Abstract
Macronutrients and trace elements are important components of living tissues that have different metabolic properties and functions. Trace elements participate in the regulation of immunity through humoral and cellular mechanisms, nerve conduction, muscle spasms, membrane potential regulation as well as mitochondrial activity and enzymatic reactions. Excessive alcohol consumption disrupts the concentrations of crucial trace elements, also increasing the risk of enhanced oxidative stress and alcohol-related liver diseases. In this review, we present the status of selected macroelements and trace elements in the serum and plasma of people chronically consuming alcohol. Such knowledge helps to understand the mechanisms of chronic alcohol-use disorder and to progress and prevent withdrawal effects, also improving treatment strategies.
Keywords:
alcoholism; alcohol use disorder; zinc; chromium; selenium; calcium; potassium; magnesium; phosphorous; sodium 1. Introduction
Alcohol dependence among adults in the United States is estimated at 14% [1]. Among heavy drinkers, alcoholic liver disease is estimated to develop in 15–30% [2]. In patients who consume excessive amounts of alcohol, various types of disturbances are observed, i.e., electrolyte, acid-base, protein-caloric malnutrition, and vitamin deficiency [3,4,5]. Chronic alcohol abuse patients are malnourished not only because of a diet low in nutrients but also because alcohol impairs the absorption of essential nutrients and elements. In addition, ethanol metabolic pathways produce toxic metabolites (acetaldehyde and free radicals) that lead to cell damage as a result of oxidative stress [6,7].
The clinical picture of the observed disorders depends on the duration and amount of alcohol consumed. Most patients who develop electrolyte imbalance, metabolic acidosis, and hyponatremia are admitted to hospital. However, clinical symptoms of chronic alcohol consumption are also decreased levels of phosphate, magnesium, potassium, sodium and calcium, and other elements in blood plasma [8,9,10]. Electrolyte abnormalities develop as a result of chronic alcohol consumption during acute alcohol intoxication, but they are particularly important during alcohol withdrawal [11,12,13,14]. It turns out that even during alcohol withdrawal, hypokalemia, hypomagnesemia, and hyponatremia are observed [15,16].
Currently, it is believed that the toxic effects of alcohol on organs are mainly associated with the activity of alcohol metabolites, induction of oxidative stress, and translocation of intestinal endotoxins into the bloodstream [17,18,19,20]. These processes lead to cell damage and stimulation of inflammatory reactions releasing a large number of cytokines, among others such as TNF-alpha and IL-6 [21,22]. With continuous alcohol abuse, which stimulates hepatocytes to secrete some interleukins, such as IL-8, activating Kupffer cells by binding to toll-like receptors, other effects are impaired including defective tissue regeneration, hepatocyte necrosis, recruitment of neutrophils and immune cells, and liver fibrosis [23].
Alcohol is metabolized in three pathways, involving enzymes such as the alcohol dehydrogenase present in the cytosol, the Microsomal Ethanol Oxidizing System (MEOS), and the catalase (CAT) contained in peroxisomes (Figure 1).
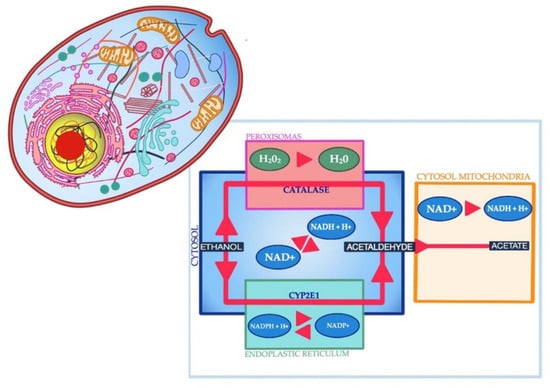
Figure 1.
Three pathways of alcohol metabolism.
Each of the three pathways is responsible for the production of free radicals [24], which can damage lipids, proteins, carbohydrates, and DNA substrates [25]. However, there are several endogenous enzymes that protect the body against the adverse effects of free radicals, including glutathione peroxidase (GPx), which neutralizes hydrogen peroxide and organic peroxides, glutathione reductase (GR), superoxide dismutase (SOD) and CAT [26]. Cofactors for antioxidant enzymes are trace elements such as selenium, manganese, copper, zinc, and iron. Selenium in the form of selenocysteine is present in the active center of GPx. In turn, manganese, copper, and zinc are components of the enzymes from the superoxide dismutase group (MnSOD, Cu/ZnSOD), which catalyze the reaction of the superoxide anion radical dismutation to hydrogen peroxide and oxygen. Iron is a part of CAT that catalyzes the decomposition of hydrogen peroxide into water and oxygen. Zinc-dependent enzymes, in turn, are alcohol dehydrogenase, RNA polymerases, and fructose 1,6-bisphosphatase, which is allosterically regulated by zinc and could influence gluconeogenesis. Therefore, trace element concentrations have a significant impact on the activity of antioxidant enzymes, and thus, on the modulation of ethanol-induced oxidative stress. Research to date has shown that alcoholism significantly changes macroelement homeostasis, i.e., calcium, phosphorous, magnesium, sodium, potassium, and trace minerals, which, as cofactors for antioxidant enzymes, play a role in the fight against oxidative stress and may affect survival (Table 1).

Table 1.
Minerals deficiency in patients of alcohol abuse.
The purpose of this paper is to present the effects of excessive and chronic alcohol consumption on magnesium, calcium, sodium, potassium, phosphorus, selenium, zinc, and chromium levels, as balanced homeostasis is crucial to prevent the dysfunctions of vital processes and functions. Besides, the authors have presented the mechanisms of the abovementioned alterations in macro- and microelement concentrations, as well as the subsequent side effects and clinical manifestations of such alterations. Further recommendations for future research and treatment options for alcohol use disorder are also evaluated.
2. Materials and Methods
Literature from PubMed, Google Scholar, and Web of Science databases was extracted and included original articles as well as review articles (including systematic reviews and meta-analyses) that were published within the time period 1952–2020. The inclusion criterion was the English language; no limits were included for the publication year. Additionally, both human and animal studies were included in the final analysis. The literature was analyzed in terms of changes in chosen macro- and microelement concentrations in cases of prolonged and excessive alcohol intake and alcohol use disorder in particular. The chosen elements were magnesium, calcium, sodium, potassium, phosphorus, selenium, zinc, and chromium, and this inclusion criterion was based on the fact that alterations of the abovementioned elements are highly prevalent among alcohol-dependent patients and usually result in clinical manifestations with a different degree of severity. The search strategy included the following keywords: (alcohol use disorder OR alcohol OR alcoholism) AND (electrolyte OR microelement OR macroelement OR mineral OR phosphorus OR hypophosphatemia OR magnesium OR hypomagnesemia OR calcium OR hypocalcemia OR potassium OR hypokalemia OR sodium OR hyponatremia OR selenium OR zinc OR chromium). In addition to the literature search on PubMed, Google Scholar, and Web of Science databases, the references included in the analyzed papers were also taken into consideration. Finally, 209 articles were assessed as relevant to the topic and included in the review.
3. Acid-Base Disturbances
Chronic alcohol users are prone to various acid-base disorders. Most patients most often have mixed disorders (78%) [44]. Alcoholic metabolic acidosis occurring in almost half of patients is manifested by an increased anion gap, which is the result of the accumulation of ketoacids, lactic acid, acetic acid, or indirect loss of bicarbonate through urine [45,46]. Almost a third of patients also have symptoms of respiratory alkalosis [47]. The development of ketoacidosis leads to an increase in the ratio of reduced NADH to oxidized nicotinamide adenine dinucleotide (NAD) and further formation of β-hydroxybutyrate. In turn, the increased ratio of NADH to NAD has an inhibitory effect on hepatic gluconeogenesis, which leads to life-threatening hypoglycemia [48,49].
4. Phosphorus Deficit
Acute hypophosphatemia associated with phosphorus deficiency develops by up to 50% in patients within the first 2–3 days after hospitalization due to problems associated with chronic alcohol abuse [50,51]. Phosphorus deficiency is most often caused by an inadequate diet low in phosphates or due to alkalizing drugs, chronic diarrhea, or vomiting that prevent efficient phosphorus absorption. At the same time, despite hypophosphatemia, increased urinary phosphate excretion is observed as a result of renal tubular dysfunction accompanied by glycosuria, amino acid urine, hypomagnesuria, and hypercalciuria [52]. It is believed that the observed disorders are the result of structural changes in the phospholipid bilayer of the renal tubules, a dysfunction of apex-located transporters, and reduced potassium-sodium ATPase activity [53,54]. In addition, increased renal excretion of phosphate in patients with metabolic acidosis results from the fact that phosphate releases from bones and is a direct effect of pH gating in proximal tubules with the NaPi-2a and NaPi-2c co-transporters [55].
Reduced phosphate reabsorption may be the result of increased parathyroid hormone (PTH) levels due to hypocalcemia induced by vitamin D deficiency or magnesium deficiency due to phosphaturia [56]. It has been documented that magnesium deficiency can lead to a decrease in the phosphate content of skeletal muscles and an increase in their excretion in urine. Thus, phosphorus deficits play a large role in the etiology of alcohol myopathy and acute rhabdomyolysis [57,58].
Magnesium deficiency can cause a state of hypoparathyroidism, thus contributing to phosphaturia. The mere normalization of pH in patients with ketoacidosis causes an intracellular phosphate shift, which in turn, leads to hypophosphatemia. This is confirmed by skeletal muscle biopsy studies that show a reduced content of phosphates and magnesium, which are accompanied by increased amounts of sodium, chloride, and calcium [59,60,61]. Hypophosphatasemia is also involved in the development of metabolic acidosis. Intracellular phosphate deficiency interferes with the production of ATP, which in turn, stimulates phosphofructokinase activity, enhancing glycolysis and lactate production. Besides, cellular phosphate deficiency in red blood cells causes tissue ischemia and increases lactic acid production.
5. Imbalances in Magnesium and Calcium Levels in Plasma
Almost a third of chronic alcohol users have hypomagnesemia [62]. Reduced magnesium content in the body is the result of insufficient consumption of products rich in magnesium, as well as impaired absorption due to chronic diarrhea associated with, among others, impaired digestion or absorption of fats, which are excreted in the form of fatty acid–magnesium complexes (steatorrhea). Magnesium concentration should be measured in serum rather than in the plasma because the anticoagulant used for plasma collection may be contaminated with magnesium or may affect the test procedure. For example, citrate binds not only calcium but also magnesium and affects the measurement result by fluorometric and colorimetric methods. It is also important to avoid hemolysis because the magnesium concentration in red blood cells is about three times higher than the serum concentration and it has been estimated that the serum magnesium concentration will increase by 0.05 mM/L for each g/L hemoglobin produced by hemolysis. Serum total magnesium may also be affected by high bilirubin, lipemic serum, and high phosphate [63]. Magnesium deficiencies are closely related to phosphate deficiencies (Table 2).
