
Implicit and Explicit Solvent Effects on the Global Reactivity and the Density Topological Parameters of the Preferred Conformers of Caespitate

 , ,
, ,
Abstract
1. Introduction
2. Computational Methodology
3. Results and Discussion
3.1. Conformational and Population Analyses
3.2. ONIOM DFT
3.3. Global Reactivity Indices
3.4. Density Topological Parameters
3.5. Validation of the Solvent Models by NMR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rolnik, A.; Olas, B. The Plants of the Asteraceae Family as Agents in the Protection of Human Health. Int. J. Mol. Sci. 2021, 22, 63009. [Google Scholar] [CrossRef] [PubMed]
- Achika, J.; Arthur, D.; Gerald, I.; Adedayo, A. A Review on the Phytoconstituents and Related Medicinal Properties of Plants in the Asteraceae Family. IOSR J. Appl. Chem. 2014, 7, 1–8. [Google Scholar] [CrossRef]
- Mathekga, A.D.M.; Meyer, J.J.M.; Horn, M.M.; Drewes, S.E. An acylated phloroglucinol with antimicrobial properties from Helichrysum caespititium. Phytochemistry 2000, 53, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Dekker, T.G.; Fourie, T.G.; Snyckers, F.O.; van der Schyf, C.J. Studies of South African medicinal plants. Part 2. Caespitin, a new phloroglucinol derivative with antimicrobial properties from Helichrysum caespititium. S. Afr. J. Chem. 1983, 36, 114–116. Available online: https://journals.co.za/doi/pdf/10.10520/AJA03794350_1036 (accessed on 19 October 2023).
- Mapaura, A.; Timberlake, J. A Checklist of Zimbabwean Vascular Plants Southern African Botanical Diversity Network Report No. 33; SABONET Publications: Pretoria, South Africa, 2004; p. 26. [Google Scholar]
- Pooley, E. A Field Guide to the Wild Flowers of KwaZulu-Natal and the Eastern Region; Natal Flora Publications Trust: Durban, South African, 1998; pp. 442–443. [Google Scholar]
- WHO. Tuberculosis. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 20 October 2023).
- Mathekga, A.D.M. Antimicrobial Activity of Helichrysum Species and the Isolation of a New Phloroglucinol from Helichrysum Caespititium. Doctoral Thesis, University of Pretoria, UPSpace International Repository, Pretoria, South Africa, 2001. Available online: https://repository.up.ac.za/handle/2263/23672 (accessed on 20 October 2023).
- Mammino, L.; Kabanda, M.M. Model structures for the study of acylated phloroglucinols and computational study of the caespitate molecule. J. Mol. Struct. THEOCHEM 2007, 805, 39–52. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. A study of the intramolecular hydrogen bond in acylphloroglucinols. J. Mol. Struct. THEOCHEM 2009, 901, 210–219. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. The conformational preferences of acylphloroglucinols—A promising class of biologically active compounds. Int. J. Comput. Chem. 2012, 112, 3691–3702. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. A Computational Study of the Effects of Different Solvents on the Characteristics of the Intramolecular Hydrogen Bond in Acylphloroglucinols. J. Phys. Chem. A 2009, 113, 15064–15077. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. The role of additional O–H···O intramolecular hydrogen bonds for acylphloroglucinols’ conformational preferences in vacuo and in solution. Mol. Simul. 2013, 39, 1–13. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. Computational study of the patterns of weaker intramolecular hydrogen bonds stabilizing acylphloroglucinols. Int. J. Quantum Chem. 2012, 112, 2650–2658. [Google Scholar] [CrossRef]
- Mammino, L.; Kabanda, M.M. The Geometric Isomers of Caespitate: A Computational Study in Vacuo and in Solution. Int. J. Biol. Biomed. Eng. 2012, 6, 114–133. Available online: http://ijdri.com/ijbbe/2012/17-931.pdf (accessed on 19 October 2023).
- Tobiason, F.L.; Vergoten, G. Chapter: GMMX Conformation Searching and Prediction of NMR Proton-Proton Coupling Constants. In Biomolecular Structure and Dynamics; NATO ASI Series; Springer: Dordrecht, The Netherlands, 1997; Volume 342, pp. 179–186. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2019; Available online: https://gaussian.com/gaussview6/ (accessed on 24 October 2023).
