Computational View toward the Inhibition of SARS-CoV-2 Spike Glycoprotein and the 3CL Protease




Abstract
1. Introduction
2. Computational Methods
2.1. Preparation of Receptor and Ligands
2.2. Molecular Docking with Autodock Vina
2.3. Analyzing the Docking Results with Chimera and BioLuminate
3. Results
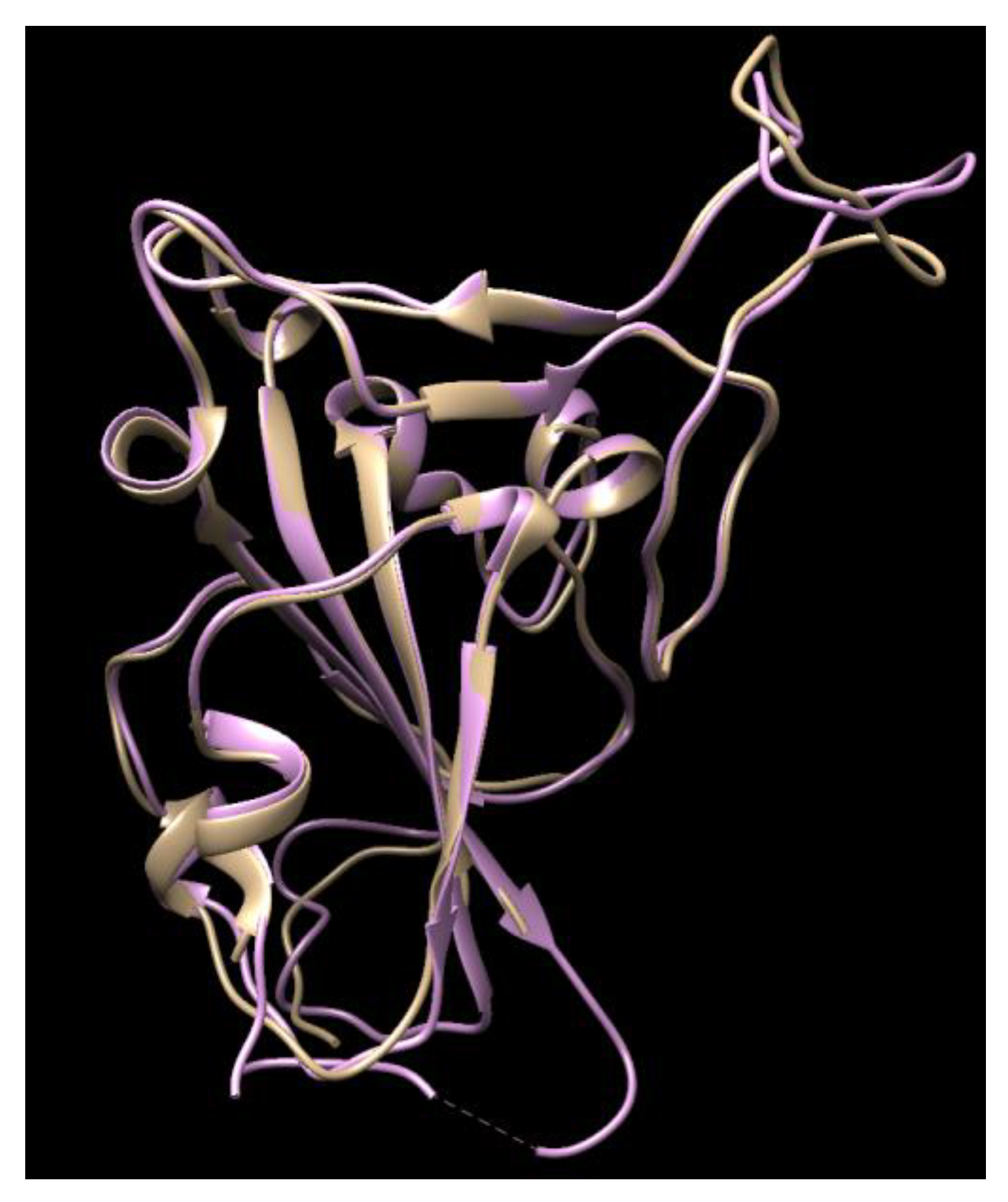
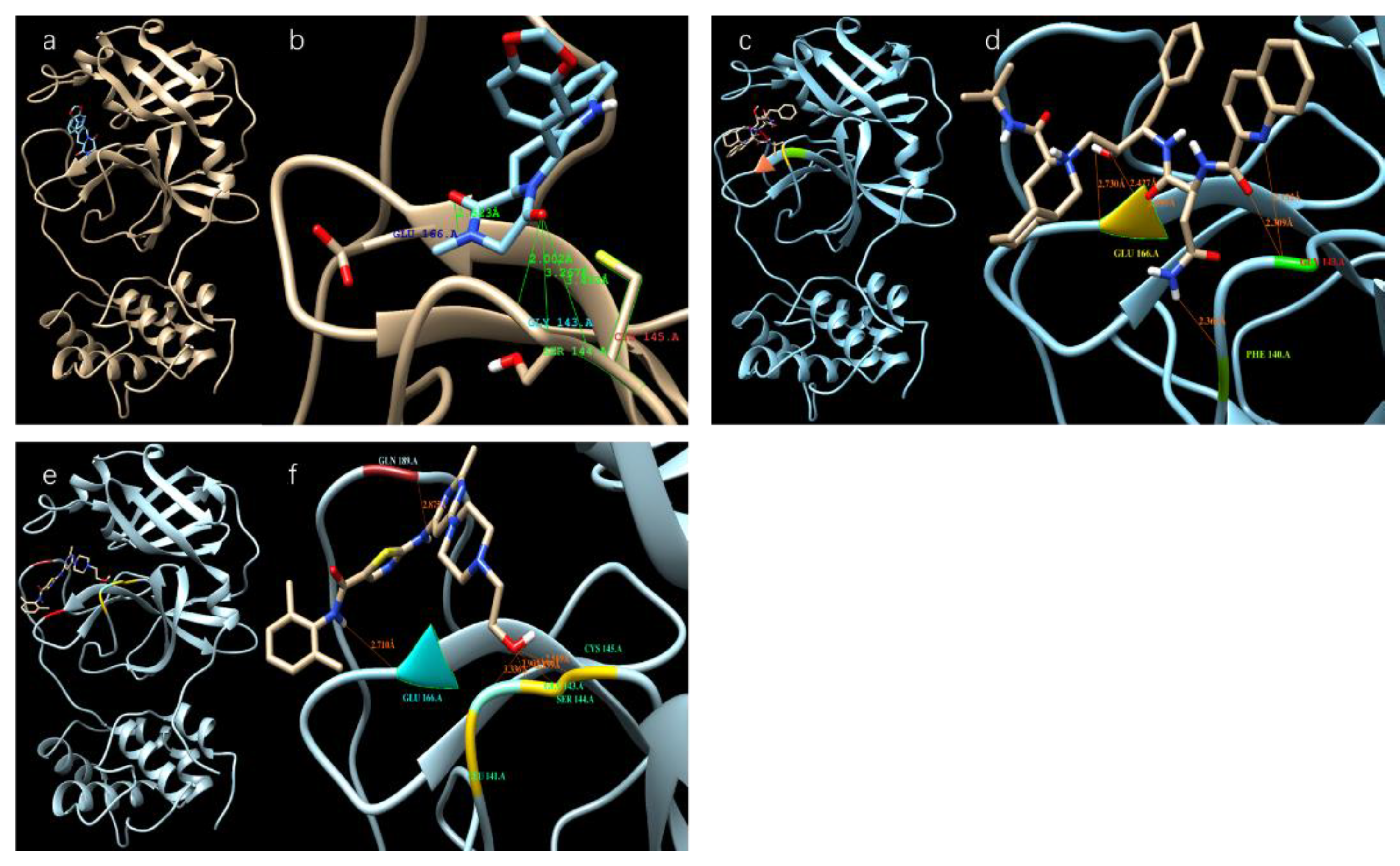
3.1. Results of the SARS-CoV-2 3CL Protease
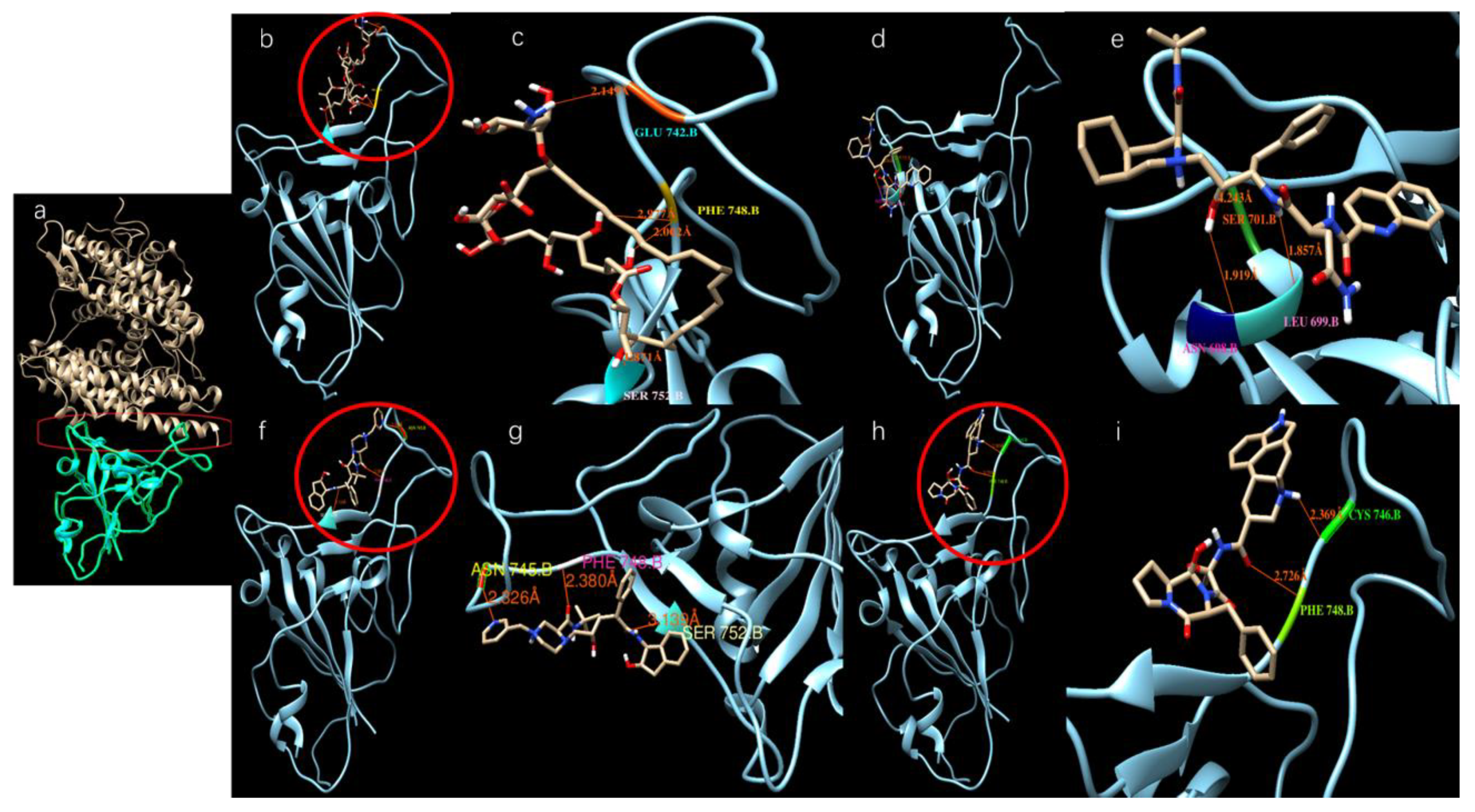
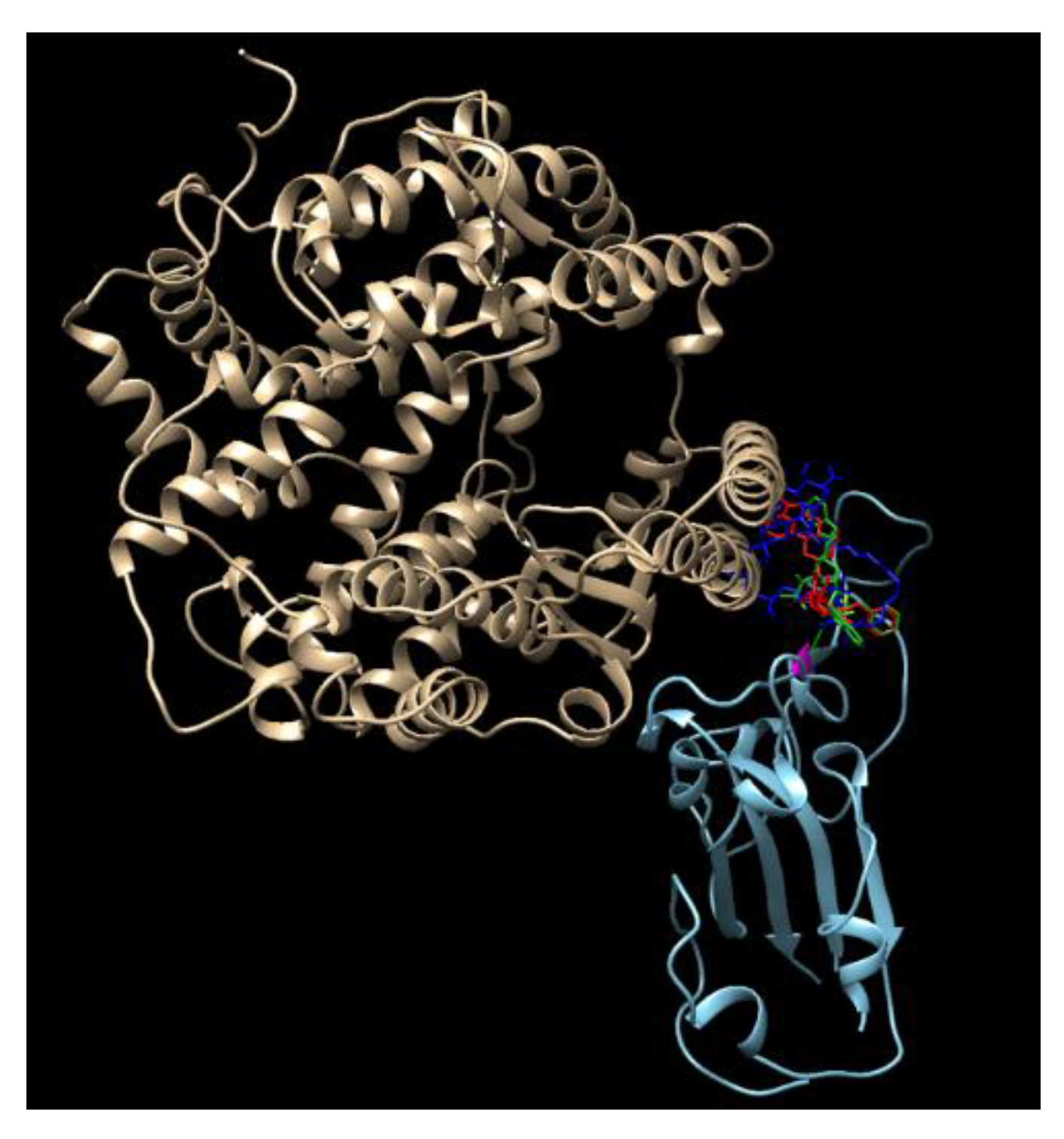
3.2. Results of SARS-CoV-2 S-Protein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Docking Score (kcal/mol) | Usage |
|---|---|---|
| Ergotamine | −8.8 | For treatment of acute migraine attacks [29] |
| Amphotericin b | −8.3 | An antifungal medication used for serious fungal infections and leishmaniasis [30] |
| Indinavir | −8.1 | A component antiretroviral therapy to treat HIV/AIDS |
| Vancomycin | −7.7 | For treatment bacterial infections [31] |
| Lonpinavir | −7.7 | An antiretroviral, often used against HIV infections |
| Zafirlukast | −7.6 | For the chronic treatment of asthma |
| Lanicor | −7.5 | Used to treat heart conditions [32] |
| PubChem ID: 54098557 | −7.5 | -- |
| Digitaline Nativelle | −7.5 | For treatment of congestive heart failure, also used as angiotensin-converting enzyme (ACE) inhibitor |
| Rivaroxaban | −7.5 | To treat and prevent blood clots [33] |
| Tadalafil | −7.5 | To treat erectile dysfunction |
| Nelfinavir | −7.3 | The treatment of HIV |
| Montelukast | −7.2 | Treatment of asthma |
| Saquinavir | −7.1 | The treatment of HIV |