Table 2.
Clinical features of minerals deficiency (hypocalcemia, hypomagnesemia, hypophosphatemia, hyponatremia, and hypokalemia).
Magnesium deficiency leads to the kidneys losing phosphate, while phosphate deficiency is associated with kidneys losing magnesium, resulting in reduced magnesium and ATP content in skeletal muscle.
The development of hypomagnesemia inhibits the release and induces peripheral resistance to parathyroid hormone, leading to hypocalcemia [94]. To reverse this process, the restoring of the physiological magnesium levels is inherent. Vitamin D deficiency is also an important risk factor for hypocalcemia, like alcohol-related steatorrhea or rhabdomyolysis.
Calcium plays an important role in the proper functioning of the body. Most calcium (99%) is found in teeth and bones, which can be considered as its reservoir [95,96]. Only 1% of total calcium is found in the serum. Adequate calcium levels in the brain activate a wide range of Ca2+-dependent processes in neurons, including neurotransmitter release, gene transcription, activation of Ca2+-dependent enzymes, and activation of some K+ and chloride channels [97,98].
The serum calcium content is regulated by calcitonin, vitamin D, and parathyroid hormone (PTH) [99,100]. According to research, acute alcohol consumption leads to an increase in calcitonin plasma levels, while chronic alcohol consumption and detoxification have varying effects [101,102]. Chronic alcohol consumption is known to lead to vitamin D deficiency, reduced intestinal calcium absorption (by up to 80%), which in turn, causes osteoporosis and osteopenia [103,104]. Reduced plasma calcium levels have been demonstrated in alcohol-dependent patients with and without cirrhosis [105]. Chronic alcohol consumption leads to an increase in phosphoinositide-dependent cytosolic calcium levels, disturbing mitochondrial Ca2+ levels and energy metabolism. In addition, Laitinen et al. [106] presented that defective parathyroid function was found to respond to hypocalcemic stimuli in alcohol-dependent patients during intoxication.
In 1952, O’Brien showed that intravenous calcium administration reduces withdrawal symptoms in alcohol-dependent patients [107]. In an animal model, it was confirmed that calcium, which is a component of the drug that reduces cravings for alcohol addicts (acamprosate calcium acetyl-homotaurine), is involved in preventing alcohol cravings in addicted patients [108,109]. This conclusion has not been confirmed by other authors [110]. Schuster et al. in 2016 conducted a plasma calcium concentration test in alcohol-dependent patients [111]. The psychometric dimension of hunger was studied using the OCDS, ADS, and ADS-HR scales. It was found, among others, the existence of a negative correlation between plasma calcium levels and alcohol cravings, a negative correlation of plasma calcium levels with alcohol concentration in the exhaled air, decreased calcitonin concentration in the group of high-risk alcoholics, and decreased vitamin D plasma concentration. The authors suggest the usefulness of calcium supplementation in reducing cravings and relapse in people addicted to alcohol. Recent studies reveal the mechanisms of disorders seen in alcoholics during abstinence. As demonstrated, voltage-sensitive calcium channels Ca2+ (CaV) in the brain are an important target for alcohol. Alcohol-induced changes in Ca2+ signaling may interfere with neuronal homeostasis, Ca2+-mediated gene transcription, and neural circuit function, leading to various neurological symptoms and neuropsychiatric disorders, including alcohol withdrawal convulsions and alcoholism [100].
6. Plasma Potassium Deficit
Hypokalemia occurs in almost 50% of patients with chronic alcohol consumption disorder [112]. Plasma potassium concentrations and incidents of hypokalemia due to alcohol intoxication might differ depending on the age of patients or coexistence of concomitant diseases. Potassium deficiency results from an inadequate diet and loss of potassium through the gastrointestinal tract due to malnutrition, diarrhea, vomiting, and increased loss of urine. Further to this, glucose infusion induces potassium shift from extracellular to intracellular compartments. The most serious symptoms of hypokalemia include myopathy, rhabdomyolysis, and cardiac effects—from asymptomatic electrocardiographic changes to potentially life-threatening cardiac arrhythmias. Central pontine myelinolysis might also be a severe consequence of hypokalemia among alcohol-dependent patients [113,114]. The above symptoms are the result of an increase in mineralocorticoid levels and an increased supply of sodium to the distal nephron. Under normal circumstances, intracellular magnesium blocks potassium channels (ROMK) in the apical membrane of the distal nephron and limits potassium losses [115,116]. Coexisting magnesium deficiency in serum causes a decrease in its level inside the cell, which results in the prevention of inhibition of ROMK channels and is responsible for potassium losses [112]. Stimulation of β2-adrenergic receptors in skeletal muscle due to respiratory alkalosis contributes to the development of hypokalemia. It is recommended to measure plasma potassium, sodium, glucose, and lactate levels among patients with alcohol intoxication [117]. Potassium and platelet levels are crucial in predicting the severity of alcohol withdrawal [118,119]. Hypokalemia along with hypochloremia are associated with a higher risk of delirium tremens. Furthermore, elevated levels of plasma catecholamines, especially during the alcohol withdrawal period, induce intracellular shift of potassium and magnesium, which explains the relationship between hypokalemia, hypomagnesemia, and further delirium tremens [15].
7. Sodium Deficit
Hyponatremia is the most common electrolyte disorder seen in people consuming excessive amounts of alcohol. Hyponatremia is diagnosed when the sodium plasma concentration is below 135 mM/L. Sodium is responsible for maintaining basic vital functions. It is involved in maintaining water and electrolyte balance [120], acid-base [121], osmotic pressure [122], transport of amino acids, carbohydrates and vitamins [123] serotonin (SERT), dopamine, noradrenaline, GABA [124,125], excitability neuromuscular [126], and conduction of nerve impulses [127]. Physiological sodium levels in the plasma range from 135 to 145 mM/L (Table 3).
Table 3.
The normal range, and deficiency conditions of trace/macroelements in the serum of adults [128,129,130,131,132].
Sodium presence has a large impact on plasma osmolality, since salts of sodium (chloride and bicarbonate) along with urea and nonelectrolyte glucose are the most inherent osmoles of plasma. Many studies describe reduced plasma sodium levels in alcohol-dependent patients compared to the control group [133,134,135,136,137].
There are three types of hyponatremia: mild (130–134 mM/L), moderate (120–129 mM/L), and severe (less than 120 mM/L); furthermore, hyponatremia can be classified as acute or chronic [138,139]. Clinical manifestations of hyponatremia depend on its severity and duration. Acute hyponatremia, for instance, is characterized by the onset of neurologic symptoms (such as seizures, impaired mental status, coma, or death) [140]. Further to this, central pontine myelinolysis might appear, which has an impact on the risk of complications and worsening prognosis of comorbidities, as well as on mental condition and quality of life, related to health. Contrarily, patients with chronic hyponatremia can be asymptomatic. Mild hyponatremia is primarily characterized by gastrointestinal symptoms (nausea, vomiting, loss of appetite) and sometimes by neurologic impairments [141]. Moderate and severe cases of hyponatremia are characterized by a higher probability of deterioration of neurologic manifestations.
Ordak et al. [142] studied the effect of hyponatremia in alcohol-dependent patients on the physical and mental state of patients. Based on verification using the BSI clinical symptom scale, Barratt impulsivity scale, SDQ-7 sleep disorder questionnaire, SF-36 quality of life questionnaire, list of five NEO-FFI factors, and MAST (Michigan Alcoholism Screening Test), the researchers confirmed that the lower the sodium concentration in plasma was, the higher was impulsiveness and neuroticism, quality of life, and mental aspect.
High alcohol consumption induces diuresis by increasing the level of vasopressin, which predisposes patients to dehydration and hypernatremia [143,144]. This is a common disorder that occurs in 17% of patients chronically consuming alcohol [145]. However, this effect may not be observed in the case of prolonged alcohol consumption. In such patients, the level of vasopressin increases, which results in an increase in urine osmolality and a decrease in free water clearance.
However, there is hyponatremia independent of vasopressin, which occurs with a long-term beer-based diet—“beer offspring”. “Beer Potomania” is a unique hyponatremia syndrome that was first described in 1972 [146,147,148,149]. The low content of nutrients in beer and the suppressing effect of alcohol on proteolysis cause secondary dilution hyponatremia, which leads to reduced removal of excess fluid from the body. Progeny includes severe hyponatremia (plasma sodium concentration, <110 mmol/L), hypokalemia, low blood urea nitrogen (indicating low protein intake), and maximum diluted urine (<100 mOsm/kg of water) [150].
The impact of hyponatremia on brain metabolism has been the subject of in-depth studies [151,152,153,154]. It turns out that hyponatremia has a complex, nonlinear relationship with the brain Glx metabolites (glutamate + glutamine), cognitive ability, and generally understood, health-related quality of life (HRQOL) [151,155]. Magnetic resonance spectroscopy (MRS) studies have shown that in a state of edema caused by hyponatremia, a decrease in the content of choline and myo-inositol (mI) in the brain is observed [156,157]. The cerebral metabolic profile of patients with hyponatremia is characterized by a low ratio of mI/Cr (creatinine) and Glx/Cr, as a result of which there may be a high risk of developing cerebral edema following a hyperammonemic stimulus. The relationship between serum sodium and Glx/Cr was nonlinear and linear with negative mI/Cr. In addition, mI/Cr ratios were significantly correlated with weak HRQOL in physical and psychosocial dimensions. At the same time, it was shown that the concentration of mI/Cr increases after the correction of hyponatremia. It should be remembered, however, that if hyponatremia is corrected too quickly, myelinolysis of the middle bridge may occur [158].
Under these circumstances, myelin glial cells are exposed to osmotic stress. However, this condition should be taken into account in any patient with alcohol dependence syndrome, even if there is no sodium flow, as these patients have a direct toxic effect of alcohol through chronic osmotic stress, which may play an important role in the pathophysiology of the observed changes.
8. Selenium Deficit
Selenium (Se) is an essential trace element, important for human health due to its anti-inflammatory, chemopreventive, and antioxidant activity realized through various selenoproteins [159]. It is an essential component of glutathione peroxidase (GPx) and phospholipid-hydroperoxide-glutathione peroxidase. It is believed that selenium counteracts the effects of oxidative stress mainly by GPx [159]. Normal serum selenium concentration is 50–120 μg/L. Low serum Se levels have been confirmed many times in patients with hepatic complications as well as in alcohol-dependent patients [151,160,161,162,163].