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A density functional with spherical atom dispersion terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.D. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639. [Google Scholar] [CrossRef]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Solvents List SCRF. Gaussian. Available online: http://gaussian.com/scrf/?tabid=7 (accessed on 10 October 2023).
- Ott, J.B.; Boerio-Goates, J. Chemical Thermodynamics: Advanced Applications, 1st ed.; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef]
- Pantazis, D.; Neese, F. All-electron scalar relativistic basis sets for the 6p elements. Theor. Chem. Acc. 2012, 131, 1292. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB-An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.E.; Rangel-Galván, M.; Morales Dávila, E.; Caballero, N.A.; Melendez, F.J. Theoretical NMR and IR spectroscopic analyses of the preferred conformers of the neurotransmitter anandamide. Int. J. Quantum Chem. 2023, 123, e27098. [Google Scholar] [CrossRef]
- Hruska, E.; Gale, A.; Huang, X.; Liu, F. AutoSolvate: A Toolkit for Automating Quantum Chemistry Design and Discovery of Solvated Molecules. J. Chem. Phys. 2022, 156, 124801. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224180. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tod, A.K. TK Gristmill Software, AIMAll Version 17.11.14; Overland Park, KS, USA, 2017. Available online: https://aim.tkgristmill.com (accessed on 14 October 2023).
- Andrienko, G.A. Chemcraft—Graphical Program for Visualization of Quantum Chemistry Computations; Version 1.8; Chemcraft Authors: Ivanovo, Russia, 2016; Available online: http://www.chemcraftprog.com (accessed on 10 October 2023).
- Parthasarathi, R.; Padmanabhan, J.; Sarkar, U.; Maiti, B.; Subramanian, V.; Chattaraj, P.K. Toxicity Analysis of Benzidine Through Chemical Reactivity and Selectivity Profiles: A DFT Approach. Internet Electron. J. Mol. Des. 2003, 2, 798–813. Available online: http://www.biochempress.com/av02_0798.html (accessed on 19 December 2023).
- Pradeep Kumar, C.B.; Raghu, M.S.; Prasad, K.N.N.; Chandrasekhar, S.; Jayanna, B.K.; Alharthi, F.A.; Prashanth, M.K.; Yoghes Kumar, K. Expatiating biological excellence of 2,3-disubstituted quinazolin-4(1H)-ones against Mycobacterium tuberculosis and DNA using docking, spectroscopic and DFT studies. New J. Chem. 2020, 45, 403–414. [Google Scholar] [CrossRef]
