
The Effect of Body Fat Distribution on Systemic Sclerosis
 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
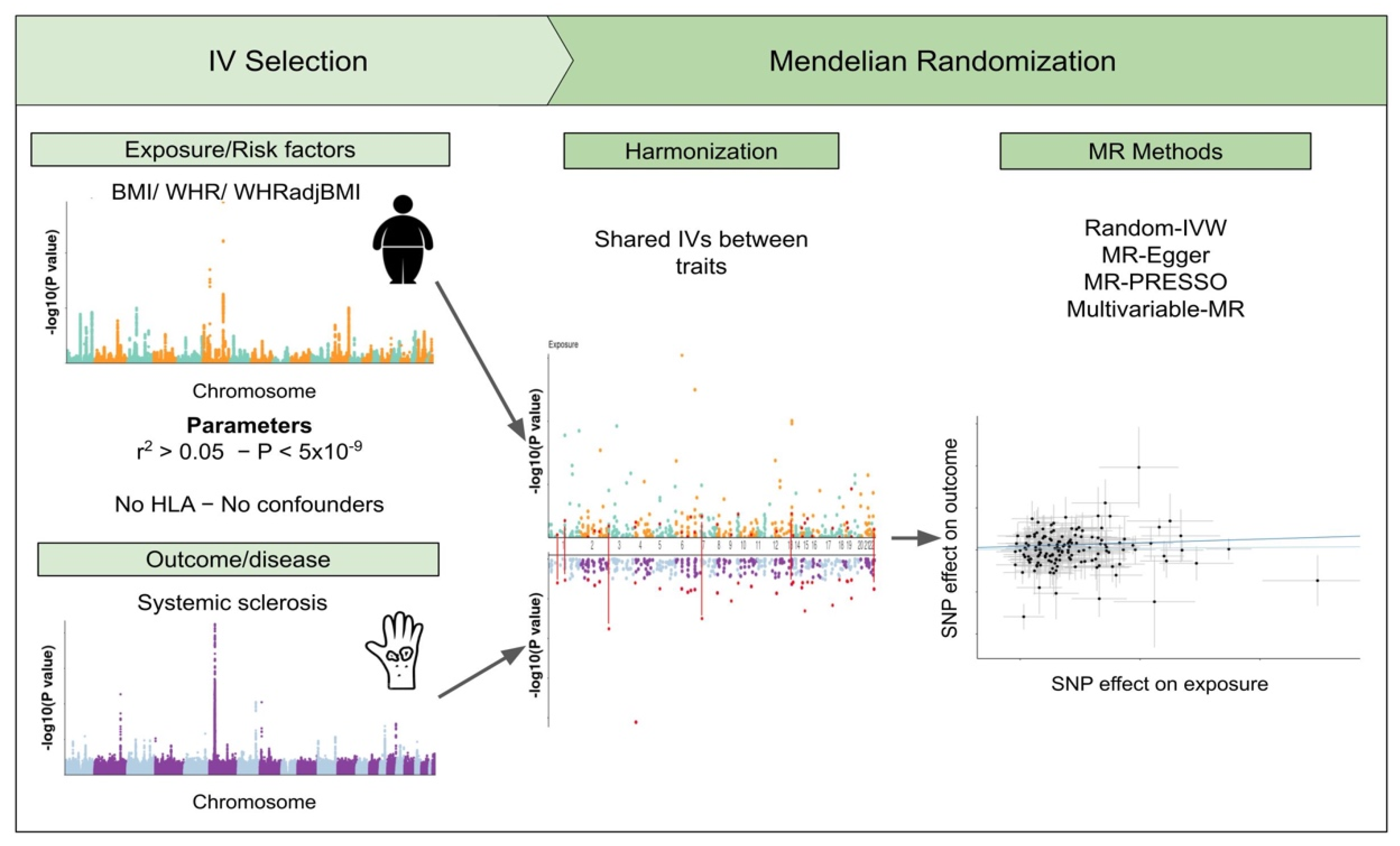
2. Materials and Methods
2.1. Instrumental Variables
2.2. Genomic Association Analysis
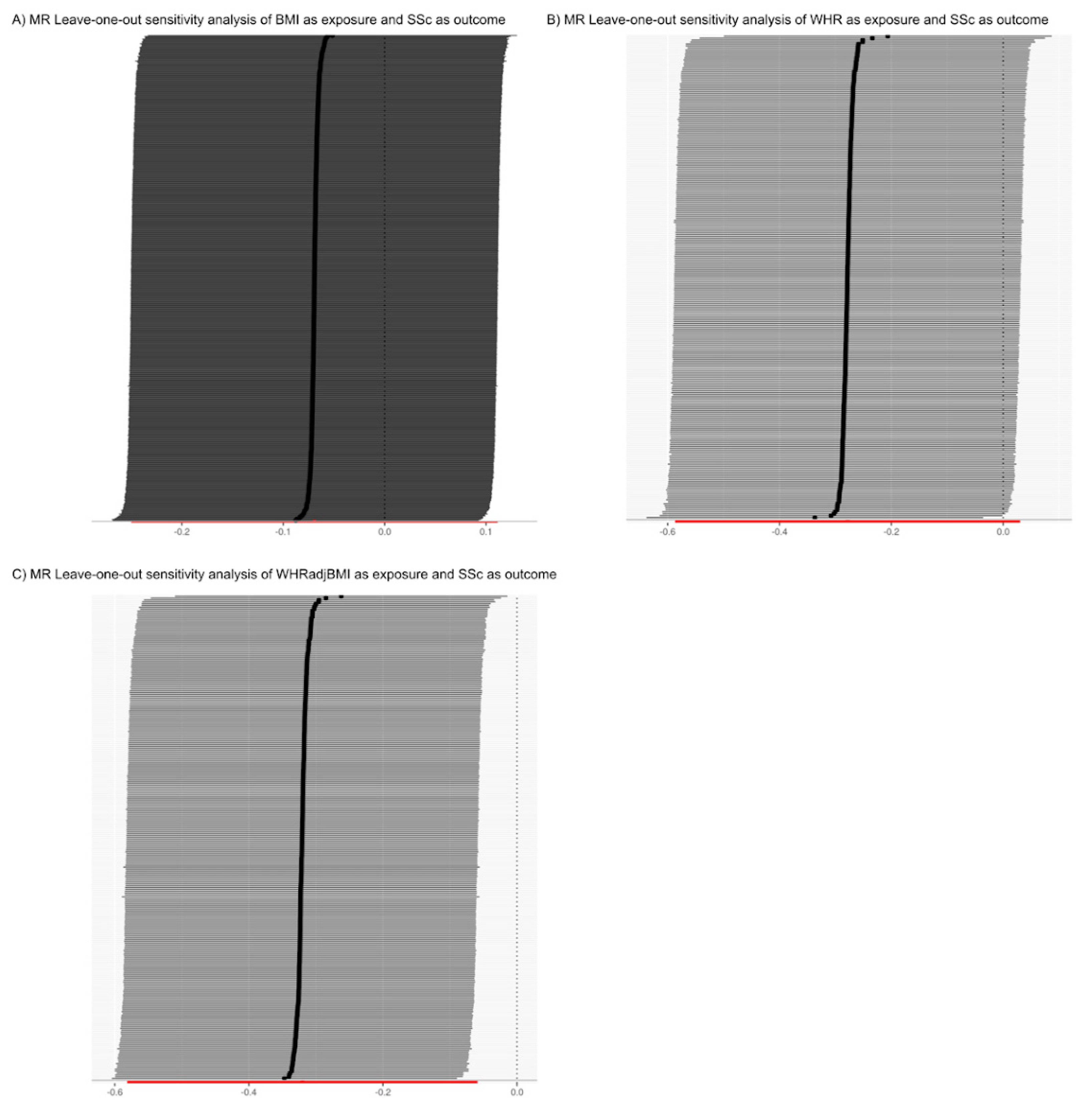
2.3. Sensitivity Analysis
3. Results
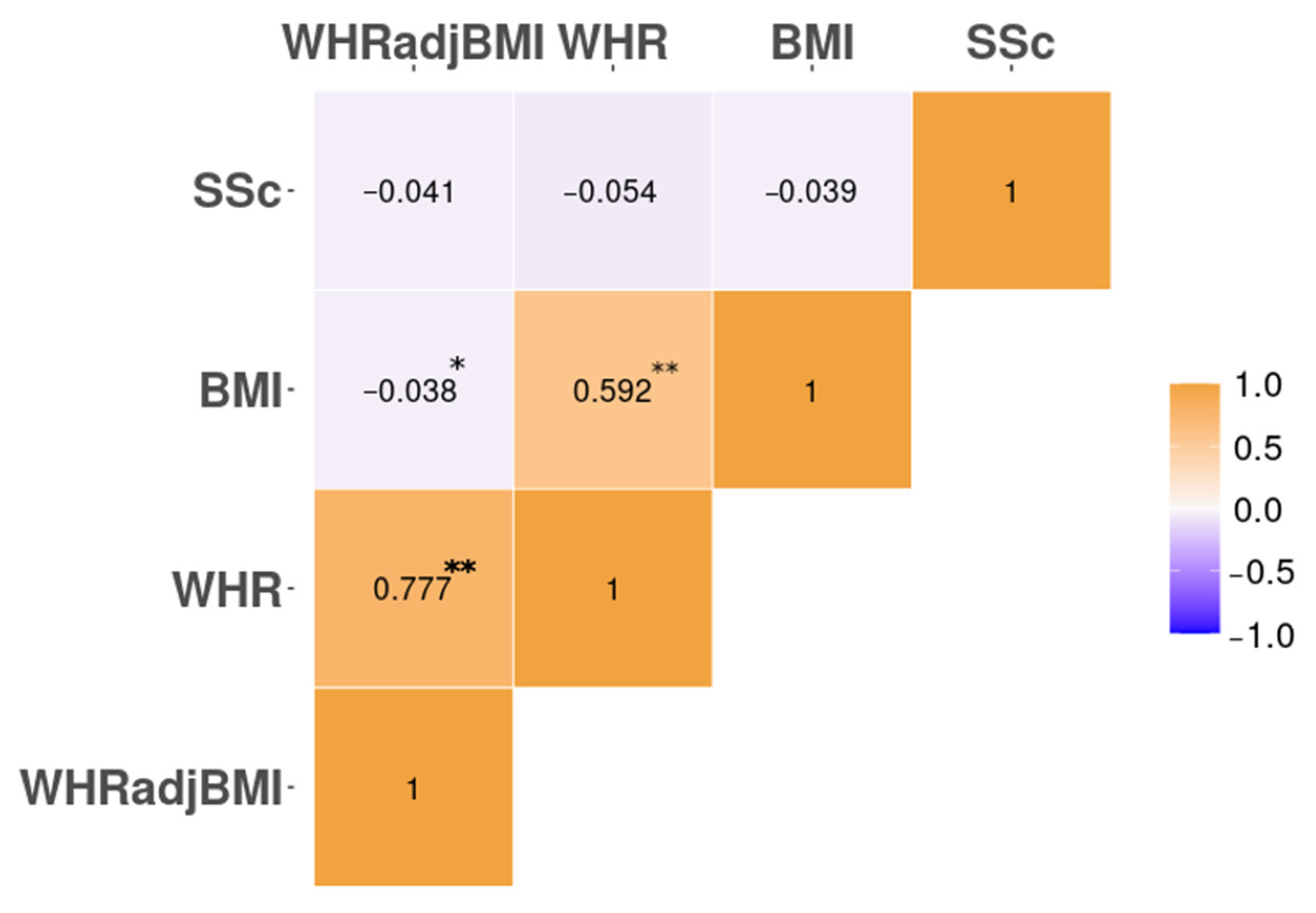
3.1. Genomic Correlation. Only the HLA Locus Harbours Local Genetic Correlation between SSc and Body Fat Distribution
3.2. The Analysis of the Causal Relationship between Obesity-Related Traits and Systemic Sclerosis Is Limited by Confounding Factors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Radstake, T.R.D.J.; Gorlova, O.; Rueda, B.; Martin, J.-E.; Alizadeh, B.Z.; Palomino-Morales, R.; Coenen, M.J.; Vonk, M.C.; Voskuyl, A.E.; Schuerwegh, A.J.; et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat. Genet. 2010, 42, 426–429. [Google Scholar] [CrossRef]
- López-Isac, E.; Acosta-Herrera, M.; Kerick, M.; Assassi, S.; Satpathy, A.T.; Granja, J.; Mumbach, M.R.; Beretta, L.; Simeón, C.P.; Carreira, P.; et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 2019, 10, 4955. [Google Scholar] [CrossRef]
- Acosta-Herrera, M.; Kerick, M.; Lopéz-Isac, E.; Assassi, S.; Beretta, L.; Simeón-Aznar, C.P.; Ortego-Centeno, N.; International SSc Group; Proudman, S.M.; Australian Scleroderma Interest Group (ASIG); et al. Comprehensive analysis of the major histocompatibility complex in systemic sclerosis identifies differential HLA associations by clinical and serological subtypes. Ann. Rheum. Dis. 2021, 80, 1040–1047. [Google Scholar] [CrossRef]
- Bossini-Castillo, L.; Villanueva-Martin, G.; Kerick, M.; Acosta-Herrera, M.; López-Isac, E.; Simeón, C.P.; Ortego-Centeno, N.; Assassi, S.; International SSc Group; Australian Scleroderma Interest Group (ASIG); et al. Genomic Risk Score Impact on Susceptibility to Systemic Sclerosis. Ann. Rheum. Dis. 2021, 80, 118–127. [Google Scholar] [CrossRef]