Rua et al. studied the effect of chronic ethanol consumption on oxidative balance and selenium (Se) levels in patients chronically consuming alcohol with or without concomitant liver disease [35]. They found that serum Se levels were lower in alcoholic patients and patients with liver disease, and particularly lower in the group of alcoholic patients with concomitant liver disease. These values were correlated with the profile of antioxidative enzymes, i.e., glutathione peroxidase (GPx), selenoproteins, glutathione reductase (GR), and superoxide dismutase (SOD). Heavy alcohol consumption and related Se deficiency, primarily lower serum GPx3 and liver GPx1 activities, as well as hepatic GPx4 and GPx1 expressions [164].
Thus, both chronic alcohol abuse and liver disease are associated with a decrease in Se levels. Therefore, there is controversy regarding the evaluation of these results. By Bergheim et al. [165] and Ojeda et al. [166], a decrease in serum and liver levels of Se can cause alcohol alone. In turn, the Se deficit further causes a type of hepatocyte necrosis similar to that observed in alcoholic patients, as demonstrated in animal studies by Simonoff and Simonoff [167]. On the other hand, it is known that liver diseases [163] also contribute to low levels of Se by disrupting the synthesis of selenoproteins involved in the transport of selenium in the serum [168]. Zhu et al. [169] and Rua et al. [35] have shown, however, that in non-alcoholic liver damage, oxidative balance differs significantly from that of chronic alcohol exposure. Alcohol consumption decreases antioxidant defense in both liver tissue and blood. These changes in the activity of antioxidant enzymes are associated with an increase in protein and lipid oxidation in alcohol patients, confirmed by MDA levels, which are particularly higher in alcohol patients [140,170,171,172,173]. The authors proposed the ratio of Se to malonic aldehyde, which is an indicator of oxidative damage (Se/MDA), as an effective diagnostic tool for assessing liver damage and its etiology. The above indicator is 37.5% lower in non-alcoholic liver disease, 65.7% in alcoholics without liver disease, and 84% in alcoholics with liver disease. Alcohol-dependent patients with concomitant liver diseases present even more decreased Se levels compared to those without coexisting diseases [43]. Selenium causes a decrease in liver fat amount and hepatocyte ballooning among patients with alcohol-related liver steatosis [174]. Considering the fact that alcohol is a pro-oxidant and Se is a mineral with antioxidant capacity, of which reserves may be depleted in patients after chronic alcohol exposure, Se supplementation may be considered as a possible antioxidant therapy, slowing down the progression of secondary alcohol diseases. A simultaneous Se and Mg supplementation provide enhanced antioxidant defense, being more effective in the prevention of oxidative stress, as well as normalization of liver functions and lipid parameters. Furthermore, the release of pro-inflammatory cytokines (TNF-α and IL-1β) is inhibited by an anti-inflammatory cytokine, IL-10, highly excreted during Se supplementation [175].
9. Zinc Concentration Disturbance
Zinc (Zn) is effectively absorbed from food. It should be noted, however, that while animal proteins improve its absorption, due to the formation of more soluble complexes with low molecular weight ligands like histidine, methionine, the phytates (myo-inositol hexaphosphate, pentaphosphate) present in plant products, they irreversibly bind zinc in the intestinal lumen, which negatively affects its absorption. Zn has many important physiological functions. Zn is a structural component of the antioxidant enzyme, Cu/Zn-SOD, also acts as a stabilizer of biological membranes, and is an important factor in controlling the transcription of genes responsible for cell proliferation and differentiation as well as intracellular signaling [176,177]. Zn is an essential structural component of about 2500 Zn-finger proteins, which constitute about 8% of the human genome [178,179]. Zn is also necessary for proper functioning of the immune system [180,181,182].
Chronic heavy alcohol consumption disturbs zinc homeostasis, decreasing serum Zn concentration by over 25% compared to healthy individuals, also affecting plasma, erythrocyte, and hepatic concentrations. Experimental studies proved that alcohol-induced decreased hepatic Zn levels are due to the alterations of hepatic zinc transporters’ downregulation of Zip5 and Zip14, and upregulation of Zip7 with Znt7 [89,183,184]. Ethanol and its metabolites stimulate Kupffer cells to release interleukins, which is further implicated in lowered plasma Zn levels. Zn deficiency is observed in approximately 30–50% of alcoholics, being the most frequent nutritional manifestation in alcohol-related liver diseases [185]. Zn deficiency in alcoholics is of a multifactorial origin, among which alcohol-related reduction in exocrine functions of the pancreas, as well as reduction in numerous ligands, are of major importance. Furthermore, Zn metabolism is directly affected by alcohol, as well as because of homeostatic alterations due to the hepatic failure itself. Chronic alcohol exposure impairs Zn homeostasis mainly due to reactive oxygen species, lipid peroxidation, and acetaldehyde. Besides, Zn deficiency decreases the activity of alcohol dehydrogenase, eventually slowing down ethanol elimination. Several studies proved that the maintenance of a balanced diet during alcohol dependence prevents altered Zn distribution [186]. There are speculations that low levels of Zn cause neuronal damage and brain dysfunctions, lymphopenia, reduced ability to respond, and susceptibility to ethanol withdrawal attacks. There is a relationship between decreased Zn and subsequent lowered vitamin A levels, and the severity of hepatic lesions. Zn supplementation inhibits oxidative stress and a following oxidative liver injury, primarily due to the inhibition of the metabolic shift from aldehyde dehydrogenase to CYP2E1 [2,187]. It also decreases alanine aminotransferase levels, which are elevated during chronic alcohol consumption. Therefore, some authors recommend daily zinc supplementation for the treatment of alcohol withdrawal symptoms, as well as alcohol-related brain dysfunctions [188,189,190]. Zima et al. [24] and Menzano and Carlen [188] described the effect of reduced serum zinc in alcoholics without liver damage on the immune system disorder and brain dysfunction.
Alcohol-modulated zinc transporter modulation has recently been shown to be one of the causes of decreased Zn levels in the lungs, liver, intestines, and brain [89]. Apart from the direct effects of alcohol on the intestinal epithelial barrier, Zn deficiency might further sensitize epithelial cells to alcohol, exaggerating symptoms, and worsening the clinical outcome of alcohol-dependent patients [191]. Deficiency of Zn in the intestine causes an increase in intestinal permeability, which ultimately leads to endotoxemia and systemic inflammation. Similarly, a deficiency of Zn in lung epithelium and alveolar macrophages reduces the function of the pulmonary barrier, causing respiratory distress syndrome. Finally, impaired bowel and liver function causes the accumulation of neurotoxic metabolites, which can play a significant role in alcoholic brain damage. There is also a hypothesis that ethanol-induced Zn deficiency may interfere with neurotransmission.
10. Chromium Deficit
Two levels of chromium oxidation are considered to be biologically relevant i.e., compounds containing chromium (6+), which are mutagenic and carcinogenic, and chromium (3+), which was a recognized essential element for mammals involved in carbohydrate, fat, and protein metabolism [93,192]. However, in the last two decades, its status has been questioned many times. In 2014, the European Food Safety Authority found no convincing evidence that chromium is an essential element [93,193].
The content of chromium in the liver is estimated at 1 μmol/kg BW, while the total content of this element is about 100 μmol/kg BW. Chromium is absorbed by passive diffusion in only about 1%. Chromium remains in the bloodstream associated with protein-transferrin and α-globulin, from where it is delivered to tissues by endocytosis [93,194]. Data on chromium content standards vary widely. In 2008, Walter et al. compared the distribution of Cr ions in whole blood, plasma, serum, and erythrocytes [195]. They concluded that most of the Cr was found in the extracellular space of the blood. Serum and plasma concentrations were highest and lowest in erythrocytes. The authors recommend using serum or plasma fractions for comparative testing of Cr levels. In the same year, De Smet et al. considered Cr > 17 μg/L as a manifestation of metallosis [196].
Chromium in its trivalent (Cr3+) chelated form (Cr-nicotinic acid-glutathione) is considered a glucose tolerance factor (GTF) and it has been postulated that it is involved in regulating the metabolism of not only carbohydrates but also lipids and potentially, protein. The best-defined task of Cr is to act as a cofactor for a biologically active molecule that enhances the effect of insulin on target tissues. Its role in the treatment of diabetes in animals was described in the 1950s, but the impact of Cr on the etiology of diabetes in humans is still controversial. Many authors confirm that patients with chromium deficiency develop severe diabetes that does not respond well to insulin, which can be corrected by chromium supplementation. It has been found that such long-term chromium deficiency develops in patients dependent on parenteral nutrition. Additionally, in people with type 2 diabetes, Cr supplementation may improve glucose metabolism [197,198,199]. A similar effect was demonstrated in patients with severe insulin resistance who were given intravenous chromium (III) [200,201]. Based on the results of Jain et al. (2012), it was stated that Cr supplementation has the potential as an adjunct therapy for patients suffering from type 2 diabetes [202].
Studies on the effects of Cr supplementation on carbohydrate and lipid metabolism in healthy individuals are often contradictory [203]. Some authors show in their studies that there are no beneficial effects in healthy people [204,205], emphasizing that Cr therapy not only does not affect insulin sensitivity but paradoxically reduces it [206]. There are also several publications describing the positive role of Cr in reducing lipid content and body weight in obese people and animals [207,208,209]. Specifically, an interaction between ethanol and chromium alters biochemical parameters such as liver total triglycerides, liver glycogen, succinate dehydrogenase, or liver glutathione.
Hypoglycemia also accompanies alcoholism, which can be a manifestation of acute hypochromism. In the medical literature, hypoglycemia in alcoholics has not been identified as a consequence of chromium deficiency, although insulin use, in this case, is not always effective. Most authors agree that chronic alcohol consumption is a potential risk factor for type 2 diabetes (T2DM), which causes insulin resistance and pancreatic β cell dysfunction.