- Mishra, V.R.; Ghanavatkar, C.W.; Mali, S.N.; Chaudhari, H.K.; Sekar, N. Schiff base clubbed benzothiazole: Synthesis, potent antimicrobial and MCF-7 anticancer activity, DNA cleavage and computational study. J. Biomol. Struct. Dyn. 2019, 38, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.N.; Pandey, A.; Thorat, B.R.; Lai, C. Multiple 3D- and 2D-quantitative structure–activity relationship models (QSAR), theoretical study and molecular modeling to identify structural requirements of imidazopyridine analogues as anti-infective agents against tuberculosis. Struct. Chem. 2022, 33, 679–694. [Google Scholar] [CrossRef]
- Eno, E.A.; Mbonu, J.I.; Louis, H.; Patrick-Inezi, F.S.; Gber, T.E.; Unimuke, T.O.; Okon, E.E.D.; Benjamin, I.; Offiong, O.E. Antimicrobial activities of 1-phenyl-3-methyl-4-trichloroacetyl-pyrazolone: Experimental, DFT studies, and molecular docking investigation. J. Indian Chem. Soc. 2022, 99, 100524. [Google Scholar] [CrossRef]
- Laurence, C.; Berthelot, M. Observations on the strength of hydrogen bonding. Perspect. Drug Discov. Des. 2000, 18, 39–60. [Google Scholar] [CrossRef]
- McDonagh, A.F.; Lightner, D.A. Influence of Conformation and Intramolecular Hydrogen Bonding on the Acyl Glucuronidation and Biliary Excretion of Acetylenic Bis-Dipyrrinones Related to Bilirubin. J. Med. Chem. 2007, 50, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.; Millan, D.S.; Perez, M.; Wakenhut, F.; Whitlock, G.A. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Med. Chem. Commun. 2011, 2, 669–674. [Google Scholar] [CrossRef]
- Meyer, J.J.M.; Lall, N.; Mathekga, A.D.M.; Jäger, A.K. In vitro inhibition of drug-resistant and drug-sensitive strains of Mycobacterium tuberculosis by Helichrysum caespititium. S. Afr. J. Bot. 2002, 68, 90–93. [Google Scholar] [CrossRef]
- Pinto, A.V.; Magalhães, A.L. Intramolecular Hydrogen Bonds in Tip-Functionalized Single-Walled Carbon Nanotubes as pH-Sensitive Gates. J. Phys. Chem. A 2020, 124, 9542–9551. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Duarte, D.J.R.; Angelina, E.L.; Peruchena, N.M. Physical meaning of the QTAIM topological parameters in hydrogen bonding. J. Mol. Model. 2014, 20, 2510. [Google Scholar] [CrossRef] [PubMed]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199. [Google Scholar] [CrossRef] [PubMed]
