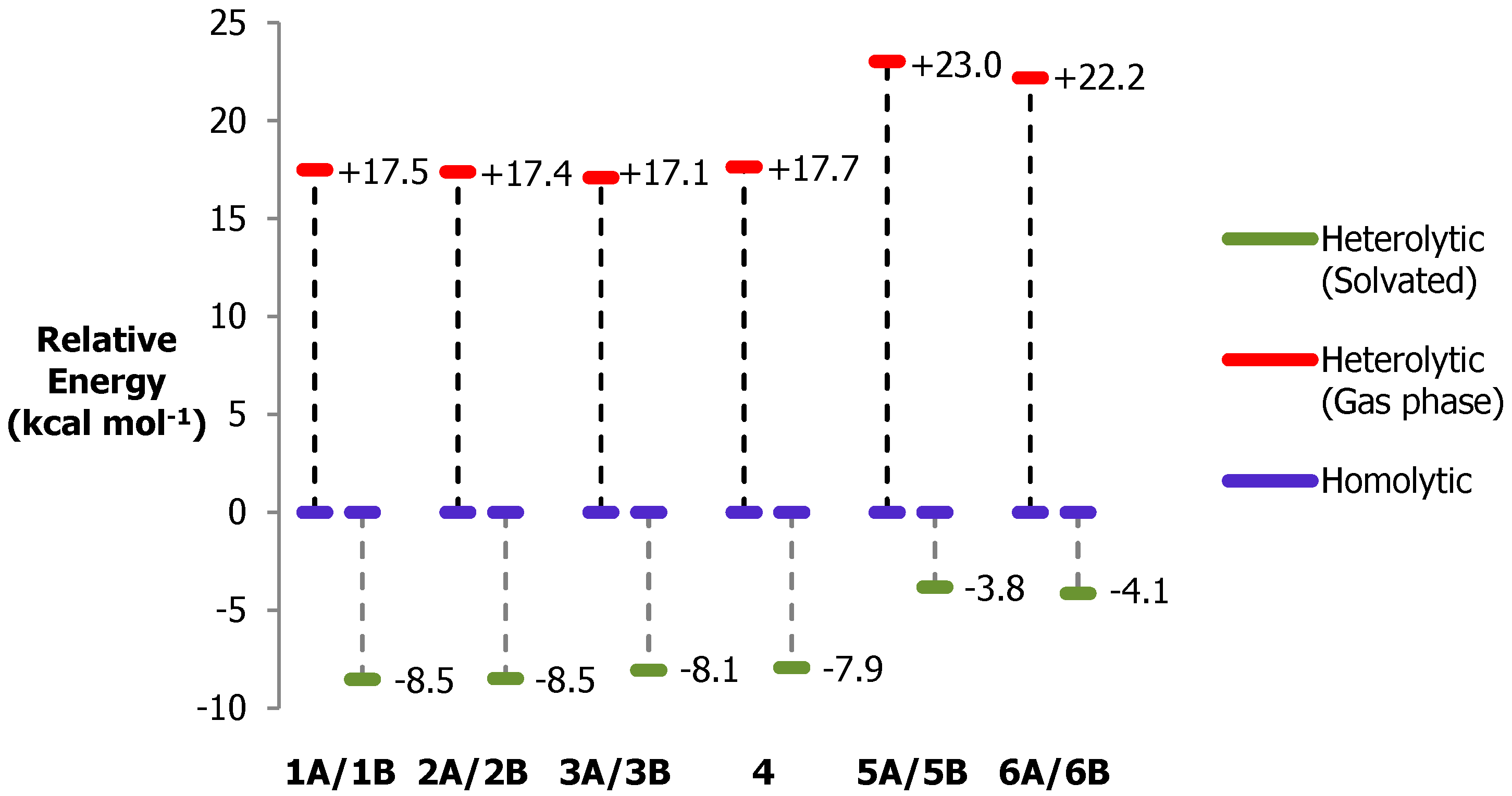
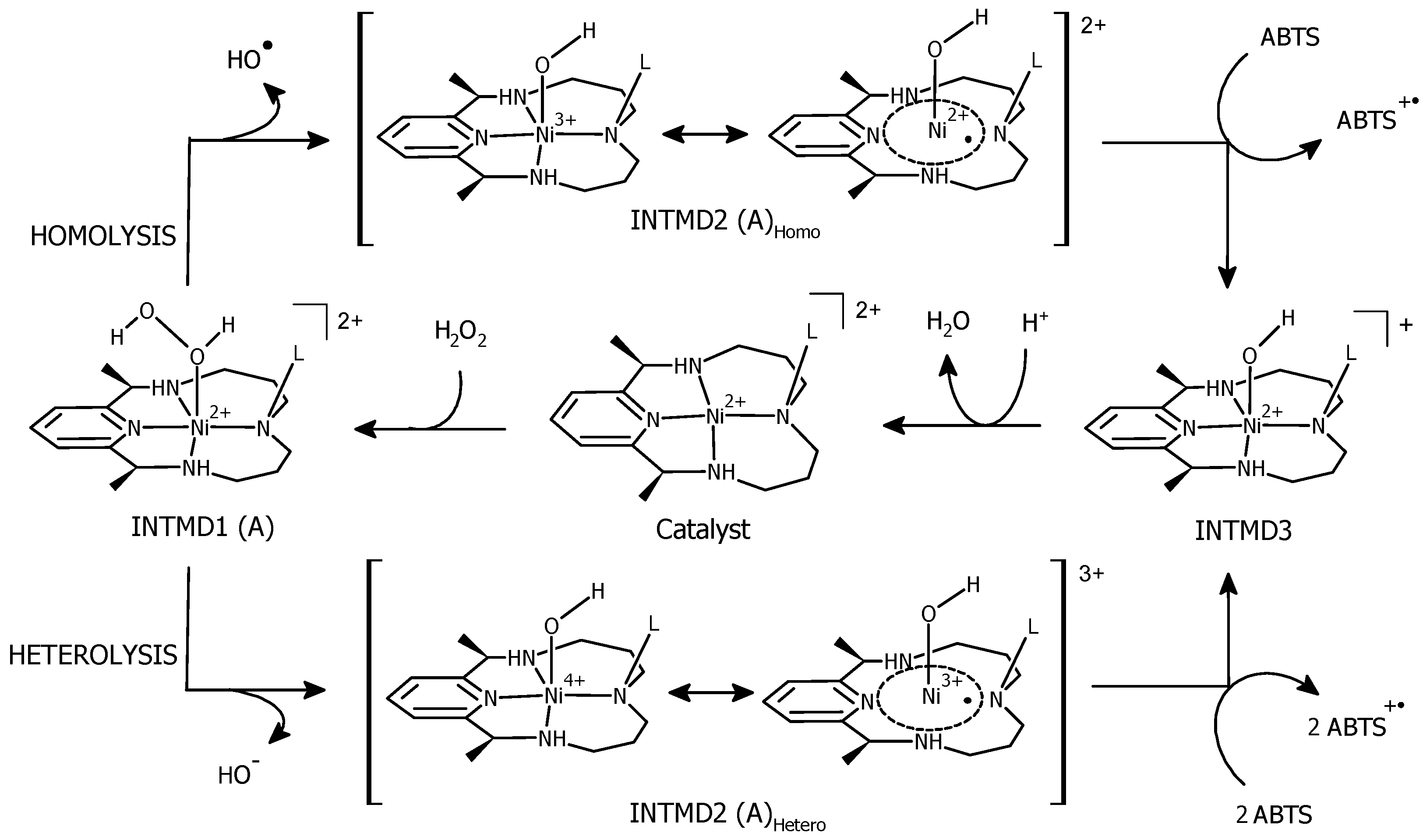
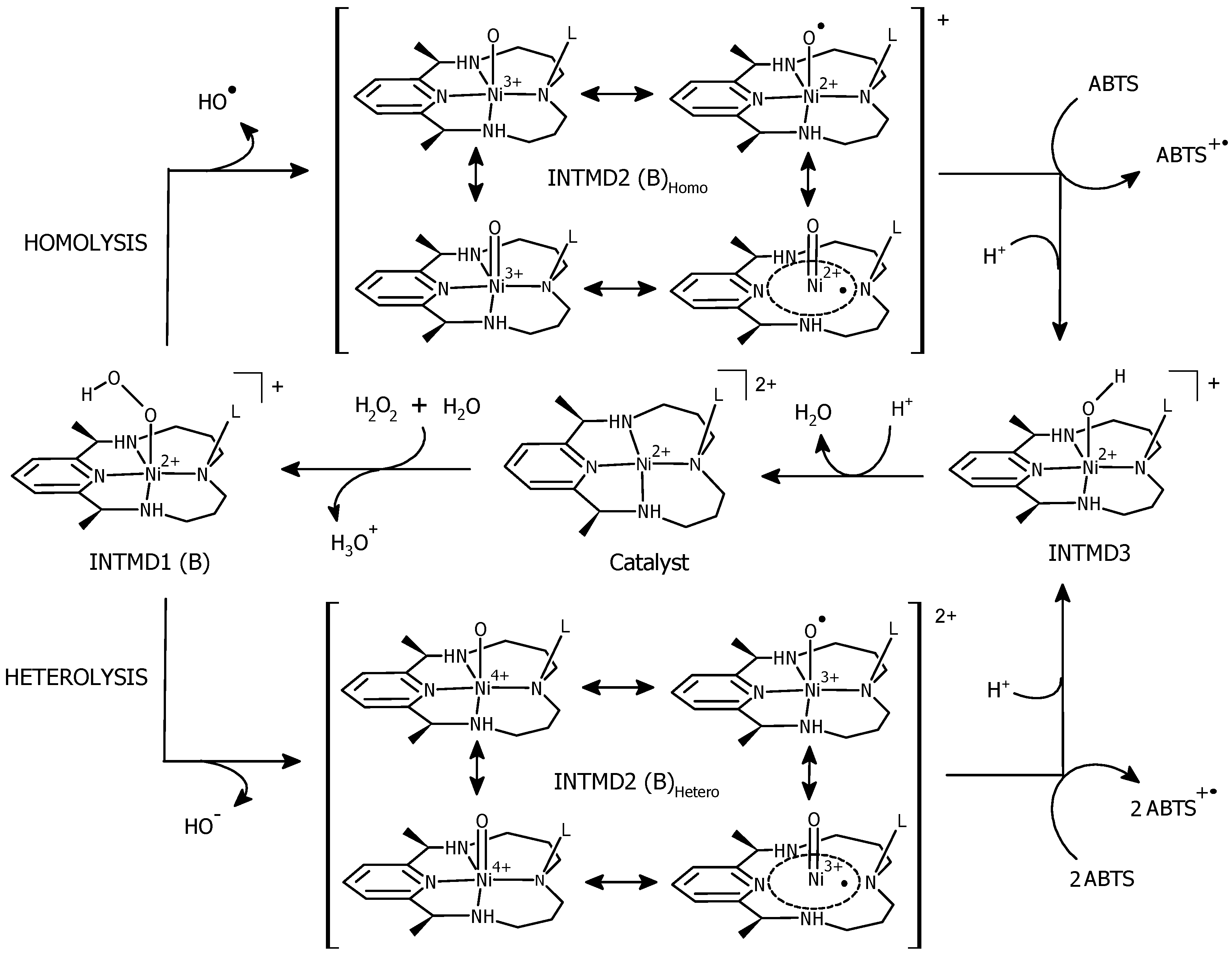
3.4. O–O Bond Cleavage: Heterolytic vs. Homolytic
3.5. Dissociation Energy of Nickel-Hydroxo O–O Bond
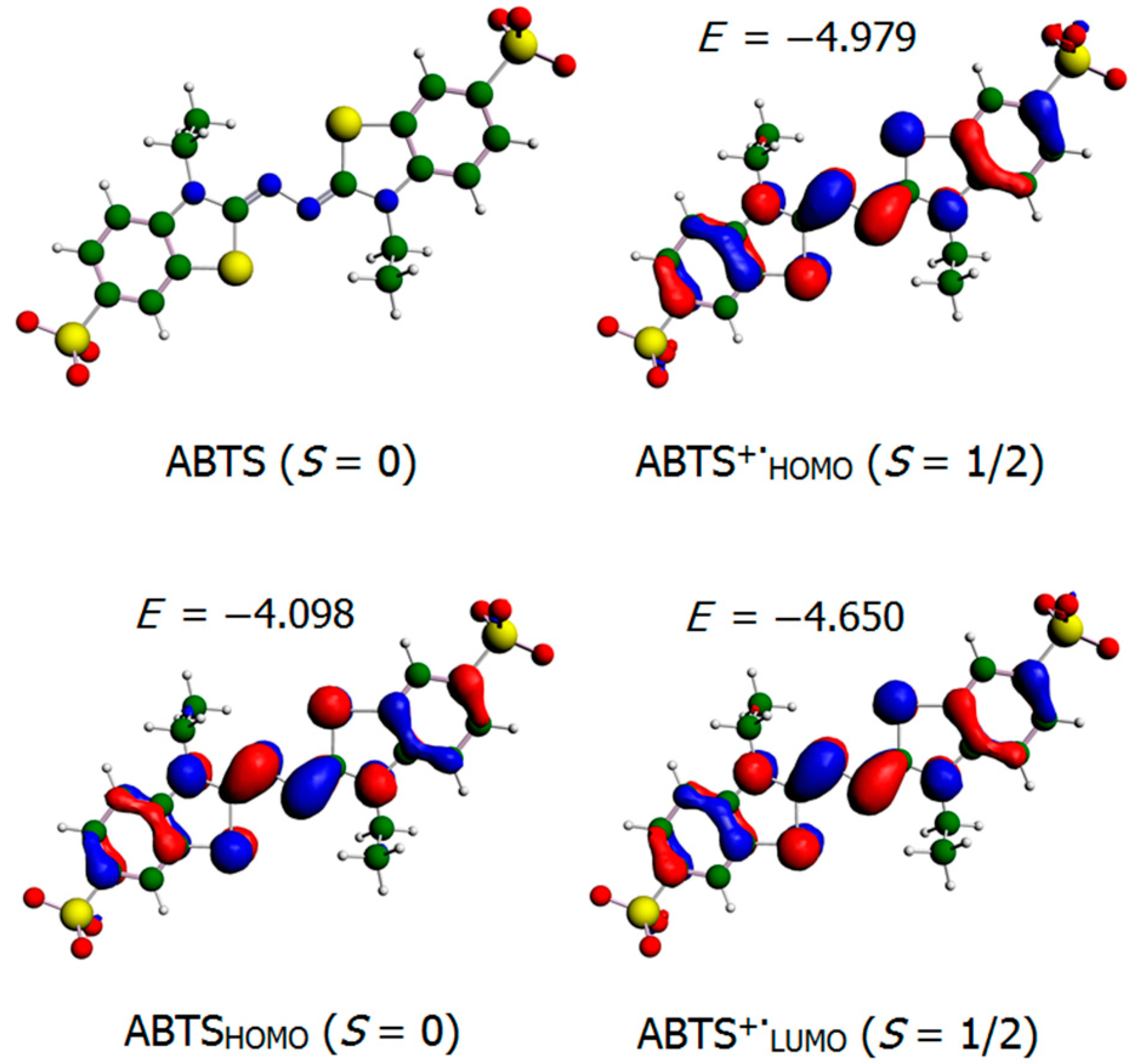
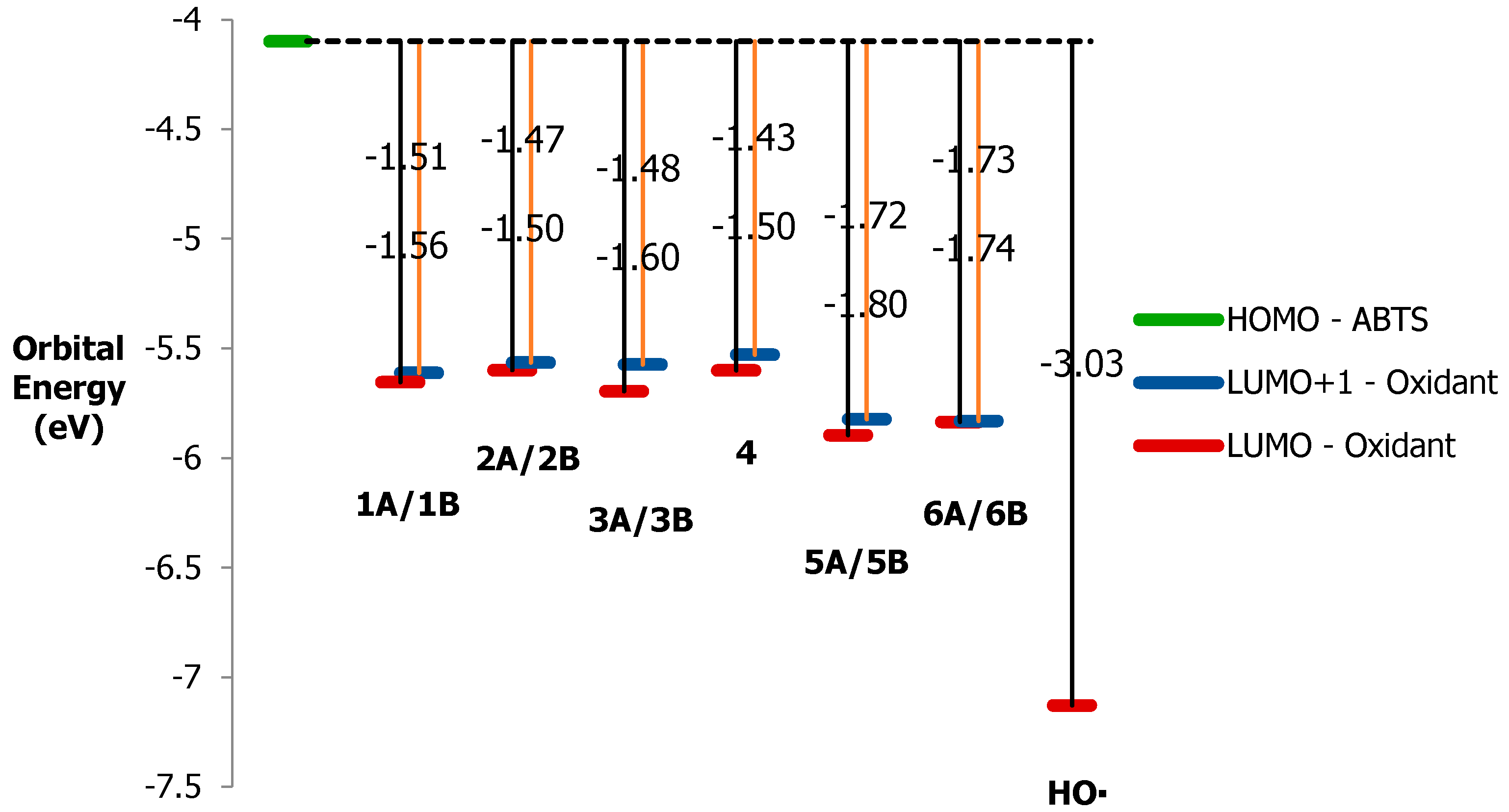
3.6. ABTS Oxidation by [(L)Ni3+–O·]2+ Active Intermediates
3.7. Formation of [(L)Ni2+–OH] and Regeneration of the Resting State
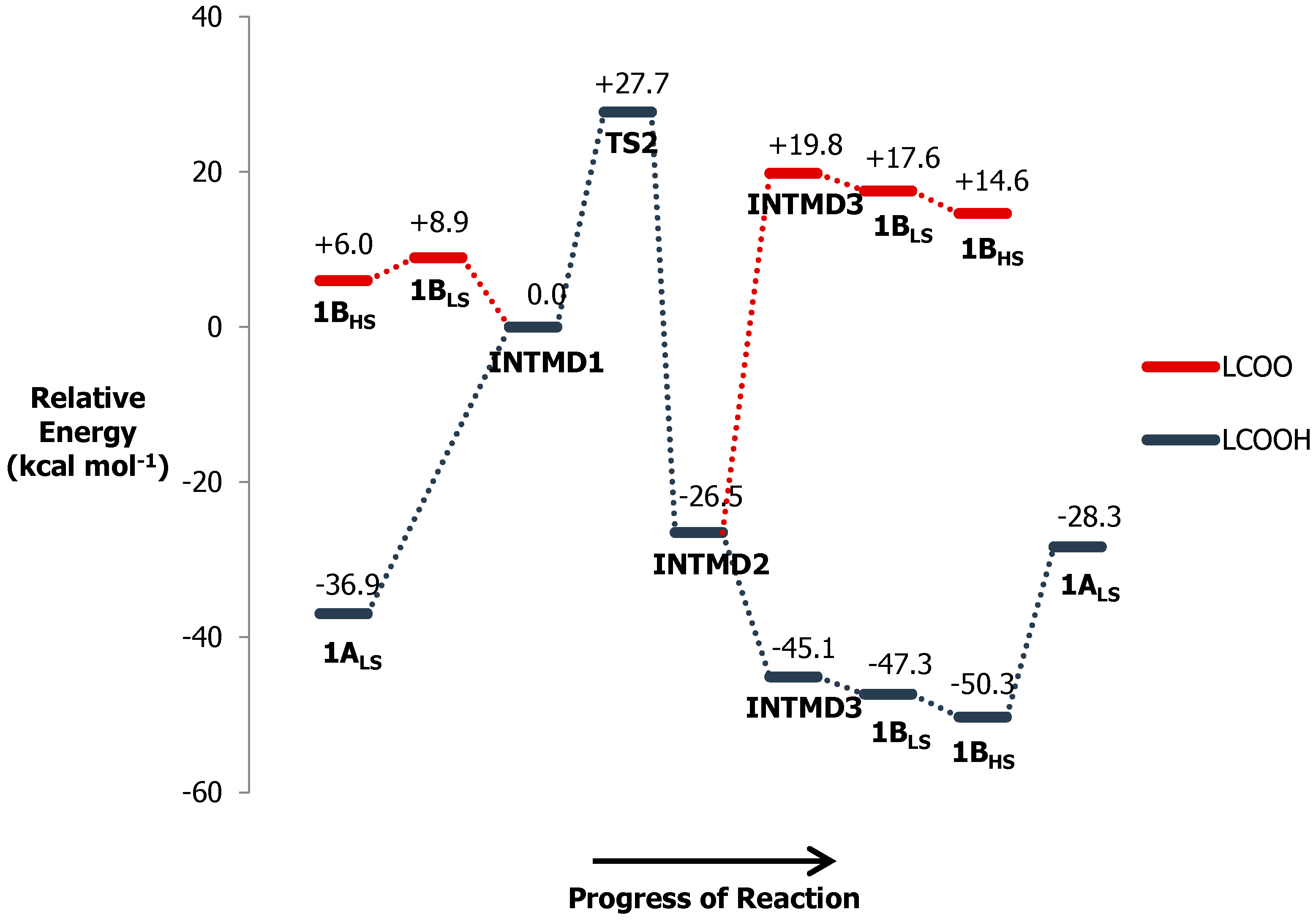
3.8. Reaction Energy Profile of Ni(II)–PyMAC Catalysis
4. Conclusions and Recommendation
Author Contributions
Funding
Conflicts of Interest
References
- Houmam, A. Electron transfer initiated reactions: Bond formation and bond dissociation. Chem. Rev. 2008, 108, 2180–2237. [Google Scholar] [CrossRef] [PubMed]
- Legros, J.; Bolm, C. Iron-catalyzed asymmetric sulfide oxidation with aqueous hydrogen peroxide. Angew. Chem. Int. Ed. 2003, 42, 5487–5489. [Google Scholar] [CrossRef] [PubMed]
- Neumann, R.; Khenkin, A.M.; Juwiler, D.; Miller, H.; Gara, M. Catalytic oxidation with hydrogen peroxide catalyzed by ‘sandwich’ type transition metal substituted polyoxometalates. J. Mol. Catal. A Chem. 1997, 117, 169–183. [Google Scholar] [CrossRef]
- Prasad, R.V.; Thakkar, N.V. Study of cobalt complexes as catalysts in the decomposition of hydrogen peroxide. J. Mol. Catal. 1994, 92, 9–20. [Google Scholar] [CrossRef]
- Shivankar, V.S.; Thakkar, N.V. Decomposition of hydrogen peroxide in presence of mixed ligand cobalt (II) and nickel (II) complexes as catalysts. J. Sci. Ind. Res. (India) 2005, 64, 496–503. [Google Scholar]
- Haber, F.; Weiss, J. Uber die Katalyse des Hydroperoxydes. Die Nat. 1932, 20, 948–950. [Google Scholar] [CrossRef]
- Kremer, M.L. Oxidation reduction step in catalytic decomposition of hydrogen peroxide by ferric ions. Trans. Faraday Soc. 1963, 59, 2535. [Google Scholar] [CrossRef]
- Stephenson, N.A.; Bell, A.T. Mechanistic insights into iron porphyrin-catalyzed olefin epoxidation by hydrogen peroxide: Factors controlling activity and selectivity. J. Mol. Catal. A Chem. 2007, 275, 54–62. [Google Scholar] [CrossRef]
- Tanaka, M.; Matsuura, K.; Yoshioka, S.; Takahashi, S.; Ishimori, K.; Hori, H.; Morishima, I. Activation of hydrogen peroxide in horseradish peroxidase occurs within ~200 μs observed by a new freeze-quench device. Biophys. J. 2003, 84, 1998–2004. [Google Scholar] [CrossRef]
- Salem, I.A.; El-maazawi, M.; Zaki, A.B. Kinetics and mechanisms of decomposition reaction of hydrogen peroxide in presence of metal complexes. Int. J. Chem. Kinet. 2000, 32, 643–666. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Teixeira, F.A.; Bokach, N.A.; Pombeiro, A.J.L.; Shul’pin, G.B. Radical decomposition of hydrogen peroxide catalyzed by aqua complexes [M(H2O)n]2+ (M=Be, Zn, Cd). J. Catal. 2014, 313, 135–148. [Google Scholar] [CrossRef]