11. Conclusions
Chronic alcohol consumption alters essential micro- and macronutrient levels leading to a significant number of metabolic disorders, among which alcohol-related liver disease and pancreatitis are of the highest prevalence. The severity of alcoholic liver disease varies from simple steatosis to liver cancer. Patients with alcoholic liver disease should be encouraged to discontinue alcohol consumption. Alcohol administration is associated with increased damage due to oxidative stress and significantly lowered trace elements and antioxidant enzyme levels. There is a multitude of causes of the abovementioned imbalances, including inappropriate nutritional status, alcohol-related gastrointestinal disorders, diarrhea, vomiting, or excessive urination. Electrolyte and trace element imbalances are observed both during alcohol administration and alcohol withdrawal because of the defective functioning of renal tubules impaired by alcohol. The most common distortions include hypophosphatemia, hypomagnesemia, hypokalemia, hypocalcemia, and hyponatremia, as well as decreased levels of selenium, chromium, and zinc. A proper understanding of the pathomechanisms of electrolyte imbalances during alcohol administration and withdrawal provides the most effective treatment strategies for such patients, significantly increasing their clinical outcome. The treatment of electrolyte disturbances in alcohol use disorder depends on the degree of deficiency and subsequent clinical manifestations. It is crucial to provide either oral or intravenous supplementation of relevant electrolytes in order to reach their proper physiological ranges. Besides, close monitoring of patients is required, as electrolyte disturbances might persist even during the alcohol withdrawal period or after the alleged initial correction of electrolyte concentrations. Further evaluation of possible treatment strategies and their improvements is crucial to minimize the potentially toxic side effects of these therapies as well as to shorten the duration of the recovery time.
Author Contributions
Conceptualization, J.B., W.F., G.T. and R.M.; methodology, G.B., K.K.; formal analysis, A.F., R.S., K.K.; resources, J.B., R.M., W.F.; writing—original draft preparation, J.B., W.F., A.F.; writing—review and editing, R.M.; visualization, J.B., W.F.; supervision, R.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Muller, A. Alcohol consumption and community hospital admissions in the United States: A dynamic regression analysis, 1950–1992. Addiction 1996, 91, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Zhou, Z. Zinc prevention and treatment of alcoholic liver disease. Mol. Asp. Med. 2005, 26, 391–404. [Google Scholar] [CrossRef]
- Grochowski, C.; Blicharska, E.; Baj, J.; Mierzwińska, A.; Brzozowska, K.; Forma, A.; Maciejewski, R. Serum iron, Magnesium, Copper, and Manganese Levels in Alcoholism: A Systematic Review. Molecules 2019, 24, 1361. [Google Scholar] [CrossRef]
- Grochowski, C.; Blicharska, E.; Bogucki, J.; Proch, J.; Mierzwińska, A.; Baj, J.; Litak, J.; Podkowinkski, A.; Flieger, J.; Teresinski, G.; et al. Increased Aluminum Content in Certain Brain Structures is Correlated with Higher Silicon Concentration in Alcoholic Use Disorder. Molecules 2019, 24, 1721. [Google Scholar] [CrossRef]
- Barrio, P.; Wurst, F.M.; Gual, A. New Alcohol Biomarkers. New challenges. Alcohol Alcohol. 2018, 53, 762–763. [Google Scholar] [CrossRef]
- Moreno Otero, R.; Cortés, J.R. Nutrition and chronic alcohol abuse. Nutr. Hosp. 2008, 23, 3–7. [Google Scholar]
- Boye, A. Metabolic derivatives of alcohol and the molecular culprits of fibro-hepatocarcinogenesis: Allies or enemies? World J. Gastroenterol. 2016, 22, 50. [Google Scholar] [CrossRef]
- Biff, F.; Palmer, M.D.; Clegg, D.J. Electrolyte Disturbances in Patients with Chronic Alcohol-Use Disorder. N. Engl. J. Med. 2017, 377, 1368–1377. [Google Scholar]
- Barrio, P.; Gual, A. Patient-centered care interventions for the management of alcohol use disorders: A systematic review of randomized controlled trials. Patient Prefer. Adher. 2016, 10, 1823–1845. [Google Scholar] [CrossRef]
- Addolorato, G.; Mirijello, A.; Barrio, P.; Gual, A. Treatment of alcohol use disorders in patients with alcoholic liver disease. J. Hepatol. 2016, 65, 618–630. [Google Scholar] [CrossRef]
- Adamson, D.J.; Laing, R.B.; Nathwani, D. Alcoholism, hyponatremia and central neurological damage. Scott. Med. J. 1992, 37, 1006–1012. [Google Scholar] [CrossRef]
- Burin, M.R.; Cook, C.C. Alcohol withdrawal and hypokalemia: A case report. Alcohol Alcohol. 2000, 35, 188–189. [Google Scholar] [CrossRef][Green Version]
- Carl, G.; Holzbach, E. Reversible hypokalemia and hypomagnesemia during alcohol withdrawal syndrome. Nervenarzt 1994, 65, 206–211. [Google Scholar]
- Cunha, D.F.; Monteiro, J.P.; Ortega, L.S.; Alves, L.G.; Cunha, S.F. Serum electrolytes in hospitalized pellagra alcoholics. Eur. J. Clin. Nutr. 2000, 54, 440–442. [Google Scholar] [CrossRef][Green Version]
- Stasiukyniene, V. Blood plasma potassium, sodiumand magnesiumlevelsin chronic alcoholic patientsduring alcohol withdrawal. Medicina 2002, 38, 892–895. [Google Scholar]
- Vetter, W.R. Hypokalemia and Electrocardiographic Abnormalities During Acute Alcohol Withdrawal. Arch. Intern. Med. 1967, 120, 536. [Google Scholar] [CrossRef]
- Grochowski, C.; Szukała, M.; Litak, J.; Budny, A.; Proch, J.; Majerek, D.; Blicharska, E.; Niedzielski, P. Correlations between Trace Elements in Selected Locations of the Human Brain in Individuals with Alcohol Use Disorder. Molecules 2020, 25, 359. [Google Scholar] [CrossRef]
- Grochowski, C.; Blicharska, E.; Krukow, P.; Jonak, K.; Maciejewski, M.; Szczepanek, D.; Jonak, K.; Flieger, J.; Maciejewski, R. Analysis of Trace Elements in Human Brain: Its Aim, Methods, and Concentration Levels. Front. Chem. 2019, 7, 115. [Google Scholar] [CrossRef]
- Skórzyńska-Dziduszko, K.E.; Kimber-Trojnar, Ż.; Patro-Małysza, J.; Olszewska, A.; Zaborowski, T.; Małecka-Massalska, T. An Interplay between Obesity and Inflammation in Gestational Diabetes Mellitus. Curr. Pharm. Biotechnol. 2016, 17, 603–613. [Google Scholar]
- Dolar-Szczasny, J.; Święch, A.; Flieger, J.; Tatarczak-Michalewska, M.; Niedzielski, P.; Proch, J.; Majerek, D.; Kawka, J.; Mackiewicz, J. Levels of Trace Elements in the Aqueous Humor of Cataract Patients Measured by the Inductively Coupled Plasma Optical Emission Spectrometry. Molecules 2019, 24, 4127. [Google Scholar] [CrossRef]
- Gonzalez-Quintela, A.; Campos, J.; Loidi, L.; Quinteiro, C.; Perez, L.-F.; Gude, F. Serum TNF-α levels in relation to alcohol consumption and common TNF gene polymorphisms. Alcohol 2008, 42, 513–518. [Google Scholar] [CrossRef]
- Hong, F.; Kim, W.-H.; Tian, Z.; Jaruga, B.; Ishac, E.; Shen, X.; Gao, B. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: Involvement of induction of Bcl-2 and Bcl-xL proteins. Oncogene 2002, 21, 32–43. [Google Scholar] [CrossRef]
- Seth, D.; D’Souza El-Guindy, N.B.; Apte, M.; Mari, M.; Dooley, S.; Neuman, M.; Haber, P.S.; Kundu, G.C.; Darwanto, A.; de Villiers, W.J.; et al. Alcohol, signaling, and ECM turnover. Alcohol Clin. Exp. Res. 2010, 34, 4–18. [Google Scholar] [CrossRef]
- Zima, T.; Fialová, L.; Mestek, O.; Janebová, M.; Crkovská, J.; Malbohan, I.; Stipek, S.; Mikulikova, L.; Popov, P. Oxidative stress, metabolism of ethanol and alcohol-related diseases. J. Biomed. Sci. 2001, 8, 59–70. [Google Scholar] [CrossRef]
- Chen, C.H.; Pan, C.H.; Chen, C.C.; Huang, M.C. Increased oxidative DNA damage in patients with alcohol dependence and its correlation with alcohol withdrawal severity. Alcohol Clin. Exp. Res. 2011, 35, 338–344. [Google Scholar] [CrossRef]
- Peng, F.C.; Tang, S.H.; Huang, M.C.; Chen, C.C.; Kuo, T.L.; Yin, S.J. Oxidative status in patients with alcohol dependence: A clinical study in Taiwan. J. Toxicol. Env. Health A 2005, 68, 1497–1509. [Google Scholar] [CrossRef]
- Vatsalya, V.; Kong, M.; Cave, M.C.; Liu, N.; Schwandt, M.L.; McClain, C.J. Association of serum zinc with markers of liver injury in very heavy drinking alcohol-dependent patients. J. Nutr. Biochem. 2018, 59, 49–55. [Google Scholar] [CrossRef]
- Bates, J.; McClain, C.J. The effect of severe zinc deficiency on serum levels of albumin, transferrin, and prealbumin in man. Am. J. Clin. Nutr. 1981, 34, 1655–1660. [Google Scholar] [CrossRef]