- Bossini-Castillo, L.; López-Isac, E.; Mayes, M.D.; Martín, J. Genetics of systemic sclerosis. Semin. Immunopathol. 2015, 37, 443–451. [Google Scholar] [CrossRef]
- Barnes, J.; Mayes, M.D. Epidemiology of Systemic Sclerosis: Incidence, Prevalence, Survival, Risk Factors, Malignancy, and Environmental Triggers. Curr. Opin. Rheumatol. 2012, 24, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Calderon, L.M.; Pope, J.E. Scleroderma epidemiology update. Curr. Opin. Rheumatol. 2021, 33, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic Sclerosis. Nat. Rev. Dis. Primers 2015, 1, 15002. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian Randomization Analysis with Multiple Genetic Variants Using Summarized Data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. “Mendelian Randomization”: Can Genetic Epidemiology Contribute to Understanding Environmental Determinants of Disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davies, N.M.; Hemani, G.; Smith, G.D. Two-sample Mendelian randomization: Avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int. J. Epidemiol. 2016, 45, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Versini, M.; Jeandel, P.-Y.; Rosenthal, E.; Shoenfeld, Y. Obesity in autoimmune diseases: Not a passive bystander. Autoimmun. Rev. 2014, 13, 981–1000. [Google Scholar] [CrossRef] [PubMed]
- Borga, M.; West, J.; Bell, J.D.; Harvey, N.C.; Romu, T.; Heymsfield, S.B.; Leinhard, O.D. Advanced body composition assessment: From body mass index to body composition profiling. J. Investig. Med. 2018, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.; Mendis, S.; Zheleznyakov, E.; Reddy, S.; Chan, J. Body Mass Index, Waist Circumference and Waist:hip Ratio as Predictors of Cardiovascular Risk--a Review of the Literature. Eur. J. Clin. Nutr. 2010, 64, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, S.; Xiong, W.; Li, Y.; Li, H.; Li, J.; Li, F. Waist-Hip Ratio as a Predictor of Myocardial Infarction Risk: A Systematic Review and Meta-Analysis. Medicine 2018, 97, e11639. [Google Scholar] [CrossRef] [PubMed]
