
Osteoarthritis: New Insight on Its Pathophysiology


 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
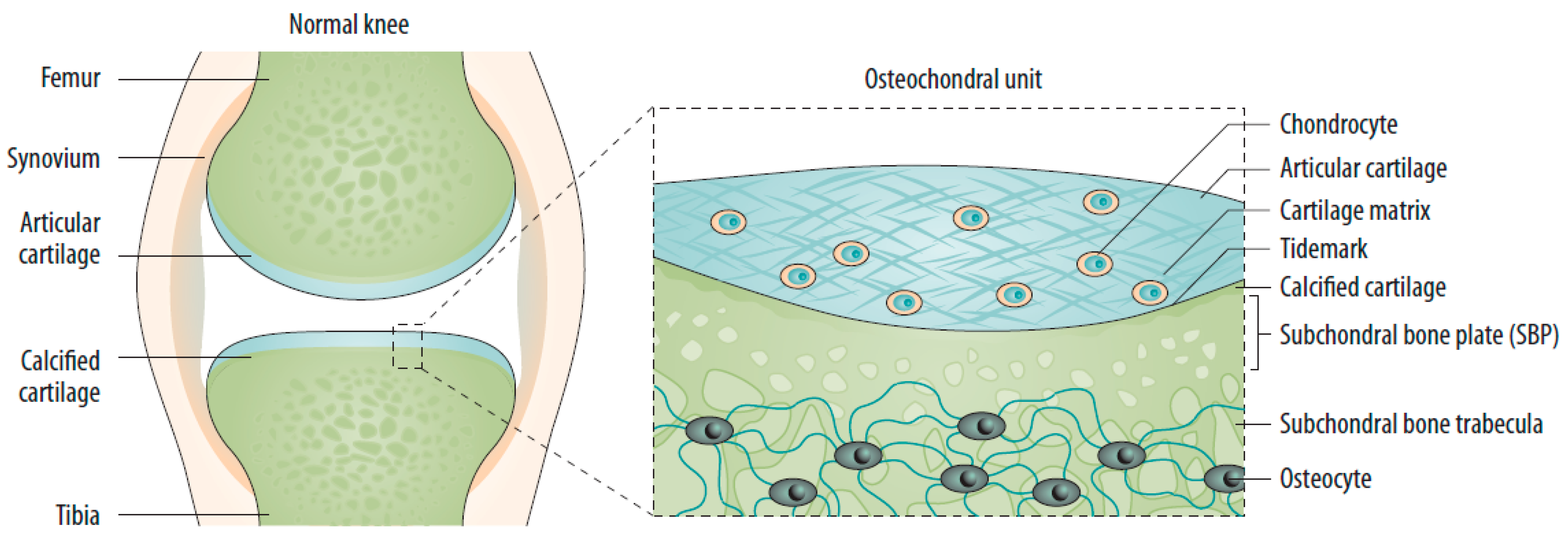
2.1. Changes in the Osteochondral Unit during Osteoarthritis: The Role of the Subchondral Bone
2.2. Risk Factors
2.2.1. Individual Risk Factors
2.2.2. Genetics
2.3. Joint-Related Factors
3. Mechanisms Underlying Joint Deterioration
3.1. The Interplay between Immunological and Biochemical Processes
3.2. Synovitis
3.3. Innate Immune System
3.4. Adaptative Immunity
3.5. Neuroinflammatory Processes
3.6. Neoangiogenesis
3.7. Osteophyte Formation and Joint Remodeling
3.8. Functional Outcomes of Osteoarthritis
3.9. Small Joints
3.10. Middle and Large Joints
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jang, S.; Lee, K.; Ju, J.H. Recent Updates of Diagnosis, Pathophysiology, and Treatment on Osteoarthritis of the Knee. Int. J. Mol. Sci. 2021, 22, 2619. [Google Scholar] [CrossRef] [PubMed]
- Sacitharan, P.K. Ageing and osteoarthritis. Subcell Biochem. 2019, 91, 123–159. [Google Scholar] [PubMed]
- Kulkarni, P.; Martson, A.; Vidya, R.; Chitnavis, S.; Harsulkar, A. Pathophysiological landscape of osteoarthritis. Adv. Clin. Chem. 2021, 100, 37–90. [Google Scholar] [PubMed]
- Abramoff, B.; Caldera, F.E. Osteoarthritis: Pathology, diagnosis, and treatment options. Med. Clin. N. Am. 2020, 104, 293–311. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, X.; Wang, S.; Jing, Y.; Su, J. Subchondral bone microenvironment in osteoarthritis and pain. Bone Res. 2021, 9, 20. [Google Scholar] [CrossRef]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, regional and national burden of osteoarthritis 1990–2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef]
- Cross, M.; Smith, E.; Hoy, D.; Nolte, S.; Ackerman, I.; Fransen, M.; Bridgett, L.; Williams, S.; Guillemin, F.; Hill, C.L.; et al. The global burden of hip and knee osteoarthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1323–1330. [Google Scholar] [CrossRef]
- Cui, A.; Li, H.; Wang, D.; Zhong, J.; Chen, Y.; Lu, H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine 2020, 29–30, 100587. [Google Scholar] [CrossRef]
- Davatchi, F.; Jamshidi, A.R.; Banihashemi, A.T.; Saidi, A.; Rashadmanesh, N.; Moghimi, S.; Ghafori, H.; Zandi, P.; Ahmadi, N.; Ghafori, H.; et al. WHO-ILAR COPCORD Study (Stage 1, Urban Study) in Iran. J. Rheumatol. 2008, 35, 1384. [Google Scholar]
- He, Y.; Li, Z.; Alexander, P.G.; Ocasio-Nieves, B.D.; Yocum, L.; Lin, H.; Tuan, R.S. Pathogenesis of osteoarthritis: Risk factors, regulatory pathways in chondrocytes, and experimental models. Biology 2020, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef] [PubMed]
- van den Bosch, M.H.J. Osteoarthritis year in review 2020: Biology. Osteoarthr. Cartil. 2021, 29, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.; Schönfeld, C. Novel insights into the pathogenesis of osteoarthritis. Curr. Rheumatol. Rev. 2018, 14, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yuan, S.; Zeng, Y.; Wang, C.; Yu, N.; Ding, C. New trends in pharmacological treatments for osteoarthritis. Front. Pharmacol. 2021, 12, 645842. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, J.; Gao, J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.; Zheng, M.H. Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res. Ther. 2013, 15, 223. [Google Scholar] [CrossRef]