- Alareeqi, S.; Bahamon, D.; Nogueira, R.P.; Vega, L.F. Understanding the relationship between the structural properties of three corrosion inhibitors and their surface protectiveness ability in different environments. Appl. Surf. Sci. 2021, 542, 148600. [Google Scholar] [CrossRef]
- Castro Sánchez, M.E.; Noriega, L.; Perez-Aguilar, J.M.; Caballero-Concha, N.A.; Merino-Montiel, P.; Romero López, A.; Melendez Bustamante, F.J. Advances in Green and Sustainable Chemistry. In Green Chemistry and Computational Chemistry: Shared Lessons in Sustainability; Elsevier: Amsterdam, The Netherlands, 2022; Chapter 8; pp. 193–214. [Google Scholar] [CrossRef]
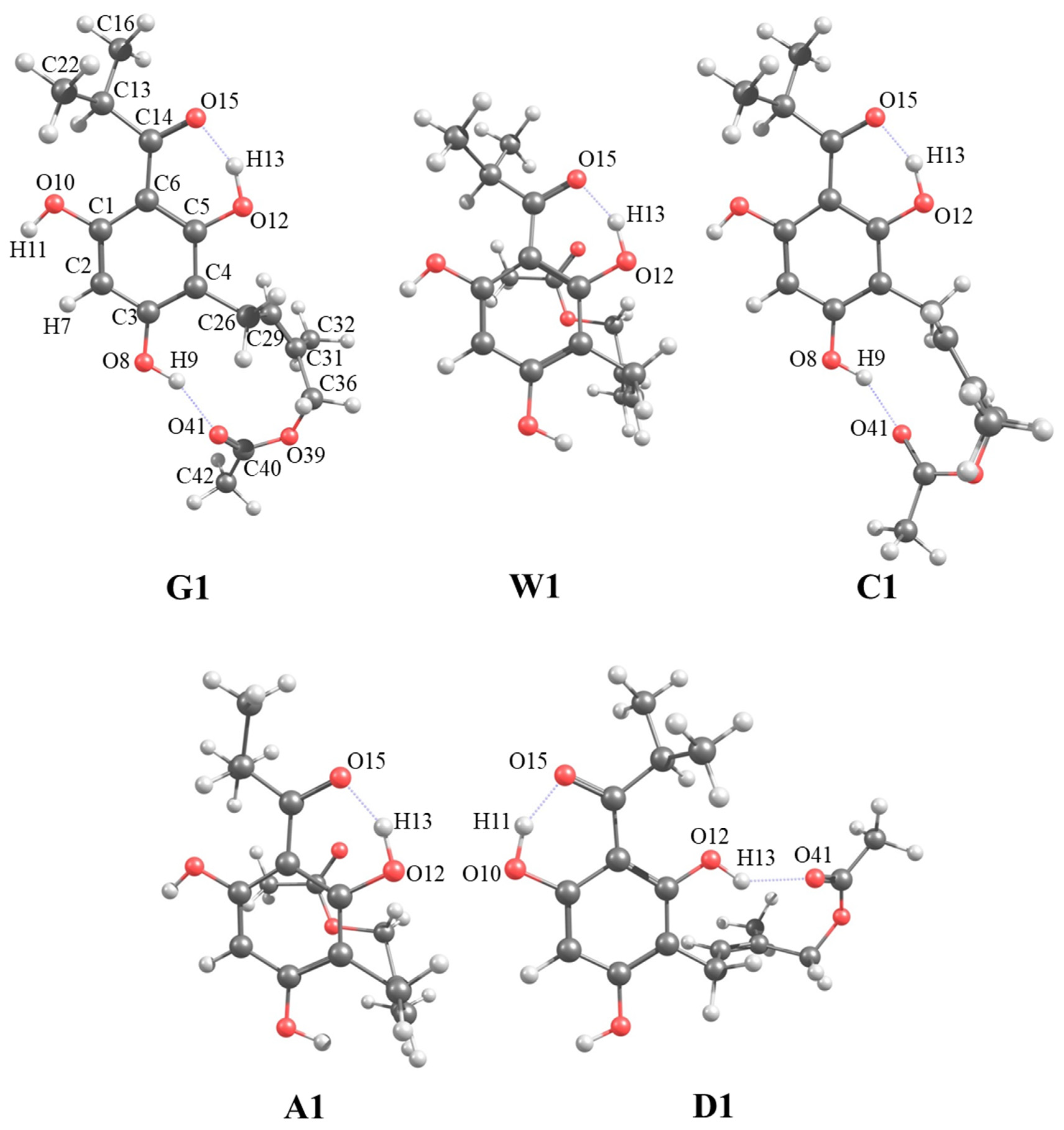







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformer | ΔG | Population | Conformation | No. of IHBs |
|---|---|---|---|---|
| Gas | ||||
| G1 | 0.0000 | 24.60 | Extended | 2 IHBs |
| G2 | 0.0063 | 23.65 | Extended | 2 IHBs |
| G3 | 0.6903 | 19.88 | Extended | 2 IHBs |
| G4 | 0.8063 | 6.73 | Hairpin | 2 IHBs |
| G5 | 0.8120 | 6.31 | Hairpin | 2 IHBs |
| Water | ||||
| W1 | 0.0000 | 26.50 | Hairpin | 1 IHB |
| W2 | 0.0454 | 17.19 | Hairpin | 2 IHBs |
| W3 | 0.0554 | 15.18 | Hairpin | 1 IHB |
| W4 | 0.2789 | 14.10 | Hairpin | 1 IHB |
| W5 | 0.3094 | 10.72 | Hairpin | 1 IHB |
| Chloroform | ||||
| C1 | 0.0000 | 49.10 | Extended | 2 IHBs |
| C2 | 0.0211 | 22.76 | Extended | 2 IHBs |
| C3 | 0.3750 | 6.37 | Hairpin | 2 IHBs |
| C4 | 0.3795 | 5.95 | Hairpin | 2 IHBs |
| C5 | 0.4444 | 5.58 | Hairpin | 2 IHBs |
| Acetonitrile | ||||
| A1 | 0.0000 | 20.25 | Hairpin | 1 IHB |
| A2 | 0.0076 | 15.54 | Extended | 2 IHBs |
| A3 | 0.0128 | 14.94 | Hairpin | 2 IHBs |
| A4 | 0.2904 | 14.34 | Extended | 2 IHBs |
| A5 | 0.3806 | 11.96 | Extended | 2 IHBs |
| DMSO | ||||
| D1 | 0.0000 | 23.62 | Hairpin | 2 IHBs |
| D2 | 0.0905 | 21.66 | Extended | 2 IHBs |
| D3 | 0.1074 | 13.10 | Extended | 2 IHBs |
| D4 | 0.3467 | 7.18 | Hairpin | 2 IHBs |
| D5 | 0.4277 | 7.09 | Hairpin | 2 IHBs |
| Explicit Solvent | Gas Phase a | Implicit Solvent b | ΔE |
|---|---|---|---|
| WG1E/W1E | −1270.265219 | −1270.265739 | −0.33 |
| CG1E/C1E | −1617.743739 | −1617.751660 | −4.79 |
| AG1E/A1E | −1382.303840 | −1382.307069 | −2.03 |
| DG1E/D1E | −1566.503477 | −1566.501234 | +1.41 |
| APFD/6-311+G(2d,p) | ||||
| Implicit solvent | ||||
| Parameters | W1 | C1 | A1 | D1 |
| O15···H13 | 1.501 | 1.493 | 1.483 | - |