| Carfilzomib | −7.1 | Anti-cancer drug as proteasome inhibitor |
| Lapatinib | −7.0 | Anti-cancer drug |
| Atovaquone | −7.0 | To treat pneumocystis pneumonia, toxoplasmosis, malaria and babesia |
| Celecoxib | −7.0 | An anti-inflammation drug |
| Vardenafil | −6.9 | For treatment of erectile dysfunction |
| Dasatinib | −6.8 | To treat certain cases of chronic myelogenous leukemia |
| Cortisone | −6.6 | Can be used for a variety of conditions |
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 20, 30251–30258. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.; Wang, X.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.; Zhu, Y.; Li, B.; Huang, C.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef]
- Chu, C.M.; Cheng, V.C.; Hung, I.F.; Wong, M.M.; Chan, K.H.; Chan, K.S.; Kao, R.Y.; Poon, L.L.; Wong, C.L.; Guan, Y.; et al. Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef]
- Nukoolkarn, V.; Lee, V.S.; Malaisree, M.; Aruksakulwong, O.; Hannongbua, S. Molecular dynamic simulations analysis of ritonavir and lopinavir as SARS-CoV 3CL(pro) inhibitors. J. Theor. Biol. 2008, 254, 861–867. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, B.; Jin, Z.; Yang, H.; Rao, Z. The crytal structure of 2019-nCoV main protease in complex with an inhibitor N3. Nature 2020. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- AndrejŠali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 2014, 82, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Salam, N.K.; Adzhigirey, M.; Sherman, W.; Pearlman, D.A. Structure-based approach to the prediction of disulfide bonds in proteins. Protein Eng. Des. Sel. 2014, 27, 365–374. [Google Scholar] [CrossRef]