- Donell, S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Fusco, M.; Skaper, S.D.; Coaccioli, S.; Varrassi, G.; Paladini, A. Degenerative Joint Diseases and Neuroinflammation. Pain Pract. 2017, 17, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, T.; Winter, D.R.; Miller, R.J.; Miller, R.E.; Malfait, A.M. Neuroimmune interactions and osteoarthritis pain: Focus on macrophages. Pain Rep. 2021, 6, e892. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage-bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef]
- Zhu, X.; Chan, Y.T.; Yung, P.S.H.; Tuan, R.S.; Jiang, Y. Subchondral bone remodeling: A therapeutic target for osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 607764. [Google Scholar] [CrossRef]
- Fan, X.; Wu, X.; Crawford, R.; Xiao, Y.; Prasadam, I. Macro, Micro, and Molecular. Changes of the osteochondral interface in osteoarthritis development. Front. Cell Dev. Biol. 2021, 9, 659654. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.A.; Parkes, M.J.; Hodgson, R.J.; Felson, D.T.; Arden, N.K.; O’Neill, T.W. Association between bone marrow lesions & synovitis and symptoms in symptomatic knee osteoarthritis. Osteoarthr. Cartil. 2020, 28, 316–323. [Google Scholar]
- Chen, L.; Yao, F.; Wang, T.; Li, G.; Chen, P.; Bulsara, M.; Zheng, J.J.Y.; Landao-Bassonga, E.; Firth, M.; Vasantharao, P.; et al. Horizontal fissuring at the osteochondral interface: A novel and unique pathological feature in patients with obesity-related osteoarthritis. Ann. Rheum Dis. 2020, 79, 811–888. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, T.W.; McCabe, P.S.; McBeth, J. Update on the epidemiology, risk factors and disease outcomes of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Varela-Eirin, M.; Loureiro, J.; Fonseca, E.; Corrochano, S.; Caeiro, J.R.; Collado, M.; Mayan, M.D. Cartilage regeneration and ageing: Targeting cellular plasticity in osteoarthritis. Ageing Res. Rev. 2018, 42, 56–71. [Google Scholar] [CrossRef]
- Georgiev, T.; Angelov, A.K. Modifiable risk factors in knee osteoarthritis: Treatment implications. Rheumatol. Int. 2019, 39, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Silverwood, V.; Blagojevic-Bucknall, M.; Jinks, C.; Jordan, J.L.; Protheroe, J.; Jordan, K.P. Current evidence on risk factors for knee osteoarthritis in older adults: A systematic review and meta-analysis. Osteoarthr. Cartil. 2015, 23, 507–515. [Google Scholar] [CrossRef]