- Pulit, S.L.; Karaderi, T.; Lindgren, C.M. Sexual dimorphisms in genetic loci linked to body fat distribution. Biosci. Rep. 2017, 37, BSR20160184. [Google Scholar] [CrossRef] [PubMed]
- Randall, J.C.; Winkler, T.W.; Kutalik, Z.; Berndt, S.I.; Jackson, A.U.; Monda, K.L.; Kilpeläinen, T.O.; Esko, T.; Mägi, R.; Li, S.; et al. Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits. PLoS Genet. 2013, 9, e1003500. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Shi, H.; Alfredsson, L.; Klareskog, L.; Padyukov, L.; Jiang, X. Obesity-Related Traits and the Development of Rheumatoid Arthritis: Evidence from Genetic Data. Arthritis Rheumatol. 2021, 73, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Torres, R.; Ibáñez-Sanz, G.; Obón-Santacana, M.; Duell, E.J.; Moreno, V. Identifying environmental risk factors for inflammatory bowel diseases: A Mendelian randomization study. Sci. Rep. 2020, 10, 19273. [Google Scholar] [CrossRef] [PubMed]
- Alwarawrah, Y.; Kiernan, K.; Maciver, N.J. Changes in Nutritional Status Impact Immune Cell Metabolism and Function. Front. Immunol. 2018, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Pulit, S.L. Summary-Level Data from Meta-Analysis of Fat Distribution Phenotypes in UK Biobank and Giant. Zenodo 2018. [Google Scholar] [CrossRef]
- Pulit, S.L.; Stoneman, C.; Morris, A.P.; Wood, A.R.; Glastonbury, C.A.; Tyrrell, J.; Yengo, L.; Ferreira, T.; Marouli, E.; Ji, Y.; et al. Meta-analysis of genome-wide association studies for body fat distribution in 694,649 individuals of European ancestry. Hum. Mol. Genet. 