- Beard, H.; Cholleti, A.; Pearlman, D.; Sherman, W.; Loving, K.A. Applying Physics-Based Scoring to Calculate Free Energies of docking for Single Amino Acid Mutations in Protein-Protein Complexes. PLoS ONE 2013, 8, e82849. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef]
- Kitchen, V.S.; Skinner, C.; Ariyoshi, K.; Lane, E.A.; Duncan, I.B.; Burckhardt, J.; Burger, H.U.; Bragman, K.; Pinching, A.J.; Weber, J.N. Safety and activity of saquinavir in HIV infection. Lancet 1995, 345, 952–955. [Google Scholar] [CrossRef]
- Baumann, M. An Overview of the Key Routes to the Best Selling 5-membered Ring Heterocyclic Pharmaceuticals. Beilstein J. Org. Chem. 2011, 7, 442–495. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; Brenner, B.; Dahl, O.E.; Eriksson, B.I.; Mouret, P.; Muntz, J.; Soglian, A.G.; Pap, A.F.; Misselwitz, F.; Haas, S.; et al. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: A double-blind, randomised controlled trial. Lancet 2008, 372, 31–39. [Google Scholar] [CrossRef]
- Terrett, N.K.; Bell, A.S.; Brown, D.; Ellis, P. Sildenafil (Viagra), a potent and selective inhibitor of type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg. Med. Chem. Lett. 1996, 6, 1819–1824. [Google Scholar] [CrossRef]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef]
- Kloner, R.A. Pharmacology and drug interaction effects of the phosphodiesterase 5 inhibitors: Focus on alpha-blocker interactions. Am. J. Cardiol. 2005, 96, 42M–46M. [Google Scholar] [CrossRef]