| O15···H11 | - | - | - | 1.503 |
| O41···H9 | - | 1.813 | - | - |
| O41···H13 | - | - | - | 1.868 |
| O15···H13–O12 | 153.17 | 153.44 | 154.00 | - |
| O15···H11–O10 | - | - | - | 152.94 |
| O41···H9–O8 | - | 150.45 | - | - |
| O41···H13–O12 | - | - | - | 142.44 |
| O15–C14–C6–C5 | −10.60 | −0.80 | −0.45 | 179.11 |
| C5–C4–C26–C29 | −115.87 | 95.57 | −123.20 | −71.74 |
| ONIOM (ωB97X-D3/Def2-TZVP:XTB2) | ||||
| Explicit solvent | ||||
| Parameters | W1E | C1E | A1E | D1E |
| O15···H13 | 1.791 | 1.642 | 1.516 | - |
| O15···H11 | - | - | - | 1.556 |
| O41···H9 | - | 1.810 | - | - |
| O41···H13 | - | - | - | 1.928 |
| O15···H13–O12 | 141.43 | 147.72 | 152.85 | - |
| O15···H11–O10 | - | - | - | 150.47 |
| O41···H9–O8 | - | 156.14 | - | - |
| O41···H13–O12 | - | - | - | 146.58 |
| O15–C14–C6–C5 | −30.10 | −24.19 | 9.84 | −171.75 |
| C5–C4–C26–C29 | −111.71 | 75.08 | −122.02 | −73.97 |
| Conformer | HOMO | LUMO | μ | χ | η | s | ω |
|---|---|---|---|---|---|---|---|
| Gas | |||||||
| G1 | −6.1177 | −1.2833 | −3.7005 | 3.7005 | 4.8344 | 0.2069 | 1.4163 |
| Implicit solvent | |||||||
| W1 | −6.2584 | −1.6068 | −3.9326 | 3.9326 | 4.6516 | 0.2150 | 1.6624 |
| C1 | −6.1542 | −1.3870 | −3.7706 | 3.7706 | 4.7672 | 0.2098 | 1.4912 |
| A1 | −6.1615 | −1.4653 | −3.8134 | 3.8134 | 4.6962 | 0.2129 | 1.5483 |
| D1 | −6.1542 | −1.4199 | −3.7870 | 3.7870 | 4.7343 | 0.2112 | 1.5147 |
| Explicit solvent | |||||||
| W1E | −6.2600 | −1.5056 | −3.8828 | 3.8828 | 4.7544 | 0.2103 | 1.5854 |
| C1E | −6.2240 | −1.3815 | −3.8028 | 3.8028 | 4.8425 | 0.2065 | 1.4931 |
| A1E | −6.1590 | −1.4414 | −3.8002 | 3.8002 | 4.7176 | 0.2120 | 1.5306 |
| D1E | −6.1971 | −1.4185 | −3.8078 | 3.8078 | 4.7786 | 0.2093 | 1.5171 |
| Conformer | BCP | (r) | (r) | G(r) | V(r) | H(r) | EH…Y | Dinter | Ainter | DI | RCP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas phase | |||||||||||
| G1 | O15H13 | 0.0792 | 0.1694 | 0.0666 | −0.0908 | 0.1574 | 28.49 | 1.50190 | 152.76 | 0.1542 | 0.0241 |
| O41H9 | 0.0289 | 0.1195 | 0.0269 | −0.0240 | 0.0509 | 7.53 | 1.81314 | 155.80 | 0.0693 | 0.0091 | |
| Implicit solvent | |||||||||||
| W1 | O15H13 | 0.0789 | 0.1700 | 0.0665 | −0.0906 | 0.1571 | 28.43 | 1.50109 | 153.17 | 0.1528 | 0.0239 |
| C1 | O15H13 | 0.0813 | 0.1701 | 0.0682 | −0.0939 | 0.1621 | 29.46 | 1.49256 | 153.44 | 0.1579 | 0.0242 |
| O41H9 | 0.0294 | 0.1205 | 0.0274 | −0.0246 | 0.0520 | 7.72 | 1.81260 | 150.45 | 0.0698 | 0.0082 | |