2019, 28, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Mancuso, N.; Spendlove, S.; Pasaniuc, B. Local Genetic Correlation Gives Insights into the Shared Genetic Architecture of Complex Traits. Am. J. Hum. Genet. 2017, 101, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Mayes, M.D.; Bossini-Castillo, L.; Gorlova, O.; Martin, J.E.; Zhou, X.; Chen, W.V.; Assassi, S.; Ying, J.; Tan, F.K.; Arnett, F.C.; et al. Immunochip Analysis Identifies Multiple Susceptibility Loci for Systemic Sclerosis. Am. J. Hum. Genet. 2014, 94, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. Interpreting Findings from Mendelian Randomization Using the MR-Egger Method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Publisher Correction: Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 1196. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018, 7, e34408. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Brion, M.-J.A.; Shakhbazov, K.; Visscher, P. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiology 2013, 42, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Oscanoa, J.; Sivapalan, L.; Gadaleta, E.; Ullah, A.Z.D.; Lemoine, N.R.; Chelala, C. SNPnexus: A web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 2020, 48, W185–W192. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Khanna, D. Systemic Sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Miller, J.B.; Gandhi, N.; Clarke, J.; McMahan, Z. Gastrointestinal Involvement in Systemic Sclerosis: An Update. J. Clin. Rheumatol. 2018, 24, 328–337. [Google Scholar] [CrossRef]
- Hughes, M.; Heal, C.; Siegert, E.; Hachulla, E.; Airó, P.; Riccardi, A.; Distler, O.; Matucci-Cerinic, M. EUSTAR Collaborators Significant Weight Loss in Systemic Sclerosis: A Study from the EULAR Scleroderma Trials and Research (EUSTAR) Database. Ann. Rheum. Dis. 2020, 79, 1123–1125. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Smith, G.D. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef] [PubMed]