- De Lepeleire, I.; Reiss, T.F.; Rochette, F.; Botto, A.; Zhang, J.; Kundu, S.; Decramer, M. Montelukast causes prolonged, potent leukotriene D4-receptor antagonism in the airways of patients with asthma. Clin. Pharmacol. Ther. 1997, 61, 83–92. [Google Scholar] [CrossRef]
- Capaldini, L. Protease inhibitors’ metabolic side effects: Cholesterol, triglycerides, blood sugar, and “Crix belly. Interview with Lisa Capaldini, M.D. Interview by John S. James”. AIDS Treatment News 1997, 277, 1–4. [Google Scholar]
- Park, N.H.; Shin, K.H.; Kang, M.K. Chapter 34. Antifungal and Antiviral Agents. In Pharmacology and Therapeutics for Dentistry, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 488–503. [Google Scholar]
- Ibraheem, J.J.; Paalzow, L.; Tfelt-Hansen, P. Low bioavailability of ergotamine tartrate after oral and rectal administration in migraine sufferers. Br. J. Clin. Pharmacol. 1983, 16, 695–699. [Google Scholar] [CrossRef]
- World Health Organization. Control of the Leishmaniasis: Report of a Meeting of the WHO Expert Committee on the Control of Leishmaniases; World Health Organization: Geneva, Switzerland, 22–26 March 2010; pp. 55–186. [Google Scholar]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical practice guidelines by the infectious diseases society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: Executive summary. Clin. Infect. Dis. 2011, 52, 285–292. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.S.; Zucker, I.H. Carotid sinus baroreceptor sensitivity in experimental heart failure. Circulation 1990, 81, 1959–1966. [Google Scholar] [CrossRef]