- Aebi, H. Peroxidase. The properties and uses of a versatile enzyme and of some related catalysts. Angew. Chemie. 1965, 77, 1144. [Google Scholar] [CrossRef]
- Veitch, N.C.; Smith, A.T. Horseradish peroxidase. Adv. Inorg. Chem. 2000, 107–162. [Google Scholar] [CrossRef]
- English, A.M.; Tsaprailis, G. Catalytic structure–function relationships in heme peroxidases. Adv. Inorg. Chem. 1995, 79–125. [Google Scholar] [CrossRef]
- Pappa, H.S.; Cass, A.E.G. A step towards understanding the folding mechanism of horseradish peroxidase Tryptophan fluorescence and circular dichroism equilibrium studies. Eur. J. Biochem. 1993, 212, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Tams, J.W.; Welinder, K.G. Deglycosylation without anisole. Anal. Biochem. 1995, 228, 48–55. [Google Scholar] [CrossRef]
- Tams, J.W.; Welinder, K.G. Glycosylation and thermodynamic versus kinetic stability of horseradish peroxidase. Fed. Eur. Biochem. Soc. Lett. 1998, 421, 234–236. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, Y.; Li, H.; Zhu, R.; Shao, Q.; Yang, S.; Xu, J. NiO nanoparticles modified with 5,10,15,20-tetrakis(4-carboxyl pheyl)-porphyrin: Promising peroxidase mimetics for H2O2 and glucose detection. Biosens. Bioelectron. 2014, 64, 147–153. [Google Scholar] [CrossRef]
- Kitamura, Y.; Mori, K.; Yamamoto, M.; Nozaki, A.; Saito, M.; Tsukamoto, I.; Mifune, M.; Saito, Y. Peroxidase-like catalytic activity of aqueous- and immobilized-Mn3+-octabromo-porphyrins on ion-exchange resin supplied as mimetic of horseradish peroxidase. Chem. Pharm. Bull. (Tokyo) 2008, 56, 1364–1366. [Google Scholar] [CrossRef][Green Version]
- Shu, J.; Qiu, Z.; Wei, Q.; Zhuang, J.; Tang, D. Cobalt-porphyrin-platinum-functionalized reduced graphene oxide hybrid nanostructures: A novel peroxidase mimetic system for improved electrochemical immunoassay. Sci. Rep. 2015, 5, 15113. [Google Scholar] [CrossRef]
- Lousada, C.M.; Yang, M.; Nilsson, K.; Jonsson, M. Catalytic decomposition of hydrogen peroxide on transition metal and lanthanide oxides. J. Mol. Catal. A Chem. 2013, 379, 178–184. [Google Scholar] [CrossRef]
- Wei, H.; Wang, E. Fe3O4 Magnetic nanoparticles as peroxidase mimetics and their applications in H2O2 and glucose detection. Anal. Chem. 2008, 80, 2250–2254. [Google Scholar] [CrossRef]
- McAteer, B.; Beattie, N.; Richens, D.T. Catalytic oxidation of cyclohexene by aqueous Iron(III)/H2O2 in mildly acidic solution: Epoxidation versus allylic oxidation. Inorg. Chem. Commun. 2013, 35, 284–289. [Google Scholar] [CrossRef]