- Vatsalya, V.; Cave, M.C.; Kumar, R.; Srivastava, S.; Khanal, S.; Jenson, A.B.; Schwandt, M.L.; Barve, S.S.; Ramchandani, V.A.; McClain, C.J. Alterations in Serum Zinc and Polyunsaturated Fatty Acid Concentrations in Treatment-Naive HIV-Diagnosed Alcohol-Dependent Subjects with Liver Injury. AIDS Res. Hum. Retrovir. 2019, 35, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Ordak, M.; Bulska, E.; Jablonka-Salach, K.; Luciuk, A.; Maj-_urawska, M.; Matsumoto, H.; Nasierowski, T.; Wojnar, M.; Matras, J.; Muszynska, E. Effect of Disturbances of Zinc and Copper on the Physical and Mental Health Status of Patients with Alcohol Dependence. Biol. Trace Elem. Res. 2018, 183, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, D.; Ahmed, Z.; Karikkineth, A.; Williams, R.; Zigel, C. Zinc-deficiency acrodermatitis in a patient with chronic alcoholism and gastric bypass: A case report. J. Community Hosp. Intern. Med. Perspect. 2014, 31, 4. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mehta, A.J.; Yeligar, S.M.; Elon, L.; Brown, L.A.; Guidot, D.M. Alcoholism Causes Alveolar Macrophage Zinc Deficiency and Immune Dysfunction. Am. J. Respir. Crit. Care Med. 2013, 188, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, H.F.; Fugleholm, A.M.; Weismann, K. Zinc deficiency and pellagra in alcohol abuse. Ugeskr Laeger 2000, 162, 6854–6856. [Google Scholar] [PubMed]
- Van Gossum, A.; Closset, P.; Noel, E.; Cremer, M.; Neve, J. Deficiency in antioxidant factors in patients with alcohol-related chronic pancreatitis. Dig. Dis. Sci. 1996, 41, 1225–1231. [Google Scholar] [CrossRef]
- Rui, M.; Rua, M.; Ojeda, L.; Nogales, F.; Rubio, J.M.; Romero-Gomez, M.; Funuyet, J.; Murillo, M.L.; Carreras, O. Serum selenium levels and oxidative balance as differential markers in hepatic damage caused by alcohol. Life Sci. 2014, 94, 158–163. [Google Scholar] [CrossRef]
- Tanner, A.R.; Bantock, I.; Hinks, L.; Lloyd, B.; Turner, N.R.; Wright, R. Depressed selenium and vitamin E levels in an alcoholic population. Possible relationship to hepatic injury through increased lipid peroxidation. Dig. Dis. Sci. 1986, 31, 1307–1312. [Google Scholar] [CrossRef]
- Dworkin, B.M.; Rosenthal, W.S.; Gordon, G.G.; Jankowski, R.H. Diminished blood selenium levels in alcoholics. Alcohol Clin. Exp. Res. 1984, 8, 535–538. [Google Scholar] [CrossRef]
- Skalny, A.V.; Berezkina, E.S.; Kiyaeva, E.V.; Alidzhanova, I.E.; Grabeklis, A.R.; Tinkov, A.A. The effect of alcohol consumption on maternal and cord blood electrolyte and trace element levels. Acta Sci. Pol. Technol. Aliment. 2016, 15, 439–445. [Google Scholar] [CrossRef]
- Kishore, B.; Thurlow, V.; Kessel, B. Hypokalaemic rhabdomyolysis. Ann. Clin. Biochem. 2007, 44 Pt 3, 308–311. [Google Scholar] [CrossRef]
- Smets, Y.F.; Bokani, N.; de Meijer, P.H.; Meinders, A.E. Tetany due to excessive use of alcohol: A possible magnesium deficiency. Ned. Tijdschr. Geneeskd. 2004, 148, 641–644. [Google Scholar]
- Mancinelli, R.; Barlocci, E.; Ciprotti, M.; Senofonte, O.; Fidente, R.M.; Draisci, R.; Attilia, M.L.; Vitali, M.; Fiore, M.; Ceccanti, M. Blood thiamine, zinc, selenium, lead and oxidative stress in a population of male and female alcoholics: Clinical evidence and gender differences. Ann. Ist. Super. Sanita 2013, 49, 65–72. [Google Scholar] [PubMed]
- Sobral-Oliveira, M.B.; Faintuch, J.; Guarita, D.R.; Oliveira, C.P.; Carrilho, F.J. Nutritional profile of asymptomatic alcoholic patients. Arq. Gastroenterol. 2011, 48, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reimers, E.; Galindo-Martin, L.; Santolaria-Fernandez, F.; Sanchez-Perez, M.J.; Alvisa-Negrin, J.; Garcia-Valdecasas-Campelo, E.; Gonzalez-Perez, J.M.; Martin-Gonzalez, M.C. Prognostic Value of Serum Selenium Levels in Alcoholics. Biol. Trace Elem. Res. 2008, 125, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Wrenn, K.D.; Slovis, C.M.; Minion, G.E.; Rutkowski, R. The syndrome of alcoholic ketoacidosis. Am. J. Med. 1991, 91, 119–128. [Google Scholar] [CrossRef]
- Halperin, M.L.; Hammeke, M.; Josse, R.G.; Jungas, R.L. Metabolic acidosis in the alcoholic: A pathophysiologic approach. Metabolism 1983, 32, 308–315. [Google Scholar] [CrossRef]
- Palmer, B.F.; Clegg, D.J. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N. Engl. J. Med. 2015, 373, 548–559. [Google Scholar] [CrossRef]
- Fulop, M.; Hoberman, H.D. Alcoholic detosis. Diabetes 1975, 24, 785–790. [Google Scholar] [CrossRef]
- Palmer, B.F.; Clegg, D.J.; Taylor, S.I.; Weir, M.R. Diabetic ketoacidosis, sodium glucose transporter-2 inhibitors and the kidney. J. Diabetes Complicat. 2016, 30, 1162–1166. [Google Scholar] [CrossRef]
- Madison, L.L.; Lochner, A.; Wulff, J. Ethanol-induced hypoglycemia. II. Mechanism of suppression of hepatic gluconeogenesis. Diabetes 1967, 16, 252–258. [Google Scholar] [CrossRef]
- Elisaf, M.S.; Siamopoulos, K.C. Mechanisms of hypophosphataemia in alcoholic patients. Int. J. Clin. Pract. 1997, 51, 501–503. [Google Scholar]
- Stein, J.H.; Smith, W.O.; Ginn, H.E. Hypophosphatemia in acute alcoholism. Am. J. Med. Sci. 1966, 252, 78–83. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, S.; Cecchin, E.; Basile, A.; Bertotti, A.; Nardini, R.; Bartoli, E. Renal tubular dysfunction in chronic alcohol abuse—Effects of abstinence. N. Engl. J. Med. 1993, 329, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Parenti, P.; Giordana, B.; Hanozet, G.M. In vitro effect of ethanol on sodium and glucose transport in rabbit renal brush border membrane vesicles. Biochim. Biophys. Acta 1991, 1070, 92–98. [Google Scholar] [CrossRef]
- Rothman, A.; Proverbio, T.; Proverbio, F. Inhibitory effect of ethanol on the Na(+)- ATPase activity of rat kidney proximal tubular cell plasma membranes. Physiol. Res. 1996, 45, 205–211. [Google Scholar] [PubMed]
- Curthoys, N.P.; Moe, O.W. Proximal tubule function and response to acidosis. Clin. J. Am. Soc. Nephrol. 2014, 9, 1627–1638. [Google Scholar] [CrossRef]
- Laitinen, K.; Tähtelä, R.; Välimäki, M. The dose-dependency of alcohol-induced hypoparathyroidism, hypercalciuria, and hypermagnesuria. Bone Miner. 1992, 19, 75–83. [Google Scholar] [CrossRef]
- Knochel, J.P. Hypophosphatemia in the Alcoholic. Arch. Intern. Med. 1980, 140, 613. [Google Scholar] [CrossRef]
- Knochel, J.P. Hypophosphatemia and rhabdomyolysis. Am. J. Med. 1992, 92, 455–457. [Google Scholar] [CrossRef]
- Anderson, R.; Cohen, M.; Haller, R.; Elms, J.; Carter, N.W.; Knochel, J.P. Skeletal muscle phosphorus and magnesium deficiency in alcoholic myopathy. Miner. Electrolyte Metab. 1980, 4, 106–112. [Google Scholar]
- Haller, R.G.; Knochel, J.P. Skeletal muscle disease in alcoholism. Med. Clin. N. Am. 1984, 68, 91–103. [Google Scholar] [CrossRef]
- Ferguson, E.R.; Blachley, J.D.; Carter, N.W.; Knochel, J.P. Derangements of muscle composition, ion transport, and oxygen consumption in chronically alcoholic dogs. Am. J. Physiol. 1984, 246, F700–F709. [Google Scholar] [CrossRef] [PubMed]
- Goff, J.P. Calcium and Magnesium Disorders. Vet. Clin. N. Am. Food Anim. Pract. 2014, 30, 359–381. [Google Scholar] [CrossRef] [PubMed]
- Elisaf, M.; Bairaktari, E.; Kalaitzidis, R.; Siamopoulos, K.C. Hypomagnesemia in alcoholic patients. Alcohol Clin. Exp. Res. 1998, 22, 134–139. [Google Scholar] [CrossRef]
- Langley, W.F.; Mann, D. Central Nervous System Magnesium Deficiency. Arch. Intern. Med. 1991, 151, 593–596. [Google Scholar] [CrossRef]
- Diringer, M. Neurologic manifestations of major electrolyte abnormalities. In Handbook of Clinical Neurology. Critical Care Neurology Part II; Elsevier: Amsterdam, The Netherlands, 2017; pp. 705–713. [Google Scholar]
- Hall, R.C.; Joffe, J.R. Hypomagnesemia. Physical and Psychiatric Symptoms. JAMA 1973, 224, 1749–1751. [Google Scholar] [CrossRef]
- Baaij, J.H.F.D.; Hoenderop, J.G.J.; Bindels, R.J.M. Magnesium in Man: Implications for Health and Disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Abrantes, C.; Brigas, D.; Casimiro, H.J.; Madeira, M. Hypocalcaemia in an Adult: The Importance of Not Overlooking the Cause. BMJ Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Newman, D.B.; Fidahussein, S.S.; Kashiwagi, D.T.; Kennel, K.A.; Kashani, K.B.; Wang, Z.; Altayar, O.; Murad, M.H. Reversible cardiac dysfunction associated with hypocalcemia: A systematic review and meta-analysis of individual patient data. Heart Fail. Rev. 2013, 19, 199–205. [Google Scholar] [CrossRef]
- Cooper, M.S.; Gittoes, N.J.L. Diagnosis and management of hypocalcaemia. BMJ 2008, 336, 1298–1302. [Google Scholar] [CrossRef]
- Peacock, M. Calcium Metabolism in Health and Disease. Clin. J. Am. Soc. Nephrol. 2010, 5 (Suppl. 1), S23–S30. [Google Scholar] [CrossRef]
- Sterns, R.H.; Silver, S.M. Complications and Management of Hyponatremia. Curr. Opin. Nephrol. Hypertens. 2016, 25, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Portales-Castillo, I.; Sterns, R.H. Allostasis and the Clinical Manifestations of Mild to Moderate Chronic Hyponatremia: No Good Adaptation Goes Unpunished. Am. J. Kidney Dis. 2019, 73, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Kogika, M.M.; Morais, H.A.D. A Quick Reference on Hypokalemia. Vet. Clin. N. Am. Small Anim. Pract. 2017, 47, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Tachi, T.; Yokoi, T.; Goto, C.; Umeda, M.; Noguchi, Y.; Yasuda, M.; Minamitani, M.; Mizui, T.; Tsuchiya, T.; Teramachi, H. Hyponatremia and hypokalemia as risk factors for falls. Eur. J. Clin. Nutr. 2014, 69, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Viera, A.J.; Wouk, N. Potassium Disorders: Hypokalemia and Hyperkalemia. Am. Fam. Physician 2015, 92, 487–495. [Google Scholar]