- Turpie, A.G. New oral anticoagulants in atrial fibrillation. Eur. Heart J. 2008, 29, 155–165. [Google Scholar] [CrossRef] [PubMed]




| Drug Name | Docking Score (kcal/mol) | Usage |
|---|---|---|
| Saquinavir | −9.5 | Antiretroviral drug to treat or prevent HIV/AIDS [20] |
| Tadalafil | −9.3 | A medication used to treat erectile dysfunction (ED), benign prostatic hyperplasia (BPH), and pulmonary arterial hypertension [21] |
| Rivaroxaban | −9.2 | An anticoagulant medication used to treat and prevent blood clots [22] |
| Sildenafil | −8.9 | A medication used to treat erectile dysfunction and pulmonary arterial hypertension [23] |
| Dasatinib | −8.8 | A targeted therapy used to treat certain cases of chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL) [24] |
| Vardenafil | −8.7 | A PDE5 inhibitor used to treat erectile dysfunction [25] |
| Montelukast | −8.5 | To treat seasonal and year-round allergies [26] |
| Indinavir | −8.3 | A component antiretroviral therapy to treat HIV/AIDS [27] |
| Lopinavir | −8.2 | Protease inhibitor |
| Cortisone | −8.2 | Can be used for a variety of conditions |
| celecoxib | −8.1 | An anti-inflammation drug |
| Atazanavir | −8.1 | An antiretroviral drug used for HIV treatment |
| Iressa | −7.9 | A drug for cancer treatment |
| Darunavir | −7.7 | An antiretroviral drug used for HIV treatment |
| Sorafenib | −7.5 | A drug for cancer treatment |
| Interaction of S-Protein and S-Protein/Drug Complex with ACE-2 | Docking Energy (kcal/mol) |
|---|---|
| SARS-CoV S-protein (for comparison) | −92.7 |