- Organo, V.G.; Filatov, A.S.; Quartararo, J.S.; Friedman, Z.M.; Rybak-Akimova, E.V. Nickel(II) complexes of monofunctionalized pyridine-azamacrocycles: Synthesis, structures, pendant arm “on-off” coordination equilibria, and peroxidase-like activity. Inorg. Chem. 2009, 48, 8456–8468. [Google Scholar] [CrossRef]
- McKenzie, S.G.; Pallucio, T.D.; Patterson, J.D.; Rybak-Akimova, E.V. Synthesis, characterization, and oxidation catalysis studies of a monofunctionalized copper pyridine-aza macrocycle. Inorg. Chim. Acta 2018, 482, 732–737. [Google Scholar] [CrossRef]
- Chng, L.L.; Chang, C.J.; Nocera, D.G. Catalytic O−O activation chemistry mediated by iron hangman porphyrins with a wide range of proton-donating abilities. Am. Chem. Soc. 2003, 5, 1403–1406. [Google Scholar] [CrossRef]
- Aniagyei, A.; Tia, R.; Adei, E. A density functional theory study of the mechanisms of oxidation of ethylene by technetium oxo complexes. Comput. Theor. Chem. 2013, 1009, 70–80. [Google Scholar] [CrossRef]
- Hou, L.-J.; Wu, B.-W.; Han, Y.-X.; Kong, C.; Chen, D.-P.; Gao, L.-G. Density functional theoretical study on the reaction mechanism of HNCS with SiHF radical. Comput. Theor. Chem. 2015, 1051, 57–61. [Google Scholar] [CrossRef]
- Lundberg, M.; Borowski, T. Oxoferryl species in mononuclear non-heme iron enzymes: Biosynthesis, properties and reactivity from a theoretical perspective. Coord. Chem. Rev. 2013, 257, 277–289. [Google Scholar] [CrossRef]
- Luo, Y.; Maeda, S.; Ohno, K. Decomposition of alkyl hydroperoxide by a copper(I) complex: Insights from density functional theory. Tetrahedron Lett. 2008, 49, 6841–6845. [Google Scholar] [CrossRef]
- Mothana, B.; Boyd, R.J. A density functional theory study of the mechanism of the Paal-Knorr pyrrole synthesis. J. Mol. Struct. THEOCHEM. 2007, 811, 97–107. [Google Scholar] [CrossRef]
- Vafaeezadeh, M.; Fattahi, A. DFT investigations for “Fischer” esterification mechanism over silica-propyl-SO3H catalyst: Is the reaction reversible? Comput. Theor. Chem. 2015, 1071, 27–32. [Google Scholar] [CrossRef]
- Hynninen, P.H.; Kaartinen, V.; Kolehmainen, E. Horseradish peroxidase-catalyzed oxidation of chlorophyll a with hydrogen peroxide Characterization of the products and mechanism of the reaction. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 531–542. [Google Scholar] [CrossRef]
- Keilin, D.; Hartree, E.F. Purification of horse-radish peroxidase and comparison of its properties with those of catalase and methaemoglobin. Biochem. J. 1951, 49, 88–106. [Google Scholar] [CrossRef]
- Gajhede, M.; Schuller, D.J.; Henriksen, A.; Smith, A.T.; Poulos, T.L. Crystal structure of horseradish peroxidase C at 2.15 Å resolution. Nat. Struct. Biol. 1997, 4, 1032–1038. [Google Scholar] [CrossRef]
- Poulos, T.; Kraut, J. A hypothetical model of the cytochrome c peroxidase. Cytochrome c electron transfer complex. J. Biol. Chem. 1980, 255, 10322–10330. [Google Scholar]