- Saberi Hosnijeh, F.; Zuiderwijk, M.E.; Versteeg, M.; Smeele, H.T.; Hofman, A.; Uitterlinden, A.G.; Agricola, R.; Oei, E.H.; Waarsing, J.H.; Bierma-Zeinstra, S.M.; et al. Cam deformity and acetabular dysplasia as risk factors for hip osteoarthritis. Arthritis Rheumatol. 2017, 69, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, N.; Muraki, S.; Oka, H.; Tanaka, S.; Kawaguchi, H.; Nakamura, K.; Akune, T. Accumulation of metabolic risk factors such as overweight, hypertension, dyslipidaemia, and impaired glucose tolerance raises the risk of occurrence and progression of knee osteoarthritis: A 3-year follow-up of the ROAD study. Osteoarthr. Cartil. 2012, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Kleyer, A.; Perricone, C.; Sahinbegovic, E.; Iagnocco, A.; Zwerina, J.; Lorenzini, R.; Aschenbrenner, F.; Berenbaum, F.; D’Agostino, M.A. Diabetes is an independent predictor for severe osteoarthritis: Results from a longitudinal cohort study. Diabetes Care 2013, 36, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Nieves-Plaza, M.; Castro-Santana, L.E.; Font, Y.M.; Mayor, A.M.; Vilá, L.M. Association of hand or knee osteoarthritis with diabetes mellitus in a population of Hispanics from Puerto Rico. J. Clin. Rheumatol. 2013, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Shin, D. Association between metabolic syndrome, radiographic knee osteoarthritis, and intensity of knee pain: Results of a national survey. J. Clin. Endocrinol. Metab. 2014, 99, 3177–3183. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Allen, J.C.; Ang, L.W.; Yuan, J.M.; Koh, W.P. Diabetes mellitus and the risk of total knee replacement among Chinese in Singapore, the Singapore Chinese Health Study. Sci. Rep. 2017, 7, 40671. [Google Scholar] [CrossRef]
- Khor, A.; Ma, C.A.; Hong, C.; Hui, L.L.; Leung, Y.Y. Diabetes mellitus is not a risk factor for osteoarthritis. RMD Open 2020, 6, e001030. [Google Scholar] [CrossRef]
- van Meurs, J.B. Osteoarthritis year in review 2016: Genetics, genomics and epigenetics. Osteoarthr. Cartil. 2017, 25, 181–189. [Google Scholar] [CrossRef]
- Tachmazidou, I.; Hatzikotoulas, K.; Southam, L.; Esparza-Gordillo, J.; Haberland, V.; Zheng, J.; Johnson, T.; Koprulu, M.; Zengini, E.; Steinberg, J. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 2019, 51, 230–236. [Google Scholar] [CrossRef]
- Styrkarsdottir, U.; Lund, S.H.; Thorleifsson, G.; Zink, F.; Stefansson, O.A.; Sigurdsson, J.K.; Juliusson, K.; Bjarnadottir, K.; Sigurbjornsdottir, S.; Jonsson, S.; et al. Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat. Genet. 2018, 50, 1681–1687. [Google Scholar] [CrossRef]
- Vincent, T.L. 2021: The year we rewrite the osteoarthritis textbooks? Function 2021, 2, zqaa043. [Google Scholar] [CrossRef] [PubMed]
- Thelin, N.; Holmberg, S.; Thelin, A. Knee injuries account for the sports-related increased risk of knee osteoarthritis. Scand. J. Med. Sci. Sports 2006, 16, 329–333. [Google Scholar] [CrossRef]
- Vigdorchik, J.M.; Nepple, J.J.; Eftekhary, N.; Leunig, M.; Clohisy, J.C. What is the association of elite sporting activities with the development of hip osteoarthritis? Am. J. Sports Med. 2017, 45, 961–964. [Google Scholar] [CrossRef]
- Driban, J.B.; Hootman, J.M.; Sitler, M.R.; Harris, K.P.; Cattano, N.M. Is participation in certain sports associated with knee osteoarthritis? A systematic review. J. Athl. Train. 2017, 52, 497–506. [Google Scholar] [CrossRef]