| SARS-CoV-2 S-protein | −82.2 |
| SARS-CoV-2 S-protein/Ergotamine | 56.4 |
| SARS-CoV-2 S-protein/Amphotericin b | 78.6 |
| SARS-CoV-2 S-protein/Indinavir | −61.9 |
| SARS-CoV-2 S-protein/Vancomycin | 81.7 |
| SARS-CoV-2 S-protein/Zafirlukast | 52.6 |
| SARS-CoV-2 S-protein/Lanicor | 4.2 |
| SARS-CoV-2 S-protein/Nelfinavir | −81.5 |
| SARS-CoV-2 S-protein/Montelukast | −71.3 |
| SARS-CoV-2 S-protein/Saquinavir | −48.2 |
| SARS-CoV-2 S-protein/Carfilzomib | −88.1 |
| SARS-CoV-2 S-protein/Lapatinib | −83.1 |
| SARS-CoV-2 S-protein/Atovaquone | −68.2 |
| SARS-CoV-2 S-protein/Celecoxib | −74.2 |
| SARS-CoV-2 S-protein/Dasatinib | −42.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, Z.; Zhang, H.; Ji, H.-F.; Chen, Q. Computational View toward the Inhibition of SARS-CoV-2 Spike Glycoprotein and the 3CL Protease. Computation 2020, 8, 53. https://doi.org/10.3390/computation8020053
Qiao Z, Zhang H, Ji H-F, Chen Q. Computational View toward the Inhibition of SARS-CoV-2 Spike Glycoprotein and the 3CL Protease. Computation. 2020; 8(2):53. https://doi.org/10.3390/computation8020053
Chicago/Turabian StyleQiao, Zhen, Hongtao Zhang, Hai-Feng Ji, and Qian Chen. 2020. "Computational View toward the Inhibition of SARS-CoV-2 Spike Glycoprotein and the 3CL Protease" Computation 8, no. 2: 53. https://doi.org/10.3390/computation8020053
APA StyleQiao, Z., Zhang, H., Ji, H.-F., & Chen, Q. (2020). Computational View toward the Inhibition of SARS-CoV-2 Spike Glycoprotein and the 3CL Protease. Computation, 8(2), 53. https://doi.org/10.3390/computation8020053