- Henriksen, A.; Smith, A.T.; Gajhede, M. The structures of the horseradish peroxidase c-ferulic acid complex and the ternary complex with cyanide suggest how peroxidases oxidize small phenolic substrates. J. Biol. Chem. 1999, 274, 35005–35011. [Google Scholar] [CrossRef]
- Stephenson, N.A.; Bell, A.T. Effects of porphyrin composition on the activity and selectivity of the iron(III) porphyrin catalysts for the epoxidation of cyclooctene by hydrogen peroxide. J. Mol. Catal. A Chem. 2007, 272, 108–117. [Google Scholar] [CrossRef]
- Song, W.J.; Ryu, Y.O.; Song, R.; Nam, W. Oxoiron(IV) porphyrin π-cation radical complexes with a chameleon behavior in cytochrome P450 model reactions. J. Biol. Inorg. Chem. 2005, 10, 294–304. [Google Scholar] [CrossRef]
- Almarsson, O.; Bruice, T.C. A homolytic mechanism of O-O bond scission prevails in the reactions of alkyl hydroperoxides with an octacationic tetraphenylporphinato-iron(III) complex in aqueous solution. J. Am. Chem. Soc. 1995, 117, 4533–4544. [Google Scholar] [CrossRef]
- Traylor, T.G.; Kim, C.; Richards, J.L.; Xu, F.; Perrin, C.L. Reactions of iron(III) porphyrins with oxidants. Structure-reactivity studies. J. Am. Chem. Soc. 1995, 117, 3468–3474. [Google Scholar] [CrossRef]
- Nam, W.; Han, H.J.; Oh, S.-Y.; Lee, Y.J.; Choi, M.-H.; Han, S.-Y.; Kim, C.; Woo, S.K.; Shin, W. New insights into the mechanisms of O−O bond cleavage of hydrogen peroxide and tert -alkyl hydroperoxides by iron(III) porphyrin complexes. J. Am. Chem. Soc. 2000, 122, 8677–8684. [Google Scholar] [CrossRef]
- Nagataki, T.; Ishii, K.; Tachi, Y.; Itoh, S. Ligand effects on NiII-catalysed alkane-hydroxylation with m-CPBA. Dalt. Trans. 2007, 1120. [Google Scholar] [CrossRef]
- Pfaff, F.F.; Heims, F.; Kundu, S.; Mebs, S.; Ray, K. Spectroscopic capture and reactivity of S = 1/2 nickel(III)–oxygen intermediates in the reaction of a Ni(II)-salt with mCPBA. Chem. Commun. 2012, 48, 3730. [Google Scholar] [CrossRef]
- Nagataki, T.; Tachi, Y.; Itoh, S. NiII(TPA) as an efficient catalyst for alkane hydroxylation with m-CPBA. Chem. Commun. 2006, 4016. [Google Scholar] [CrossRef]
- Schröder, D.; Schwarz, H. C-H and C-C bond activation by bare transition-metal oxide cations in the gas phase. Angew. Chemie Int. Ed. English. 1995, 34, 1973–1995. [Google Scholar] [CrossRef]
- Fenton, H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; Van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Boerrigter, P.M.; Te Velde, G.; Baerends, J.E. Three-dimensional numerical integration for electronic structure calculations. Int. J. Quantum Chem. 1988, 33, 87–113. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; Te Velde, G.; Baerends, E.J. Towards an order- N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- Schreckenbach, G.; Ziegler, T. The calculation of NMR shielding tensors based on density functional theory and the frozen-core approximation. Int. J. Quantum Chem. 1996, 60, 753–766. [Google Scholar] [CrossRef]