- Qin, J.; Barbour, K.E.; Nevitt, M.C.; Helmick, C.G.; Hootman, J.M.; Murphy, L.B.; Cauley, J.A.; Dunlop, D.D. Objectively measured physical activity and risk of knee osteoarthritis. Med. Sci. Sports Exerc. 2018, 50, 277–283. [Google Scholar] [CrossRef]
- Barreto, G.; Manninen, M.; KEklund, K. Osteoarthritis and Toll-like receptors: When innate immunity meets chondrocyte apoptosis. Biology 2020, 9, 65. [Google Scholar] [CrossRef]
- Zahan, O.M.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med. Pharm. Rep. 2020, 93, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed. Pharmacother. 2020, 129, 110452. [Google Scholar] [CrossRef]
- Altay, M.A.; Ertürk, C.; Bilge, A.; Yaptı, M.; Levent, A.; Aksoy, N. Evaluation of prolidase activity and oxidative status in patients with knee osteoarthritis: Relationships with radiographic severity and clinical parameters. Rheumatol. Int. 2015, 35, 1725–1731. [Google Scholar] [CrossRef]
- Ertürk, C.; Altay, M.A.; Selek, S.; Koçyiğit, A. Paraoxonase-1 activity and oxidative status in patients with knee osteoarthritis and their relationship with radiological and clinical parameters. Scand. J. Clin. Lab. Investig. 2012, 72, 433–439. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Sharma, N.; Drobinski, P.; Kayed, A.; Chen, Z.; Kjelgaard-Petersen, C.F.; Gantzel, T.; Karsdal, M.A.; Michaelis, M.; Ladel, C.; Bay-Jensen, A.C.; et al. Inflammation and joint destruction may be linked to the generation of cartilage metabolites of ADAMTS-5 through activation of toll-like receptors. Osteoarthr. Cartil. 2020, 28, 658–668. [Google Scholar] [CrossRef]
- Lambert, C.; Zappia, J.; Sanchez, C.; Florin, A.; Dubuc, J.E.; Henrotin, Y. The damage-associated molecular patterns (DAMPs) as potential targets to treat osteoarthritis: Perspectives from a review of the literature. Front. Med. 2021, 7, 607186. [Google Scholar] [CrossRef] [PubMed]
- Culemann, S.; Grüneboom, A.; Nicolás-Ávila, J.Á.; Weidner, D.; Lämmle, K.F.; Rothe, T.; Quintana, J.A.; Kirchner, P.; Krljanac, B.; Eberhardt, M.; et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019, 572, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.N.; Furman, B.D.; Zeitlin, J.; Kimmerling, K.A.; Wu, C.L.; Guilak, F.; Olson, S.A. Intra-articular depletion of macrophages increases acute synovitis and alters macrophage polarity in the injured mouse knee. Osteoarthr. Cartil. 2020, 28, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Mei, J.; Han, X.; Li, H.; Yang, S.; Wang, M.; Chu, L.; Qiao, H.; Tang, T. Kinsenoside attenuates osteoarthritis by repolarizing macrophages through inactivating NF-κB/MAPK signaling and protecting chondrocytes. Acta Pharm. Sin. B 2019, 9, 973–985. [Google Scholar] [CrossRef]
- Zhou, F.; Mei, J.; Yang, S.; Han, X.; Li, H.; Yu, Z.; Qiao, H.; Tang, T. Modified ZIF-8 nanoparticles attenuate osteoarthritis by reprogramming the metabolic pathway of synovial macrophages. ACS Appl. Mater. Interfaces 2020, 12, 2009–2022. [Google Scholar] [CrossRef]
- Mahon, O.R.; Kelly, D.J.; McCarthy, G.M.; Dunne, A. Osteoarthritis-associated basic calcium phosphate crystals alter immune cell metabolism and promote M1 macrophage polarization. Osteoarthr. Cartil. 2020, 28, 603–612. [Google Scholar] [CrossRef]