- Julià, A.; Martínez-Mateu, S.H.; Domènech, E.; Cañete, J.D.; Ferrándiz, C.; Tornero, J.; Gisbert, J.P.; Fernández-Nebro, A.; Daudén, E.; Barreiro-de Acosta, M.; et al. Food groups associated with immune-mediated inflammatory diseases: A Mendelian randomization and disease severity study. Eur. J. Clin. Nutr. 2021, 75, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- González-Serna, D.; Villanueva-Martin, G.; Acosta-Herrera, M.; Márquez, A.; Martín, J. Approaching Shared Pathophysiology in Immune-Mediated Diseases through Functional Genomics. Genes 2020, 11, 1482. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Żółkiewicz, J.; Stochmal, A.; Rudnicka, L. The Role of Adipokines in Systemic Sclerosis: A Missing Link? Arch. Dermatol. Res. 2019, 311, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Iannone, F.; Praino, E.; Rotondo, C.; Natuzzi, D.; Bizzoca, R.; Lacarpia, N.; Fornaro, M.; Cacciapaglia, F. Body mass index and adipokines/cytokines dysregulation in systemic sclerosis. Clin. Exp. Immunol. 2021, 206, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Attolini, C.S.-O.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shao, M.; Hepler, C.; Zi, Z.; Zhao, S.; An, Y.A.; Zhu, Y.; Ghaben, A.L.; Wang, M.-Y.; Li, N.; et al. Dermal adipose tissue has high plasticity and undergoes reversible dedifferentiation in mice. J. Clin. Investig. 2019, 129, 5327–5342. [Google Scholar] [CrossRef] [PubMed]
- Taki, Z.; Gostjeva, E.; Thilly, W.; Yaseen, B.; Lopez, H.; Mirza, M.; Hassuji, Z.; Vigneswaran, S.; Abdi, B.A.; Hart, A.; et al. Pathogenic Activation of Mesenchymal Stem Cells Is Induced by the Disease Microenvironment in Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 1361–1374. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index and secondary SNPs (p < 5 × 10−9) | ||||||