- Versluis, L.; Ziegler, T. The determination of molecular structures by density functional theory. The evaluation of analytical energy gradients by numerical integration. J. Chem. Phys. 1988, 88, 322. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943. [Google Scholar] [CrossRef]
- Head, J.D.; Zerner, M.C. A Broyden-Fletcher-Goldfarb-Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Fan, L.; Ziegler, T. Nonlocal density functional theory as a practical tool in calculations on transition states and activation energies. Applications to elementary reaction steps in organic chemistry. J. Am. Chem. Soc. 1992, 114, 10890–10897. [Google Scholar] [CrossRef]
- Fan, L.; Ziegler, T. Application of density functional theory to infrared absorption intensity calculations on main group molecules. J. Chem. Phys. 1992, 96, 9005. [Google Scholar] [CrossRef]
- Bérces, A.; Dickson, R.M.; Fan, L.; Jacobsen, H.; Swerhone, D.; Ziegler, T. An implementation of the coupled perturbed Kohn-Sham equations: Perturbation due to nuclear displacements. Comput. Phys. Commun. 1997, 100, 247–262. [Google Scholar] [CrossRef]
- Jacobsen, H.; Bérces, A.; Swerhone, D.P.; Ziegler, T. Analytic second derivatives of molecular energies: A density functional implementation. Comput. Phys. Commun. 1997, 100, 263–276. [Google Scholar] [CrossRef]
- Alexiadis, A.; Kassinos, S. On the use of the BLYP functional for the DFT calculation of graphite-hydrogen systems. J. Nuc. Mat. 2010, 396, 307–308. [Google Scholar] [CrossRef]
- Yankov, E.P.; Bakalska, R.I.; Horkel, E.; Svatunek, D.; Delchev, V.B. Experimental and theoretical study of the excited-state tautomerism of 6-azauracil in water surroundings. Chem. Phys. 2018, 515, 663–671. [Google Scholar] [CrossRef]
- de Oliveira, A.Z.; Jorge, F.E. Structural, electronic, electrical, and magnetic properties of Rhn (1≤ n ≤13) clusters. Comput. Theo. Chem. 2020, 1177, 112765. [Google Scholar] [CrossRef]
- Delchev, V.B.; Horkel, E.; Svatunek, D. Excited-state photocyclodimerization of 6-azauracil to oxazetidine cyclodimer: A mechanism elucidation in water surroundings. J. Mol. Struc. 2020, 1205, 127571. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like Screening Model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224. [Google Scholar] [CrossRef]
- Klamt, A.; Jones, V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996, 105, 9972. [Google Scholar] [CrossRef]