- Kulkarni, P.; Harsulkar, A.; Märtson, A.G.; Suutre, S.; Märtson, A.; Koks, S. Mast cells differentiated in synovial fluid and resident in osteophytes exalt the inflammatory pathology of osteoarthritis. Int. J. Mol. Sci. 2022, 23, 541. [Google Scholar] [CrossRef]
- Wang, Q.; Lepus, C.M.; Raghu, H.; Reber, L.L.; Tsai, M.M.; Wong, H.H.; von Kaeppler, E.; Lingampalli, N.; Bloom, M.S.; Hu, N.; et al. IgE-mediated mast cell activation promotes inflammation and cartilage destruction in osteoarthritis. eLife 2019, 8, e39905. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, Y.; Li, X.; Deng, Z.; Zheng, M.; Zheng, Q. The immune cell landscape in different anatomical structures of knee in osteoarthritis: A gene expression-based study. Biomed. Res. Int. 2020, 2020, 9647072. [Google Scholar] [CrossRef]
- de Lange-Brokaar, B.J.; Kloppenburg, M.; Andersen, S.N.; Dorjée, A.L.; Yusuf, E.; Herb-van Toorn, L.; Kroon, H.M.; Zuurmond, A.M.; Stojanovic-Susulic, V.; Bloem, J.L.; et al. Characterization of synovial mast cells in knee osteoarthritis: Association with clinical parameters. Osteoarthr. Cartil. 2016, 24, 664–671. [Google Scholar] [CrossRef]
- Thomson, A.; Hilkens, C.M.U. Synovial Macrophages in Osteoarthritis: The Key to Understanding Pathogenesis? Front. Immunol. 2021, 12, 678757. [Google Scholar] [CrossRef] [PubMed]
- Haubruck, P.; Pinto, M.M.; Moradi, B.; Little, C.B.; Gentek, R. Monocytes, macrophages, and their potential niches in synovial joints—therapeutic targets in post-traumatic osteoarthritis? Front. Immunol. 2021, 12, 763702. [Google Scholar] [CrossRef] [PubMed]
- Arendt-Nielsen, L. Pain sensitisation in osteoarthritis. Clin. Exp. Rheumatol. 2017, 35, S68–S74. [Google Scholar]
- Arendt-Nielsen, L.; Nie, H.; Laursen, M.B.; Laursen, B.S.; Madeleine, P.; Simonsen, O.H.; Graven-Nielsen, T. Sensitization in patients with painful knee osteoarthritis. Pain 2010, 149, 573–581. [Google Scholar] [CrossRef]
- Imamura, M.; Imamura, S.T.; Kaziyama, H.H.; Targino, R.A.; Hsing, W.T.; de Souza, L.P.; Cutait, M.M.; Fregni, F.; Camanho, G.L. Impact of nervous system hyperalgesia on pain, disability, and quality of life in patients with knee osteoarthritis: A controlled analysis. Arthritis Rheum. 2008, 59, 1424–1431. [Google Scholar] [CrossRef]
- Thacker, M.A.; Clark, A.K.; Marchand, F.; McMahon, S.B. Pathophysiology of peripheral neuropathic pain: Immune cells and molecules. Anesth. Analg. 2007, 105, 838–847. [Google Scholar] [CrossRef]
- Nieto, F.R.; Clark, A.K.; Grist, J.; Chapman, V.; Malcangio, M. Calcitonin gene-related peptide-expressing sensory neurons and spinal microglial reactivity contribute to pain states in collagen-induced arthritis. Arthritis Rheumatol. 2015, 67, 1668–1677. [Google Scholar] [CrossRef]
- MacDonald, I.J.; Liu, S.-C.; Su, C.-M.; Wang, Y.H.; Tsai, C.H.; Tang, C.H. Implications of angiogenesis involvement in arthritis. Int. J. Mol. Sci. 2018, 19, 2012. [Google Scholar] [CrossRef]