| MR Approach | nSNPs | OR (95% CI) | p | padj | pfor Heterogeneity or Pleiotropy | |
| BMI | MR-Egger | 533 | 1.0575(0.6403–1.7466) | 0.827 | 0.8273 | 0.6005 |
| Random-effects IVW | 0.9326(0.7787–1.117) | 0.449 | 0.4485 | <0.001 | ||
| MR-PRESSO (1) * | 0.943(0.7892–1.1269) | 0.5189 | NA | NA | ||
| WHR | MR-Egger | 247 | 0.2698(0.0914–0.7965) | 0.0185 | 0.0384 | 0.0519 |
| Random-effects IVW | 0.7564(0.5567–1.0278) | 0.0743 | 0.11145 | <0.001 | ||
| MR-PRESSO (3) * | 0.7809(0.5907–1.0324) | 0.0838 | NA | NA | ||
| WHRadjBMI | MR-Egger | 262 | 0.4251(0.2014–0.8971) | 0.0256 | 0.0384 | 0.1344 |
| Random-effects IVW | 0.7269(0.5603–0.9431) | 0.0163 | 0.0489 | <0.001 | ||
| MR-PRESSO (1) * | 0.77(0.6015–0.9857) | 0.039 | NA | NA | ||
| Index and Secondary SNPs (p < 5 × 10−9) | ||||||
| MR Approach | nSNPs | OR (95% CI) | p | padj | pfor Heterogeneity or Pleiotropy | |
| BMI | MR-Egger | 483 | 1.422(0.721–2.803) | 0.3103 | 0.3103 | 0.1769 |
| Random-effects IVW | 0.909(0.741–1.115) | 0.3598 | 0.3598 | 0.0011 | ||
| MR-PRESSO (1) * | 0.922(0.754–1.128) | 0.4288 | NA | NA | ||
| WHR | MR-Egger | 221 | 0.301(0.086–1.060) | 0.0629 | 0.09435 | 0.1391 |
| Random-effects IVW | 0.752(0.535–1.057) | 0.1007 | 0.15105 | < 0.001 | ||
| MR-PRESSO (2) * | 0.764(0.559–1.044) | 0.0927 | NA | NA | ||
| WHRadjBMI | MR-Egger | 237 | 0.335(0.137–0.819) | 0.0172 | 0.0516 | 0.0772 |
| Random-effects IVW | 0.716(0.534–0.961) | 0.0261 | 0.0783 | < 0.001 | ||
| MR-PRESSO (1) * | 0.769(0.582–1.015) | 0.0651 | NA | NA | ||
| Before Confounder SNP Removal. | After Confounder SNP Removal | ||||||||
| Index and secondary SNPs (p < 5 × 10−9) | Index and secondary SNPs (p < 5 × 10−9) | ||||||||
| Outcome | Exposure | nSNP | OR (95% CI) | p | Outcome | Exposure | nSNP | OR (95% CI) | p |
| SSc | BMI | 666 | 1.026(0.79–1.331) | 0.849 | SSc | BMI | 610 | 1.027(0.760–1.387) | 0.863 |