- Ye, S.; Neese, F. Accurate Modeling of Spin-State Energetics in Spin-Crossover Systems with Modern Density Functional Theory. Inorg. Chem. 2010, 49, 772–774. [Google Scholar] [CrossRef]
- Boguslawski, K.; Jacob, C.R.; Reiher, M. Can DFT accurately predict spin densities? Analysis of discrepancies in iron nitrosyl complexes. J. Chem. Theory Comput. 2011, 7, 2740–2752. [Google Scholar] [CrossRef]
- Kepp, K.P. Consistent descriptions of metal–ligand bonds and spin-crossover in inorganic chemistry. Coord. Chem. Rev. 2013, 257, 196–209. [Google Scholar] [CrossRef]
- Fouqueau, A.; Mer, S.; Casida, M.E. Comparison of density functionals for energy and structural differences between the high- [5T2g: (t2g)4(eg)2] and low- [1A1g: (t2g)6(eg)0] spin states of the hexaquoferrous cation [Fe(H2O)6]2+. J. Chem. Phys. 2004, 120, 9473–9486. [Google Scholar] [CrossRef] [PubMed]
- Reiher, M.; Salomon, O.; Hess, B.A. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar] [CrossRef]
- Lawson Daku, L.M.; Vargas, A.; Hauser, A.; Fouqueau, A.; Casida, M.E. Assessment of Density Functionals for the High-Spin/Low-Spin Energy Difference in the Low-Spin Iron(II) Tris(2,2′-bipyridine) Complex. Chem. Phys. Chem. 2005, 6, 1393–1410. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.M.; Staples, R.J.; Kryatov, S.V.; Nazarenko, A.Y.; Rybak-Akimova, E.V. Nickel(II) and copper(II) complexes with pyridine-containing macrocycles bearing an aminopropyl pendant arm: Synthesis, characterization, and modifications of the pendant amino group. Dalton Trans. 2003, 846–856. [Google Scholar] [CrossRef]
- Shiren, K.; Ogo, S.; Fujinami, S.; Hayashi, H.; Suzuki, M.; Uehara, A.; Watanabe, Y.; Moro-oka, Y. Synthesis, structures, and properties of bis(μ-oxo)nickel(III) and bis(μ-superoxo)nickel(II) complexes: An unusual conversion of a NiIII2(μ-O)2 core into a NiII2(μ-OO)2 core by H2O2 and oxygenation of ligand. J. Am. Chem. Soc. 2000, 122, 254–262. [Google Scholar] [CrossRef]
- Sankaralingam, M.; Balamurugan, M.; Palaniandavar, M.; Vadivelu, P.; Suresh, C.H. Nickel(II) complexes of pentadentate N5 ligands as catalysts for alkane hydroxylation by using m-CPBA as oxidant: A combined experimental and computational study. Chem.-A Eur. J. 2014, 20, 11346–11361. [Google Scholar] [CrossRef]
- Sandhiya, L.; Zipse, H. O-O Bond Homolysis in Hydrogen Peroxide. J. Comput. Chem. 2017, 38, 2186–2192. [Google Scholar] [CrossRef]
- Scott, S.L.; Wen-Jang, C.; Bakac, A.; Espenson, J.H. Spectroscopic Parameters, Electrode Potentials, Acid Ionization Constants, and Electron Exchange Rates of the 2,2′-Azinobis(3-ethylbenzothiazoline-6-sulfonate) Radicals and Ions. J. Phys. Chem. 1993, 97, 6710–6714. [Google Scholar] [CrossRef]