| A1 | O15H13 | 0.0833 | 0.1701 | 0.0698 | −0.0971 | 0.1669 | 30.47 | 1.48306 | 154.00 | 0.1605 | 0.0243 |
| D1 | O15H11 | 0.0793 | 0.1704 | 0.0668 | −0.0910 | 0.1580 | 28.54 | 1.50269 | 152.94 | 0.1555 | 0.0240 |
| O41H13 | 0.0263 | 0.1095 | 0.0242 | −0.0211 | 0.0450 | 6.62 | 1.86758 | 142.44 | 0.0501 | 0.0079 | |
| Explicit solvent | |||||||||||
| W1E | O15H13 | 0.0379 | 0.1299 | 0.0331 | −0.0338 | 0.0669 | 10.605 | 1.516 | 152.84 | 0.0839 | 0.0198 |
| C1E | O15H13 | 0.0550 | 0.1561 | 0.0476 | −0.0562 | 0.1038 | 17.633 | 1.642 | 147.71 | 0.1178 | 0.0221 |
| O41H9 | 0.0293 | 0.1197 | 0.0271 | −0.0244 | 0.0515 | 7.655 | 1.809 | 156.13 | 0.0712 | 0.0098 | |
| A1E | O15H13 | 0.0762 | 0.1731 | 0.0651 | −0.0870 | 0.1521 | 27.296 | 1.516 | 152.84 | 0.1480 | 0.0238 |
| D1E | O15H11 | 0.0689 | 0.1688 | 0.0591 | −0.0760 | 0.1351 | 23.845 | 1.556 | 150.46 | 0.1398 | 0.0231 |
| O41H13 | 0.0220 | 0.0949 | 0.0202 | −0.0166 | 0.0368 | 5.208 | 1.927 | 146.58 | 0.0532 | 0.0085 | |
| Experimental | Gas Phase | Implicit Solvent | Explicit Solvent | ||||
|---|---|---|---|---|---|---|---|
| Assignment | δExp. | δCalc. | δRecalc. a | δCalc. | δRecalc. b | δCalc. | δRecalc. c |
| 3H, d, C3H–C1 | 1.17 | 1.092 | 1.520 | 0.906 | 1.014 | 1.111 | 1.202 |
| 3H, d, C3H–C2 | 1.17 | 1.209 | 1.614 | 1.075 | 1.183 | 1.124 | 1.216 |
| 3H, s, =C5–C4 | 1.73 | 1.782 | 2.077 | 1.722 | 1.836 | 1.637 | 1.731 |
| 3H, s, CO2–C6 | 2.12 | 2.169 | 2.391 | 1.934 | 2.049 | 2.038 | 2.133 |
| 2H, brd, C9–C7 | 3.40 | 3.760 | 3.678 | 3.339 | 3.464 | 3.309 | 3.410 |
| 1H, septupl, C3H | 3.96 | 3.899 | 3.790 | 3.606 | 3.733 | 3.853 | 3.956 |
| 2H, s, H–C8 | 4.79 | 4.875 | 4.580 | 4.803 | 4.938 | 4.507 | 4.613 |
| 1H, brt, H–C9 | 5.49 | 5.506 | 5.090 | 5.957 | 6.101 | 5.292 | 5.401 |
| 1H, s, H–C10 | 5.98 | 5.913 | 5.419 | 5.705 | 5.847 | 6.014 | 6.126 |
| 2H, bs, on H–O11; H–O12 | 7.90 | 7.583 | 6.770 | 7.442 | 7.596 | 7.782 | 7.902 |
| 1H, bs, on H–O13 | 12.90 | 16.128 | 13.682 | 12.658 | 12.848 | 12.778 | 12.918 |
| Experimental | Gas Phase | Implicit Solvent | Explicit Solvent | ||||
|---|---|---|---|---|---|---|---|
| Assignment | δExp | δCalc. | δRecalc a | δcalc | δrecalc b | δcalc | δrecalc c |
| Me–C1 | 19.80 | 22.720 | 21.425 | 19.944 | 19.597 | 20.925 | 19.598 |
| Me–C2 | 19.80 | 18.115 | 16.974 | 19.405 | 19.607 | 21.161 | 19.827 |