| WHR | 666 | 0.804(0.573–1.128) | 0.207 | WHR | 610 | 0.812(0.552–1.195) | 0.291 | ||
| Index SNPs (p < 5 × 10−9) | Index SNPs (p < 5 × 10−9) | ||||||||
| Outcome | Exposure | nSNP | OR (95% CI) | p | Outcome | Exposure | nSNP | OR (95% CI) | p |
| SSc | BMI | 581 | 0.99(0.749–1.309) | 0.946 | SSc | BMI | 524 | 1.013(0.726–1.412) | 0.941 |
| WHR | 581 | 0.876(0.607–1.263) | 0.477 | WHR | 524 | 0.881(0.574–1.352) | 0.561 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanueva-Martin, G.; Acosta-Herrera, M.; Kerick, M.; López-Isac, E.; Simeón, C.P.; Callejas, J.L.; Assassi, S.; Beretta, L.; SSc Group, I.; , A.S.I.G.; et al. The Effect of Body Fat Distribution on Systemic Sclerosis. J. Clin. Med. 2022, 11, 6014. https://doi.org/10.3390/jcm11206014
Villanueva-Martin G, Acosta-Herrera M, Kerick M, López-Isac E, Simeón CP, Callejas JL, Assassi S, Beretta L, SSc Group I, ASIG, et al. The Effect of Body Fat Distribution on Systemic Sclerosis. Journal of Clinical Medicine. 2022; 11(20):6014. https://doi.org/10.3390/jcm11206014
Chicago/Turabian StyleVillanueva-Martin, Gonzalo, Marialbert Acosta-Herrera, Martin Kerick, Elena López-Isac, Carmen P. Simeón, José L. Callejas, Shervin Assassi, Lorenzo Beretta, International SSc Group, Australian Scleroderma Interest Group (ASIG), and et al. 2022. "The Effect of Body Fat Distribution on Systemic Sclerosis" Journal of Clinical Medicine 11, no. 20: 6014. https://doi.org/10.3390/jcm11206014
APA StyleVillanueva-Martin, G., Acosta-Herrera, M., Kerick, M., López-Isac, E., Simeón, C. P., Callejas, J. L., Assassi, S., Beretta, L., SSc Group, I., , A. S. I. G., Allanore, Y., Proudman, S. M., Nikpour, M., Fonseca, C., Denton, C. P., Radstake, T. R. D. J., Mayes, M. D., Jiang, X., Martin, J., & Bossini-Castillo, L. (2022). The Effect of Body Fat Distribution on Systemic Sclerosis. Journal of Clinical Medicine, 11(20), 6014. https://doi.org/10.3390/jcm11206014