- Miessler, G.L.; Fischer, P.J.; Tarr, D.A. Inorganic Chemistry, 5th ed.; Pearson Education, Inc.: Upper Saddle River, NJ, USA, 2014; pp. 185–188. [Google Scholar]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP Decolorization Assay of Antioxidant Capacity Reaction Pathways. Int. J. Mol. Sci. 2020, 21, 1131. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
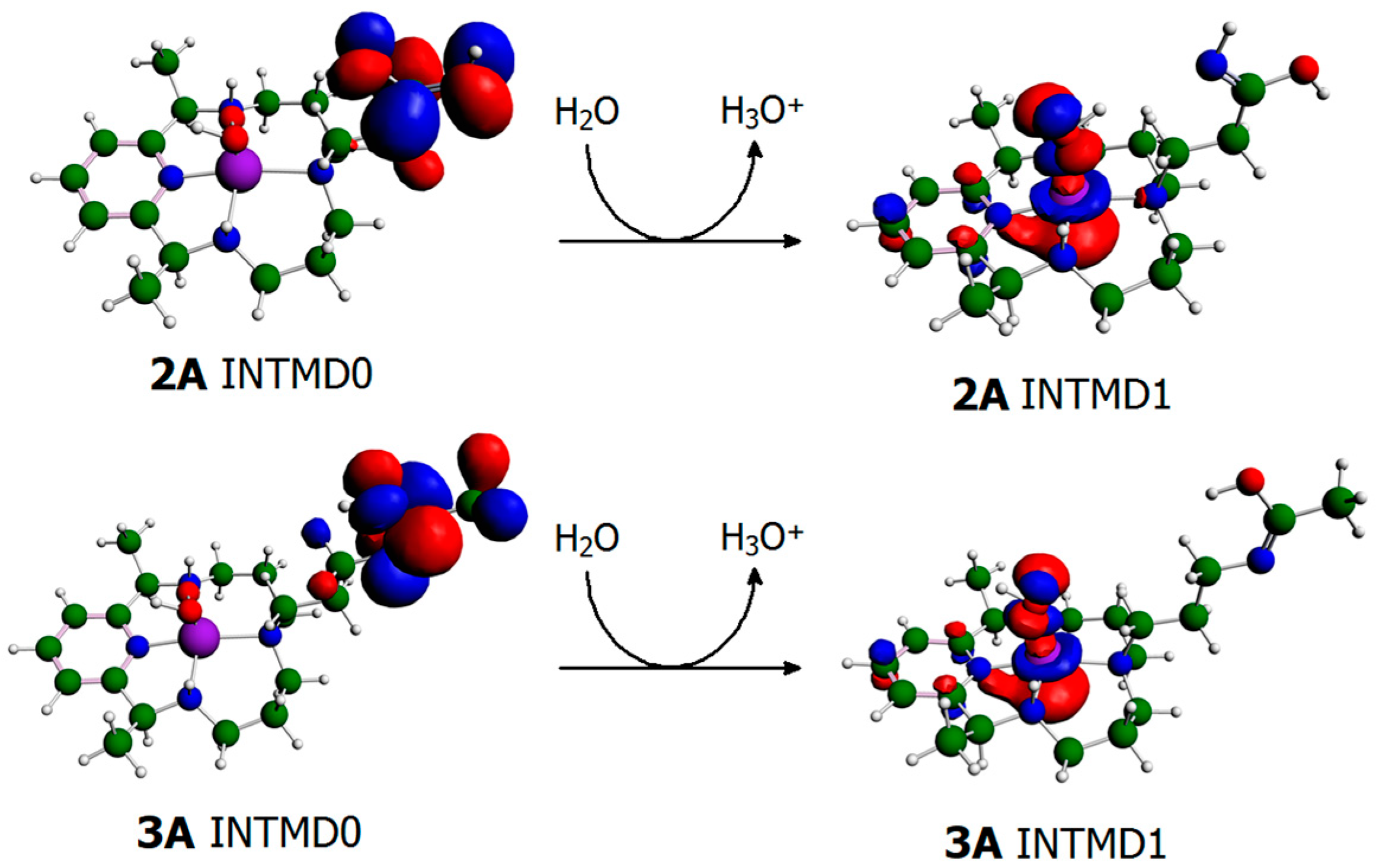{kind=link}
{kind=link}
| XC Functional | HF Exchange Admixture | Bond Energy Splitting (ELS-EHS) |
|---|---|---|
| BLYP | 0 | −0.9 |
| B3LYP* | 0.15 | 7.7 |
| B3LYP | 0.20 | 11.5 |
| Catalyst | U (kcal mol−1) | H (kcal mol−1) | S (cal mol−1 K−1) | G (kcal mol−1) |
|---|---|---|---|---|
| 1ALS | −6462.93 | −6462.34 | 149.98 | −6507.05 |
| 1BLS | −6551.99 | −6551.40 | 154.40 | −6597.43 |
| 1BHS | −6560.72 | −6560.12 | 154.90 | −6606.31 |
| 2ALS | −6577.25 | −6576.66 | 146.71 | −6620.40 |
| 2BLS | −6647.97 | −6647.38 | 153.69 | −6693.20 |
| 2BHS | −6661.41 | −6660.82 | 155.29 | −6707.12 |
| 3ALS | −7260.42 | −7259.83 | 154.60 | −7305.93 |
| 3BLS | −7320.64 | −7320.04 | 161.11 | −7368.08 |
| 3BHS | −7332.76 | −7332.16 | 163.90 | −7381.03 |
| 4LS | −5637.29 | −5636.70 | 143.20 | −5679.39 |
| 5ALS | −6395.05 | −6394.46 | 148.87 | −6438.84 |
| 5BLS | −6544.61 | −6544.02 | 153.36 | −6589.74 |
| 5BHS | −6546.03 | −6545.44 | 147.94 | −6589.55 |
| 6ALS | −7781.61 | −7781.02 | 170.97 | −7831.99 |
| 6BLS | −7938.21 | −7937.62 | 167.01 | −7987.41 |
| Atoms | 1A [1B] | 2A [2B] | 3A [3B] | 4 | 5A [5B] | 6A [6B] |
|---|---|---|---|---|---|---|
| Bond Length (Å) | ||||||
| Ni–N(1) | 1.888 [2.023] | 1.887 [2.017] | 1.892 [2.054] | 1.881 | 1.886 [2.037] | 1.887 [1.901] |
| Ni–N(2) | 2.009 [2.157] | 2.006 [2.170] | 2.018 [2.190] | 2.005 | 2.010 [2.201] | 2.006 [2.026] |
| Ni–N(3) | 2.054 [2.123] | 2.053 [2.137] | 2.056 [2.178] | 2.050 | 2.099 [2.134] | 2.091 [2.070] |
| Ni–N(4) | 2.009 [2.157] | 2.005 [2.170] | 2.011 [2.190] | 2.005 | 2.010 [2.201] | 2.003 [2.024] |
| Ni–Laxial | - [1.979] | - [1.993] | - [2.048] | - | - [2.103] | - [-] |
| N(1)–N(3) | 3.941 [4.117] | 3.940 [4.116] | 3.949 [4.101] | 3.930 | 3.985 [4.120] | 3.978 [3.970] |
| N(2)–N(4) | 3.978 [4.253] | 3.972 [4.278] | 3.989 [4.300] | 3.973 | 3.983 [4.329] | 3.973 [4.008] |
| Bond Angle (°) | ||||||
| N(1)–Ni–N(2) | 83.48 [80.36] | 83.57 [80.33] | 83.34 [79.25] | 83.69 | 83.49 [79.77] | 83.49 [83.28] |
| N(1)–Ni–N(3) | 179.08 [166.29] | 178.71 [164.42] | 179.38 [151.37] | 178.68 | 179.98 [162.02] | 179.62 [178.55] |
| N(1)–Ni–N(4) | 83.48 [80.36] | 83.54 [80.33] | 83.55 [79.25] | 83.69 | 83.49 [79.77] | 83.58 [83.26] |
| N(2)–Ni–N(3) | 96.44 [99.55] | 96.42 [99.15] | 96.42 [98.54] | 96.21 | 96.51 [99.16] | 96.49 [96.60] |
| N(2)–Ni–N(4) | 163.63 [160.66] | 163.97 [160.62] | 163.89 [158.20] | 164.49 | 164.38 [159.22] | 164.56 [163.64] |