- Roelofs, A.J.; Kania, K.; Rafipay, A.J.; Sambale, M.; Kuwahara, S.T.; Collins, F.L.; Smeeton, J.; Serowoky, M.A.; Rowley, L.; Wang, H.; et al. Identification of the skeletal progenitor cells forming osteophytes in osteoarthritis. Ann. Rheum. Dis. 2020, 79, 1625–1634. [Google Scholar] [CrossRef]
- Rockel, J.; Kapoor, M. The metabolome and osteoarthritis: Possible contributions to symptoms and pathology. Metabolites 2018, 8, 92. [Google Scholar] [CrossRef]
- Kizaki, K.; Uchida, S.; Yamashita, F.; Tsukamoto, M.; Azuma, K. Microstructure of osteophytes in medial knee osteoarthritis. Clin. Rheumatol. 2018, 37, 2893–2896. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Kokubun, T.; Onitsuka, K.; Oka, Y.; Kano, T.; Morishita, Y.; Ozone, K.; Kuwabara, N.; Nishimoto, J.; Isho, T.; et al. Controlling joint instability after anterior cruciate ligament transection inhibits transforming growth factor-beta-mediated osteophyte formation. Osteoarthr. Cartil. 2019, 27, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ishijima, M.; Kaneko, H.; Sadatsuki, R.; Hada, S.; Kinoshita, M.; Aoki, T.; Futami, I.; Yusup, A.; Arita, H.; et al. The MRI-detected osteophyte score is a predictor for undergoing joint replacement in patients with end-stage knee osteoarthritis. Mod. Rheumatol. 2017, 27, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Schofield, D.; Callander, E. The individual and socioeconomic impact of osteoarthritis. Nat. Rev. Rheumatol. 2014, 10, 437–441. [Google Scholar] [CrossRef]
- Kouraki, A.; Bast, T.; Ferguson, E.; Valdes, A.M. The association of socio-economic and psychological factors with limitations in day-to-day activity over 7 years in newly diagnosed osteoarthritis patients. Sci. Rep. 2022, 12, 943. [Google Scholar] [CrossRef]
- Andersson, J.K.; Hagert, E.; Brittberg, M. Cartilage injuries and posttraumatic osteoarthritis in the wrist: A review. Cartilage 2021, 13, 156S–168S. [Google Scholar] [CrossRef]
- Shahabpour, M.; Abid, W.; Van Overstraeten, L.; De Maeseneer, M. Wrist trauma: More than bones. J. Belg. Soc. Radiol. 2021, 105, 90. [Google Scholar] [CrossRef]
- Laulan, J.; Marteau, E.; Bacle, G. Wrist osteoarthritis. Orthop. Traumatol. Surg. Res. 2015, 101, S1–S9. [Google Scholar] [CrossRef]
- Giddins, G. Functional outcomes after surgery for thumb carpometacarpal joint arthritis. J. Hand Surg. Eur. Vol. 2020, 45, 64–70. [Google Scholar] [CrossRef]
- Whittaker, J.L.; Truong, L.K.; Dhiman, K.; Beck, C. Osteoarthritis year in review 2020: Rehabilitation and outcomes. Osteoarthr. Cartil. 2021, 29, 190–207. [Google Scholar] [CrossRef]
- Katz, J.N.; Arant, K.R.; Loeser, R.F. Diagnosis and treatment of hip and knee osteoarthritis: A review. JAMA 2021, 325, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Deveza, L.A.; Nelson, A.E.; Loeser, R.F. Phenotypes of osteoarthritis: Current state and future implications. Clin. Exp. Rheumatol. 2019, 37, 64–72. [Google Scholar] [PubMed]
- Cope, P.J.; Ourradi, K.; Li, Y.; Sharif, M. Models of osteoarthritis: The good, the bad and the promising. Osteoarthr. Cartil. 2019, 27, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Camilloni, A.; Nati, G.; Maggiolini, P.; Romanelli, A.; Carbone, G.; Giannarelli, D.; Terrenato, I.; De Marinis, M.G.; Rossi, A.; D’Angelo, D.; et al. Chronic non-cancer pain in primary care: An Italian cross-sectional study. Signa Vitae 2021, 17, 54–62. [Google Scholar] [CrossRef]