| H2–C7 | 21.50 | 23.651 | 22.326 | 23.812 | 23.394 | 25.086 | 23.634 |
| Me–C4 | 21.70 | 22.941 | 21.639 | 23.409 | 22.998 | 23.527 | 22.122 |
| Me–C6 | 21.70 | 22.579 | 21.289 | 21.008 | 21.623 | 22.625 | 21.248 |
| 2Me–C3 | 39.60 | 43.799 | 41.800 | 40.653 | 39.920 | 42.830 | 40.843 |
| H2–C8 | 64.80 | 66.789 | 64.021 | 64.508 | 63.330 | 65.386 | 62.718 |
| H–C10 | 95.70 | 96.263 | 92.509 | 95.194 | 93.444 | 96.499 | 92.893 |
| C14 | 104.50 | 105.240 | 101.187 | 107.617 | 105.636 | 108.476 | 104.509 |
| C15 | 106.50 | 109.381 | 105.188 | 105.164 | 103.229 | 107.483 | 103.546 |
| H–C9 | 129.50 | 138.954 | 133.773 | 137.302 | 134.767 | 137.963 | 133.106 |
| C5 | 130.10 | 138.559 | 133.391 | 132.657 | 130.209 | 137.626 | 132.780 |
| C11 | 160.20 | 164.646 | 158.605 | 162.111 | 159.114 | 163.896 | 158.257 |
| C12 | 161.60 | 168.873 | 162.691 | 165.919 | 162.851 | 166.556 | 160.837 |
| C13 | 164.00 | 172.110 | 165.819 | 166.913 | 163.826 | 167.933 | 162.173 |
| OC=O | 173.60 | 182.444 | 175.808 | 176.349 | 173.086 | 181.642 | 175.468 |
| C=O | 211.80 | 215.724 | 207.975 | 215.309 | 211.320 | 220.177 | 212.841 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Ceballos, A.; Castro, M.E.; Caballero, N.A.; Mammino, L.; Melendez, F.J. Implicit and Explicit Solvent Effects on the Global Reactivity and the Density Topological Parameters of the Preferred Conformers of Caespitate. Computation 2024, 12, 5. https://doi.org/10.3390/computation12010005
Moreno-Ceballos A, Castro ME, Caballero NA, Mammino L, Melendez FJ. Implicit and Explicit Solvent Effects on the Global Reactivity and the Density Topological Parameters of the Preferred Conformers of Caespitate. Computation. 2024; 12(1):5. https://doi.org/10.3390/computation12010005
Chicago/Turabian StyleMoreno-Ceballos, Andrea, María Eugenia Castro, Norma A. Caballero, Liliana Mammino, and Francisco J. Melendez. 2024. "Implicit and Explicit Solvent Effects on the Global Reactivity and the Density Topological Parameters of the Preferred Conformers of Caespitate" Computation 12, no. 1: 5. https://doi.org/10.3390/computation12010005
APA StyleMoreno-Ceballos, A., Castro, M. E., Caballero, N. A., Mammino, L., & Melendez, F. J. (2024). Implicit and Explicit Solvent Effects on the Global Reactivity and the Density Topological Parameters of the Preferred Conformers of Caespitate. Computation, 12(1), 5. https://doi.org/10.3390/computation12010005