| N(3)–Ni–N(4) | 96.44 [99.55] | 96.25 [99.15] | 96.59 [98.54] | 96.21 | 96.51 [99.16] | 96.38 [96.62] |
| pKa | 3.03 a | 11.36 a | 11.30 a | - | 6.75 b | 9.36 a |
| Group | Complex | Spin State | Deprotonation of H2O2 | Binding of HOO− to Ni | |
|---|---|---|---|---|---|
| Gas Phase | Aqueous | ||||
| G1 | 1A, 4, 5A, 6A | 1A, 2A, 3A, 4, 5A, 6A | Low-Spin | No | Yes |
| G2 | 2A, 3A, 6B | 6B | Low-Spin | Yes | Yes |
| G3 | 1B, 5B | 1B, 2B, 3B, 5B | High-Spin Low-Spin | No Yes | No Yes |
| G4 | 2B, 3B | - | High-Spin Low-Spin | Yes Yes | No Yes |
| Catalyst | Homolysis | Heterolysis | ||||
|---|---|---|---|---|---|---|
| High-Spin (S = 3/2) | Low-Spin (S = 1/2) | High-Spin (S = 1) | ||||
| Ni | O | Ni | O | Ni | O | |
| 1A/1B | 1.5123 [1.5421] | 1.0991 [1.0394] | 0.1737 [0.7512] | 1.0821 [−0.0319] | 0.6587 [0.6955] | 1.3137 [1.2724] |
| 2A/2B | 1.5097 [1.5406] | 1.1041 [1.0416] | 0.1726 [0.7044] | 1.0887 [0.0094] | 0.6559 [0.6922] | 1.3183 [1.2761] |
| 3A/3B | 1.5158 [1.5434] | 1.0903 [1.0353] | 1.1987 [1.1716] | −0.5527 [−0.5365] | 0.8605 [0.7324] | 1.2500 [1.2461] |
| 4 | 1.5239 [1.5540] | 1.0890 [1.0281] | 0.1850 [0.7622] | 1.0756 [−0.0504] | 0.6571 [0.6916] | 1.3165 [1.2775] |
| 5A/5B | 1.5276 [1.5457] | 1.0736 [1.0294] | 0.3890 [0.4053] | 0.6143 [0.5946] | 0.6822 [0.7125] | 1.3076 [1.2471] |
| 6A/6B | 1.5268 [1.5468] | 1.0775 [1.0318] | 1.1828 [0.2419] | −0.5288 [1.0087] | 0.6854 [0.7080] | 1.2774 [1.2515] |
| Catalyst | TS2 | INTMD2 | ||||
|---|---|---|---|---|---|---|
| ΔE (kcal mol−1) | vi (cm−1) | r (Å) | ΔE (kcal mol−1) | r (Å) | ||
| O1–O2 | O1–O2 | Ni–O1 | Ni–O1 | |||
| 1A/1B | +27.7 | −142 | 2.116 | 1.987 | −26.5 | 1.910 |
| 2A/2B | +27.0 | −105 | 2.109 | 2.000 | −25.7 | 1.910 |
| 3A/3B | +26.2 | −291 | 2.119 | 1.985 | −27.0 | 1.885 |
| 4 | +25.9 | −100 | 2.121 | 1.973 | −28.3 | 1.917 |
| 5A/5B | +27.2 | −242 | 2.124 | 1.973 | −21.6 | 1.878 |
| 6A/6B | +27.5 | −210 | 2.122 | 1.994 | −22.5 | 1.891 |
| H2O2 | +41.8 | −254 | 2.100 | - | +53.0 a | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taping, J.J.E.; Billones, J.B.; Organo, V.G. Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory. Computation 2020, 8, 52. https://doi.org/10.3390/computation8020052
Taping JJE, Billones JB, Organo VG. Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory. Computation. 2020; 8(2):52. https://doi.org/10.3390/computation8020052
Chicago/Turabian StyleTaping, Jerwin Jay E., Junie B. Billones, and Voltaire G. Organo. 2020. "Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory" Computation 8, no. 2: 52. https://doi.org/10.3390/computation8020052
APA StyleTaping, J. J. E., Billones, J. B., & Organo, V. G. (2020). Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory. Computation, 8(2), 52. https://doi.org/10.3390/computation8020052