- Marti, G.; Schwarz, C.; Leichtle, A.B.; Fiedler, G.-M.; Arampatzis, S.; Exadaktylos, A.K.; Lindner, G. Etiology and symptoms of severe hypokalemia in emergency department patients. Eur. J. Emerg. Med. 2014, 21, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, N.; Nogi, M.; Ando, A.; Watanabe, H.; Umekawa, S. Cardiovascular Consequences of Hypophosphatemia. Panminerva Med. 2017, 59, 230–240. [Google Scholar]
- Ariyoshi, N.; Nogi, M.; Ando, A.; Watanabe, H.; Umekawa, S. Hypophosphatemia-induced Cardiomyopathy. Am. J. Med. Sci. 2016, 352, 317–323. [Google Scholar] [CrossRef]
- Pesta, D.H.; Tsirigotis, D.N.; Befroy, D.E.; Caballero, D.; Jurczak, M.J.; Rahimi, Y.; Cline, G.W.; Dufour, S.; Birkenfeld, A.L.; Rothman, D.L.; et al. Hypophosphatemia promotes lower rates of muscle ATP synthesis. FASEB J. 2016, 30, 3378–3387. [Google Scholar] [CrossRef]
- Leung, J.; Crook, M. Disorders of phosphate metabolism. J. Clin. Pathol. 2019, 72, 741–747. [Google Scholar] [CrossRef]
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Orho-Melander, M.; Struck, J.; Bergmann, A.; Melander, O. Selenoprotein-P Deficiency Predicts Cardiovascular Disease and Death. Nutrients 2019, 11, 1852. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, M.; Filippini, T.; Wise, L.A. Environmental Selenium and Human Health: An Update. Curr. Environ. Health Rep. 2018, 5, 464–485. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Rose, A.H.; Hoffmann, P.R. The Role of Selenium in Inflammation and Immunity: From Molecular Mechanisms to Therapeutic Opportunities. Redox Signal. 2012, 16, 705–743. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, Y.; Hornick, J.-L.; Istasse, L.; Dufrasne, I. Selenium in the Environment, Metabolism and Involvement in Body Functions. Molecules 2013, 18, 3292–3311. [Google Scholar] [CrossRef] [PubMed]
- Mangiapane, E.; Pessione, A.; Pessione, E. Selenium and Selenoproteins: An Overview on Different Biological Systems. Curr. Protein Pept. Sci. 2014, 15, 598–607. [Google Scholar] [CrossRef]
- Babür, E.; Tan, B.; Yousef, M.; Cinbaş, S.; Süer, C.; Dursun, N. Deficiency but Not Supplementation of Selenium Impairs the Hippocampal Long-Term Potentiation and Hippocampus-Dependent Learning. Biol. Trace Elem. Res. 2019, 192, 252–262. [Google Scholar] [CrossRef]
- Skalny, A.V.; Skalnaya, M.G.; Grabeklis, A.R.; Skalnaya, A.A.; Tinkov, A.A. Zinc deficiency as a mediator of toxic effects of alcohol abuse. Eur. J. Nutr. 2017, 57, 2313–2322. [Google Scholar] [CrossRef]
- Elitt, C.M.; Fahrni, C.J.; Rosenberg, P.A. Zinc homeostasis and zinc signaling in white matter development and injury. Neurosci. Lett. 2019, 707, 134247. [Google Scholar] [CrossRef]
- Sandstead, H.H.; Freeland-Graves, J.H. Dietary phytate, zinc and hidden zinc deficiency. J. Trace Elem. Med. Biol. 2014, 28, 414–417. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kinoshita, M.; Shimada, S.; Kawamura, T. Zinc and Skin Disorders. Nutrients 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.B.; Lukaski, H.C. Chromium. Adv. Nutr. 2018, 9, 505–506. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.J.; González, E.A.; Slatopolsky, E. Clinical Consequences and Management of Hypomagnesemia. J. Am. Soc. Nephrol. 2008, 20, 2291–2995. [Google Scholar] [CrossRef] [PubMed]
- Areco, V.; Rivoira, M.A.; Rodriguez, V.; Marchionatti, A.M.; Carpentieri, A.; Tolosa de Talamoni, N. Dietary and pharmacological compounds altering intestinal calcium absorption in humans and animals. Nutr. Res. Rev. 2015, 28, 83–99. [Google Scholar] [CrossRef]
- Emkey, R.D.; Emkey, G.R. Calcium metabolism and correcting calcium deficiencies. Endocrinol. Metab. Clin. N. Am. 2012, 41, 527–556. [Google Scholar] [CrossRef]
- Siesjö, B.K. Calcium in the Brain under Physiological and Pathological Conditions. Eur. Neurol. 1990, 30, 3–9. [Google Scholar] [CrossRef]
- Frazier, H.N.; Maimaiti, S.; Anderson, K.L.; Brewer, L.D.; Gant, J.C.; Porter, N.M.; Thibault, O. Calciums role as nuanced modulator of cellular physiology in the brain. Biochem. Biophys. Res. Commun. 2017, 483, 981–987. [Google Scholar] [CrossRef]
- Fleet, J.C.; Schoch, R.D. Molecular mechanisms for regulation of intestinal calcium absorption by vitamin D and other factors. Crit. Rev. Clin. Lab. Sci. 2010, 47, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Koopmann, A.; Grosshans, M.; Reinhard, I.; Spanagel, R.; Kiefer, F. Association of plasma calcium concentrations with alcohol craving: New data on potential pathways. Eur. Neuropsychopharmacol. 2017, 27, 42–47. [Google Scholar] [CrossRef]
- Ilias, I.; Paparrigopoulos, T.; Tzavellas, E.; Karaiskos, D.; Meristoudis, G.; Liappas, A.; Liappas, I. Inpatient alcohol detoxification and plasma calcitonin (with original findings). Hell. J. Nucl. Med. 2011, 14, 177–178. [Google Scholar]
- Vantyghem, M.C.; Danel, T.; Marcelli-Tourvieille, S.; Moriau, J.; Leclerc, L.; Cardot-Bauters, C.; Docao, C.; Carnaille, B.; Wemeau, J.L.; D’Herbomez, M. Calcitonin levels do not decrease with weaning in chronic alcoholism. Thyroid 2007, 17, 213–217. [Google Scholar] [CrossRef]
- Beto, J.A. The role of calcium in human aging. Clin. Nutr. Res. 2015, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Prince, R.L. Lifestyle and osteoporosis. Curr. Osteoporos. Rep. 2015, 13, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Vodoz, J.F.; Luisier, M.; Donath, A.; Courvoisier, B.; Garcia, B. Decrease of intestinal absorption of 47-calcium in chronic alcoholism. Med. Wochenschr. 1977, 107, 1525–1529. [Google Scholar]
- Laitinen, K.; Tahtela, R.; Luomanmaki, K.; Valimaki, M.J. Mechanisms of hypocalcemia and markers of bone turnover in alcohol-intoxicated drinkers. Bone Min. 1994, 24, 171–179. [Google Scholar] [CrossRef]
- O’Brien, C.C. Experimental evidence in the treatment of alcoholism by intensive calcium therapy. J. Am. Osteopath. Assoc. 1952, 51, 393–394. [Google Scholar]
- Spanagel, R.; Vengeliene, V.; Jandeleit, B.; Fischer, W.N.; Grindstaff, K.; Zhang, X.; Gallop, M.A.; Krstew, E.V.; Lawrence, A.J.; Kiefer, F. Acamprosate produces its anti-relapse effects via calcium. Neuropsychopharmacology 2014, 39, 783–791. [Google Scholar] [CrossRef]
- Kalk, N.J.; Lingford-Hughes, A.R. The clinical pharmacology of acamprosate. Br. J. Clin. Pharmacol. 2014, 77, 315–323. [Google Scholar] [CrossRef]
- Mann, K.; Hoffmann, S.; Pawlak, C.R. Does acamprosate really produce its anti-relapse effects via calcium? No support from the PREDICT study in human alcoholics. Neuropsychopharmacology 2016, 41, 659–660. [Google Scholar] [CrossRef][Green Version]
- N’Gouemo, P. Voltage-Sensitive Calcium Channels in the Brain: Relevance to Alcohol Intoxication and Withdrawal. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2018; Volume 248, pp. 263–280. [Google Scholar]
- Elisaf, M.; Liberopoulos, E.; Bairaktari, E.; Siamopoulos, K. Hypokalaemia in alcoholic patients. Drug Alcohol Rev. 2002, 21, 73–76. [Google Scholar] [CrossRef]
- Bahr, M.; Sommer, N.; Petersen, D.; Wietholtre, H.; Dichgans, J. Central Pontine Myelinosis Associated with Low Potassium Levels in Alcoholism. J. Neurol. 1990, 237, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.V.; Jude, E.B. Central pontine myelinolysis: Electrolytes and beyond. Case Rep. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Physiology and pathophysiology of potassium homeostasis. Adv. Physiol. Educ. 2016, 40, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.L.; Kuo, E. Mechanism of hypokalemia in magnesium deficiency. J. Am. Soc. Nephrol. 2007, 18, 2649–2652. [Google Scholar] [CrossRef] [PubMed]
- Tõnisson, M.; Tillmann, V.; Kuudeberg, A.; Väli, M. Plasma glucose, lactate, sodium, and potassium levels in children hospitalized with acute alcohol intoxication. Alcohol 2010, 44, 565–571. [Google Scholar] [CrossRef]
- Benedict, N.J.; Wong, A.; Cassidy, E.; Lohr, B.R.; Pizon, A.F.; Smithburger, P.L.; Falcione, B.A.; Kirisci, L.; Kane-Gill, S.L. Predictors of resistant alcohol withdrawal (RAW): A retrospective case-control study. Drug Alcohol Depend. 2018, 192, 303–308. [Google Scholar] [CrossRef]
- Goodson, C.M.; Clark, B.J.; Douglas, I.S. Predictors of Severe Alcohol Withdrawal Syndrome: A Systematic Review and Meta-Analysis. Alcohol. Clin. Exp. Res. 2014, 38, 2664–2677. [Google Scholar] [CrossRef]
- Sterns, R.H. Disorders of plasma sodium—Causes, consequences, and correction. N. Engl. J. Med. 2015, 372, 55–65. [Google Scholar] [CrossRef]
- Geoffrey, C.; Aiken, A. History of medical understanding and misunderstanding of acid base balance. J. Clin. Diagn. Res. 2013, 7, 2038–2041. [Google Scholar]