- Mandl, L.A. Osteoarthritis year in review 2018: Clinical. Osteoarthr. Cartil. 2019, 27, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Rekatsina, M.; Paladini, A.; Piroli, A.; Zis, P.; Pergolizzi, J.V.; Varrassi, G. Pathophysiology and therapeutic perspectives of oxidative stress and neurodegenerative diseases: A narrative review. Adv. Ther. 2020, 37, 113–139. [Google Scholar] [CrossRef]
- Rios-Arce, N.D.; Murugesh, D.; Hum, N.R.; Sebastian, A.; Jbeily, E.H.; Christiansen, B.A.; Loots, G.G. Preexisting type 1 diabetes mellitus blunts the development of posttraumatic osteoarthritis. JBMR Plus 2022, 6, e10625. [Google Scholar] [CrossRef]
- Charen, D.A.; Solomon, D.; Zubizarreta, N.; Poeran, J.; Colvin, A.C. Examining the association of knee pain with modifiable cardiometabolic risk factors. Arthritis Care Res. 2021, 73, 1777–1783. [Google Scholar] [CrossRef]
- Harasymowicz, N.S.; Azfer, A.; Burnett, R.; Simpson, H.; Salter, D.M. Chondrocytes from osteoarthritic cartilage of obese patients show altered adiponectin receptors expression and response to adiponectin. J. Orthop. Res. 2021, 39, 2333–2339. [Google Scholar] [CrossRef]
- Hurley, M.; Dickson, K.; Hallett, R.; Grant, R.; Hauari, H.; Walsh, N.; Stansfield, C.; Oliver, S. Exercise interventions and patient beliefs for people with hip, knee or hip and knee osteoarthritis: A mixed methods review. Cochrane Database Syst. Rev. 2018, 4, CD010842. [Google Scholar] [CrossRef]
- Kraus, V.B.; Sprow, K.; Powell, K.E.; Buchner, D.; Bloodgood, B.; Piercy, K.; George, S.M.; Kraus, W.E. 2018 Physical Activity Guidelines Advisory Committee* Effects of physical activity in knee and hip osteoarthritis: A systematic umbrella review. Med. Sci. Sports Exerc. 2019, 51, 1324–1339. [Google Scholar] [CrossRef] [PubMed]
- VillafaÑe, J.H.; Bishop, M.D.; Pedersini, P.; Berjano, P. Physical activity and osteoarthritis: Update and perspectives. Pain Med. 2019, 20, 1461–1463. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coaccioli, S.; Sarzi-Puttini, P.; Zis, P.; Rinonapoli, G.; Varrassi, G. Osteoarthritis: New Insight on Its Pathophysiology. J. Clin. Med. 2022, 11, 6013. https://doi.org/10.3390/jcm11206013
Coaccioli S, Sarzi-Puttini P, Zis P, Rinonapoli G, Varrassi G. Osteoarthritis: New Insight on Its Pathophysiology. Journal of Clinical Medicine. 2022; 11(20):6013. https://doi.org/10.3390/jcm11206013
Chicago/Turabian StyleCoaccioli, Stefano, Piercarlo Sarzi-Puttini, Panagiotis Zis, Giuseppe Rinonapoli, and Giustino Varrassi. 2022. "Osteoarthritis: New Insight on Its Pathophysiology" Journal of Clinical Medicine 11, no. 20: 6013. https://doi.org/10.3390/jcm11206013
APA StyleCoaccioli, S., Sarzi-Puttini, P., Zis, P., Rinonapoli, G., & Varrassi, G. (2022). Osteoarthritis: New Insight on Its Pathophysiology. Journal of Clinical Medicine, 11(20), 6013. https://doi.org/10.3390/jcm11206013