- Carrillo, J.M.Y.; Dobrynin, A.V. Salt effect on osmotic pressure of polyelectrolyte solutions: Simulation study. Polymers 2014, 6, 1897–1913. [Google Scholar] [CrossRef]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed]
- Hilber, B.; Scholze, P.; Dorostkar, M.M.; Sandtner, W.; Holy, M.; Boehm, S.; Singer, E.A.; Sitte, H.H. Serotonin-transporter mediated efflux: A pharmacological analysis of amphetamines and non-amphetamines. Neuropharmacology 2005, 49, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Kanner, B.I.; Zomot, E. Sodium-coupled neurotransmitter transporters. Chem. Rev. 2008, 108, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Catterall, W.A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003, 4, 207. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Martin-Eauclaire, M.F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef]
- Kratz, A.; Ferraro, M.; Sluss, P.M.; Lewandrowski, K.B. Laboratory reference values. N. Engl. J. Med. 2004, 351, 1548–1563. [Google Scholar] [CrossRef]
- Tietz, N.W. Clinical Guide to Laboratory Tests, 3rd ed.; W. B. Saunders: Philadelphia, PA, USA, 1995. [Google Scholar]
- Sunde, R.A. Selenium. In Modern Nutrition in Health and Disease, 11th ed.; Ross, A.C., Caballero, B., Cousins, R.J., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; p. 225. [Google Scholar]
- Versieck, J.; De Rudder, J.; Barbier, F. Serum Chromium Levels. JAMA 1979, 242, 1613. [Google Scholar] [CrossRef]
- Wiernsperger, N.; Rapin, J. Trace elements in glucometabolic disorders: An update. Diabetol. Metab. Syndr. 2010, 2, 70. [Google Scholar] [CrossRef]
- Liamis, G.L.; Milionis, H.J.; Rizos, E.C.; Siamopoulos, K.C.; Elisaf, M.S. Mechanisms of hynatraemia in alcohol patients. Alcohol Alcohol. 2000, 35, 612–616. [Google Scholar] [CrossRef]
- Papazachariou, I.M.; Martinez-Isla, A.; Efthimiou, E.; Williamson, R.C.N.; Girgis, S.I. Magnesium deficiency in patients with chronic pancreatitis identified by an intravenous loading test. Clin. Chim. Acta 2000, 302, 145–154. [Google Scholar] [CrossRef]
- Aagaard, N.K.; Andersen, H.; Vilstrup, H.; Clausen, T.; Jakobsen, J.; Dżrup, I. Muscle strength, Na, K-pumps magnesium and potassium in patients with alcoholic liver cirrhosis—Relation to spironolactone. J. Intern. Med. 2002, 252, 56–63. [Google Scholar] [CrossRef]
- Lindsay, A. Profound hyponatremia in cirrhosis: A case report. Cases J. 2010, 3, 77. [Google Scholar] [CrossRef]
- Majdanik, S.; Borowiak, K.; Orowicz, W. Stezenia wybranych biopierwiastkow w moczu osob znajdujacych sie w chwili smierci w stanie upojenia alkoholowego. Biul. Magnezol. 2001, 6, 594–598. [Google Scholar]
- Burst, V. Etiology and Epidemiology of Hyponatremia. Disorders of Fluid and Electrolyte Metabolism. Front. Horm. Res. 2019, 24–35. [Google Scholar] [PubMed]
- Rondon-Berrios, H.; Agaba, E.I.; Tzamaloukas, A.H. Hyponatremia: Pathophysiology, classification, manifestations and management. Int. Urol. Nephrol. 2014, 46, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-C.; Chen, C.-H.; Peng, F.-C.; Tang, S.-H.; Chen, C.-C. Alterations in Oxidative Stress Status during Early Alcohol Withdrawal in Alcoholic Patients. J. Formos. Med Assoc. 2009, 108, 560–569. [Google Scholar] [CrossRef]
- Sahay, M.; Sahay, R. Hyponatremia: A practical approach. Indian J. Endocrinol. Metab. 2014, 18, 760. [Google Scholar] [CrossRef]
- Ordak, M.; Maj-Zurawska, M.; Matsumoto, H.; Bujalska Zadrozny, M.; Nasierowski, T.; Muszynska, E.; Wojnar, M. Hyponatremia effect in patients with alcohol dependence on their physical and mental health status. Alcohol 2016, 57, 49–53. [Google Scholar]
- Eisenhofer, G.; Johnson, R.H. Effect of ethanol ingestion on plasma vasopressin and water balance in humans. Am. J. Physiol. 1982, 242, R522–R527. [Google Scholar] [CrossRef] [PubMed]
- Helderman, J.H.; Vestal, R.E.; Rowe, J.W.; Tobin, J.D.; Andres, R.; Robertson, G.L. The response of arginine vasopressin to intravenous ethanol and hypertonic saline in man: The impact of aging. J. Gerontol. 1978, 33, 39–47. [Google Scholar] [CrossRef]
- Elisaf, M.; Kalaitzidis, R. Metabolic abnormalities in alcoholic patients: Focus on acid base and electrolyte disorders. J. Alcohol Drug Depend. 2015, 3, 185–190. [Google Scholar]
- Gwinup, G.; Chelvam, R.; Jabola, R.; Meister, L. Beer drinker’s hyponatremia. Inappropriate concentration of the urine during ingestion of beer. Calif. Med. 1972, 116, 78–81. [Google Scholar] [PubMed]
- Ouellette, L.; Michel, K.; Riley, B.; Jones, J. Beer potomania: Atypical cause of severe hyponatremia in older alcoholics. Am. J. Emerg. Med. 2018, 36, 1303. [Google Scholar] [CrossRef]
- Bhttarai, N.; Kafle, P.; Panda, M. Beer Potomania: A Case Report. BMJ Case Rep. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, S.R.; Kellerman, P.S.; Nanovic, L. Beer Potomania: An Unusual Cause of Hyponatremia at High Risk of Complications from Rapid Correction. Am. J. Kidney Dis. 2007, 50, 673–680. [Google Scholar] [CrossRef]
- Lodhi, M.; Saleem, T.; Kuzel, A.R.; Khan, D.; Syed, I.A.; Rahim, U.; Iqbal, H.I.; Rahim, M. “Beer Potomania”—A Syndrome of Severe Hyponatremia with Unique Pathophysiology: Case Studies and Literature Review. Cureus 2017, 9, e2000. [Google Scholar] [CrossRef]
- Ahluwalia, V.; Wade, J.B.; Thacker, L.; Kraft, K.A.; Sterling, R.K.; Stravitz, R.T.; Fuchs, M.; Bouneva, I.; Puri, P.; Luketic, V.; et al. Differential Impact of Hyponatremia and Hepatic Encephalopathy on Health-Related Quality of Life and Brain Metabolite Abnormalities in Cirrhosis. J. Hepatol. 2013, 59, 467–473. [Google Scholar] [CrossRef]
- Fujisawa, H.; Sugimura, Y.; Takagi, H.; Mizoguchi, H.; Takeuchi, H.; Izumida, H.; Nakashima, K.; Ochiai, H.; Takeuchi, S.; Kiyota, A.; et al. Chronic Hyponatremia Causes Neurologic and Psychologic Impairments. J. Am. Soc. Nephrol. 2015, 27, 766–780. [Google Scholar] [CrossRef]
- Costa, K.N.; Nakamura, H.M.; Cruz, L.R.D.; De Miranda, L.S.V.F.; Santos-Neto, R.C.D.; Cosme, S.D.L.; Casulari, L.A. Hyponatremia and brain injury: Absence of alterations of serum brain natriuretic peptide and vasopressin. Arq. Neuro-Psiquiatr. 2009, 67, 1037–1044. [Google Scholar] [CrossRef][Green Version]
- Podestà, M.A.; Faravelli, I.; Cucchiari, D.; Reggiani, F.; Oldani, S.; Fedeli, C.; Graziani, G. Neurological Counterparts of Hyponatremia: Pathological Mechanisms and Clinical Manifestations. Curr. Neurol. Neurosci. Rep. 2015, 15, 18. [Google Scholar] [CrossRef]
- Adrogué, H.J. Consequences of Inadequate Management of Hyponatremia. Am. J. Nephrol. 2005, 25, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Hellem, T.; Shi, X.; Latendresse, G.; Renshaw, P.F. The Utility of Magnetic Resonance Spectroscopy for Understanding Substance Use Disorders. J. Am. Psychiatr. Nurses Assoc. 2015, 21, 244–275. [Google Scholar] [CrossRef] [PubMed]
- Demnitz, N.; Topiwala, A.; Zsoldos, E.; Stagg, C.J.; Emir, U.E.; Johansen-Berg, H.; Ebmeier, K.P.; Sexton, C.E. Alcohol consumption is associated with reduced creatine levels in the hippocampus of older adults. Psychiat. Res.-Neuroim. 2020, 295, 111019. [Google Scholar] [CrossRef] [PubMed]
- McNamara, P.H.; Williams, J.; McCabe, D.J.H.; Walsh, R.A. Striking Central Pontine Myelinolysis in a Patient with Alcohol Dependence Syndrome without Hyponatremia. JAMA Neurol. 2015, 73, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef]
- Johansson, U.; Johnsson, F.; Joelsson, B.; Berglund, M.; Akesson, B. Selenium status in patients with liver cirrhosis and alcoholism. Br. J. Nutr. 1986, 55, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef]
- Tinggi, U. Selenium: Its role as antioxidant in human health. Environ. Health Prev. Med. 2008, 13, 102–108. [Google Scholar] [CrossRef]
- Martinez-Peinado, M.; Nogueras-Lopez, F.; Arcos-Cebrian, A.; Agil, A.; Navarro-Alarcon, M. Serum selenium levels in cirrhotic patients are not influenced by the disease severity index. Nutr. Res. 2010, 30, 574–578. [Google Scholar] [CrossRef]
- Rossi, R.E.; Conte, D.; Massironi, S. Diagnosis and treatment of nutritional deficiencies in alcoholic liver disease: Overview of available evidence and open issues. Dig. Liver Dis. 2015, 47, 819–825. [Google Scholar] [CrossRef]
- Bergheim, I.; Parlesak, A.; Dierks, C.; Bode, J.C.; Bode, C. Nutritional deficiencies in German middle-class male alcohol consumers: Relation to dietary intake and severity of liver disease. Eur. J. Clin. Nutr. 2003, 57, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, M.L.; Vazquez, B.; Nogales, F.; Murillo, M.L.; Carreras, O. Ethanol consumption by Wistar rat dams affects selenium bioavailability and antioxidant balance in their progeny. Int. J. Environ. Res. Public Health 2009, 6, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Simonoff, M.; Simonoff, G. Le sélénium et la vie; Masson: Paris, France, 1991; p. 242. [Google Scholar]
- Burk, R.F.; Hill, K.E. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, Y.; Zhang, L.; Guo, Q. Oxidative stress and liver disease. Hepatol. Res. 2012, 42, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Misra, H.; Li, Y.R. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: Updated experimental and clinical evidence. J. Dig. Dis. 2012, 13, 133–142. [Google Scholar] [CrossRef]
- Tuma, D.J. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radic. Biol. Med. 2002, 32, 303–308. [Google Scholar] [CrossRef]
- Singh, M.; Gupta, S.; Singhal, U.; Pandey, R.; Aggarwal, S.L. Evaluation of the Oxidative Stress in Chronic Alcoholics. J. Clin. Diag. Res. 2013, 7, 1568–1571. [Google Scholar]
- Perez-Hernandez, O.; Gonzalez-Reimers, E.; Quintero-Platt, G.; Abreu-Gonzalez, P.; de la Vega-Prieto, M.J.; Sanchez-Perez, M.J.; Martin-Gonzalez, C.; Martinez-Riera, A.; Santolaria-Fernandez, F. Malondialdehyde as a Prognostic Factor in Alcoholic Hepatitis. Alcohol Alcohol. 2017, 52, 305–310. [Google Scholar] [CrossRef][Green Version]
- Gonzalez-Reimers, E.; Monedero-Prieto, M.J.; Gonzalez-Perez, J.M.; Duran-Castellon, M.C.; Galindo-Martin, L.; Abreu-Gonzalez, P.; Sanchez-Perez, M.J.; Santolaria-Fernandez, F. Relative and Combined Effects of Selenium, Protein Deficiency and Ethanol on Hepatocyte Ballooning and Liver Steatosis. Biol. Trace Elem. Res. 2013, 154, 281–287. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, Y.; Qiu, C.; Deng, G.; Guo, M. Protective Action of Se-Supplement Against Acute Alcoholism Is Regulated by Selenoprotein P (SelP) in the Liver. Biol. Trace Elem. Res. 2016, 175, 375–387. [Google Scholar] [CrossRef]
- Livingstone, C. Zinc: Physiology, Deficiency, and Parenteral Nutrition. Nutr. Clin. Pract. 2015, 30, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Marger, L.; Schubert, C.; Bertrand, D. Zinc: An underappreciated modulatory factor of brain function. Biochem. Pharmacol. 2014, 91, 426–435. [Google Scholar] [CrossRef]
- Najafabadi, H.S.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.N.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Altemose, N.; Noor, N.; Bitoun, E.; Tumian, A.; Imbeault, M.; Chapman, J.R.; Aricescu, A.R.; Myers, S.R. A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis. eLife 2017, 6, e28383. [Google Scholar] [CrossRef] [PubMed]
- Cousins, R.J. Zinc. In Present Knowledge in Nutrition, 7th ed.; Ziegler, E.E., Filer, I.J., Jr., Eds.; ILSI Press: Washington, DC, USA, 1996; pp. 293–306. [Google Scholar]
- Fraker, P.J.; Jardieu, P.; Cook, J. Zinc deficiency and immune function. Arch. Dermatol. 1987, 123, 1699–1701. [Google Scholar] [CrossRef]
- Zhou, Z. Zinc and Alcoholic Liver Disease. Dig. Dis. 2010, 28, 745–750. [Google Scholar] [CrossRef]
- Sun, Q.; Li, Q.; Zhong, W.; Zhang, J.; Sun, X.; Tan, X.; Yin, X.; Sun, X.; Zhang, X.; Zhou, Z. Dysregulation of hepatic zinc transporters in a mouse model of alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G313–G322. [Google Scholar] [CrossRef]
- Sun, Q.; Zhong, W.; Zhang, W.; Li, Q.; Sun, X.; Tan, X.; Sun, X.; Dong, D.; Zhou, Z. Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G757–G766. [Google Scholar] [CrossRef]
- Moghe, A. Histone modifications and alcohol-induced liver disease: Are altered nutrients the missing link? World J. Gastroenterol. 2011, 17, 2465. [Google Scholar] [CrossRef]
- Gonzalez-Reimers, E.; Martinez-Riera, A.; Santolaria-Fernandez, F.; Mas-Pascual, A.; Rodriguez-Moreno, F.; Galindo-Martin, L.; Molina-Perez, M.; Barros-Lopez, N. Relative and Combined Effects of Ethanol and Protein Deficiency on Zinc, Iron, Copper, and Manganese Contents in Different Organs and Urinary and Fecal Excretion. Alcohol 1998, 16, 7–12. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, L.; Song, Z.; Saari, J.T.; Mcclain, C.J.; Kang, Y.J. Zinc Supplementation Prevents Alcoholic Liver Injury in Mice through Attenuation of Oxidative Stress. Am. J. Pathol. 2005, 166, 1681–1690. [Google Scholar] [CrossRef]
- Menzano, E.; Carlen, P.L. Zinc deficiency and corticosteroids in the pathogenesis of alcoholic brain dysfunction—A review. Alcohol. Clin. Exp. Res. 1994, 18, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Ham, B.J.; Choi, I.G. Psychiatric Implications of Nutritional Deficiencies in Alcoholism. Psychiatry Investig. 2005, 2, 44–59. [Google Scholar]
- Gueguen, S.; Pirollet, P.; Leroy, P.; Guilland, J.C.; Arnaud, J.; Paille, F.; Siest, G.; Visvikis, S.; Hercberg, S.; Herbeth, B. Changes in Serum Retinol, α-Tocopherol, Vitamin C.; Carotenoids, Zinc and Selenium after Micronutrient Supplementation during Alcohol Rehabilitation. J. Am. Coll. Nutr. 2003, 22, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Mcclain, C.J.; Cave, M.; Kang, Y.J.; Zhou, Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G625–G633. [Google Scholar] [CrossRef]
- Anderson, R.A. Nutritional role of chromium. Sci. Total Environ. 1981, 17, 13–29. [Google Scholar] [CrossRef]
- European Food Safety Authority. Scientific opinion on dietary reference values for chromium. EFSA J. 2014, 12, 3845. [Google Scholar] [CrossRef]
- Vincent, J.B. The Bioinorganic Chemistry of Chromium; John Wiley & Sons: Chichester, UK, 2013. [Google Scholar]
- Walter, L.R.; Marel, E.; Harbury, R.; Wearne, J. Distribution of Chromium and Cobalt ions in various blood fractions after resurfacing hip arthroplasty. J. Arthroplast. 2008, 23, 814–821. [Google Scholar] [CrossRef]
- De Smet, K.; De Haan, R.; Calistri, A.; Campbell, P.A.; Embramzadeh, E.; Pattyn, C.; Gill, H.S. Metal ion measurement as a diagnostic tool to identify problems with metal-on-metal hip resurfacing. J. Bone Jt. Surg. A 2008, 90, 202–208. [Google Scholar] [CrossRef]
- Costello, R.B.; Dwyer, J.T.; Bailey, R.L. Chromium supplements for glycemic control in type 2 diabetes: Limited evidence of effectiveness. Nutr. Rev. 2016, 74, 455–468. [Google Scholar] [CrossRef]
- Cefalu, W.T.; Rood, J.; Pinsonat, P.; Qin, J.; Sereda, O.; Levitan, L.; Anderson, R.; Zhang, X.H.; Martin, J.M.; Martin, C.; et al. Characterization of the metabolic and physiologic response to chromium supplementation in subjects with type 2 diabetes mellitus. Metabolism 2010, 59, 755–762. [Google Scholar] [CrossRef]
- Sharma, S.; Agrawal, R.P.; Choudhary, M.; Jain, S.; Goyal, S.; Agarwal, V. Beneficial effect of chromium supplementation on glucose, HbA1C and lipid variables in individuals with newly onset type-2 diabetes. J. Trace Elem. Med. Biol. 2011, 25, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Drake, T.C.; Rudser, K.D.; Seaquist, E.R.; Saeed, A. Chromium infusion in hospitalized patients with severe insulin resistance: A retrospective analysis. Endocr. Pract. 2012, 18, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Surani, S.R.; Ratnani, I.; Guntupalli, B.; Bopparaju, S. Severe insulin resistance treatment with intravenous chromium in septic shock patient. World J. Diabetes 2012, 3, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Kahlon, G.; Morehead, L.; Dhawan, R.; Lieblong, B.; Stapleton, T.; Caldito, G.; Hoeldtke, R.; Levine, S.N.; Bass, P.F., III. Effect of chromium dinicocysteinate supplementation on circulating levels of insulin, TNF-α, oxidative stress, and insulin resistance in type 2 diabetic subjects: Randomized, double-blind, placebo-controlled study. Mol. Nutr. Food Res. 2012, 56, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Lewicki, S.; Zdanowski, R.; Krzy_owska, M.; Lewicka, A.; D_bski, B.; Niemcewicz, M.; Goniewicz, M. The role of Chromium III in the organism and its possible use in diabetes and obesity treatment. Ann. Agric. Environ. Med. 2014, 21, 331–335. [Google Scholar] [CrossRef]
- Balk, E.M.; Tatsioni, A.; Lichtenstein, A.H.; Lau, J.; Pittas, A.G. Effect of chromium supplementation on glucose metabolism and lipids: A systematic review of randomized controlled trials. Diabetes Care 2007, 30, 2154–2163. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Cefalu, W.T. Current concepts about chromium supplementation in type 2 diabetes and insulin resistance. Curr. Diab. Rep. 2010, 10, 145–151. [Google Scholar] [CrossRef]
- Masharani, U.; Gjerde, C.; McCoy, S.; Maddux, B.A.; Hessler, D.; Goldfine, I.D.; Youngren, J.F. Chromium supplementation in non-obese non-diabetic subjects is associated with decline in insulin sensitivity. BMC Endocr. Disord. 2012, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Kuryl, T.; Debski, B.; Martinik, K. The effect of microelements supplementation on beta-oxidation activity in healthy and type 1 diabetic rats. Cent. Eur. J. Public Health 2008, 16, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Lau, F.C.; Bagchi, M.; Sen, C.K.; Bagchi, D. Nutrigenomic basis of beneficial effects of chromium(III) on obesity and diabetes. Mol. Cell Biochem. 2008, 317, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tuzcu, M.; Sahin, N.; Orhan, C.; Agca, C.A.; Akdemir, F.; Tuzcu, Z.; Komorowski, J.; Sahin, K. Impact of chromium histidinate on high fat diet induced obesity in rats. Nutr. Metab. 2011, 8, 28. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).