
Defining NASH from a Multi-Omics Systems Biology Perspective

 , ,
, ,
Abstract
1. Introduction
2. State-of-the-Art Proteomics, Metabolomics, and Lipidomics Technologies
2.1. MS-Based Proteomics
2.2. Proteomics Platforms beyond MS
2.3. MS-Based Metabolomics and Lipidomics
3. Proteomics-Based Biomarker Discovery Studies in Liver Disease
4. Metabolomics-Based Biomarker Discovery Studies in NASH
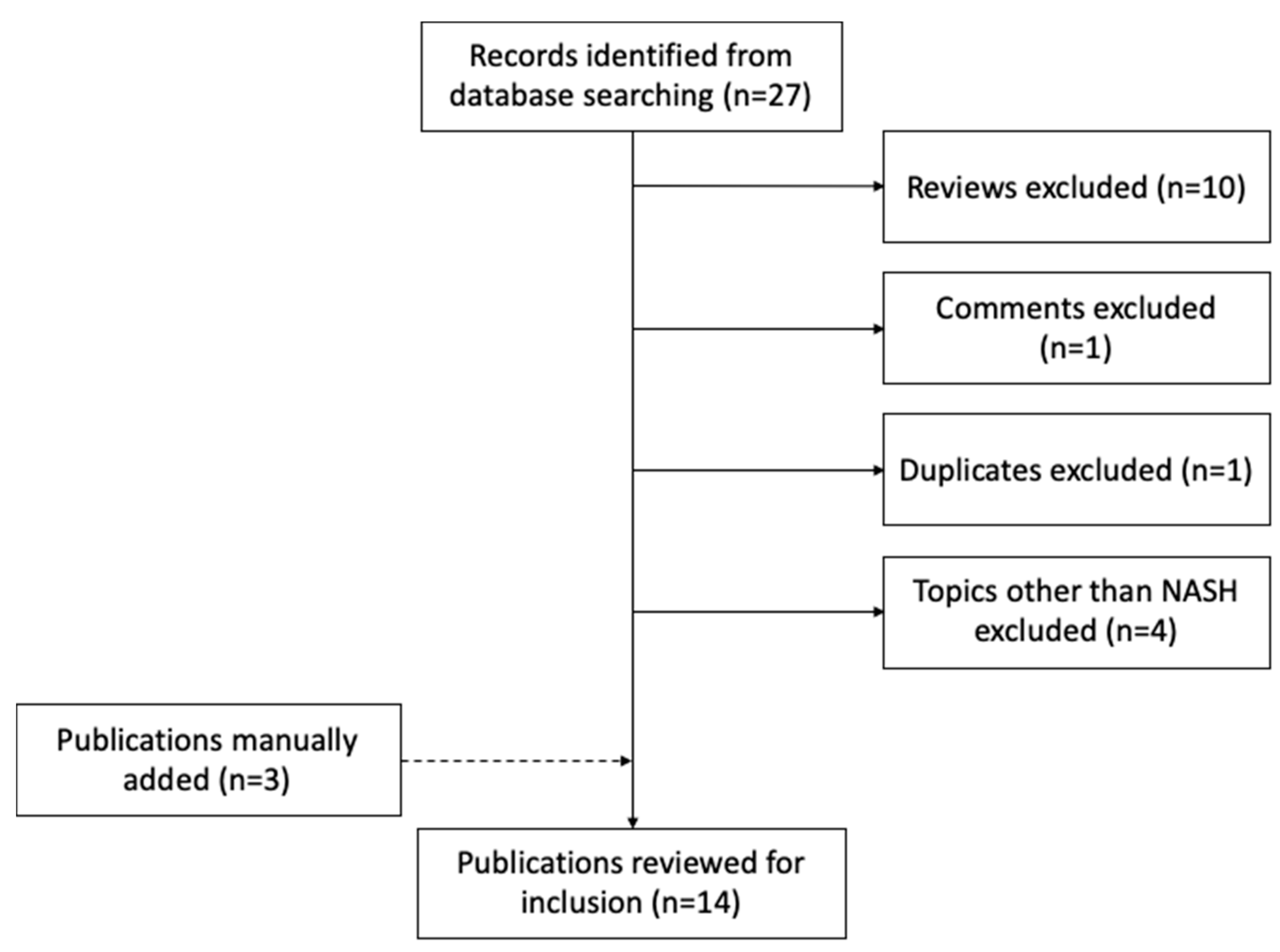
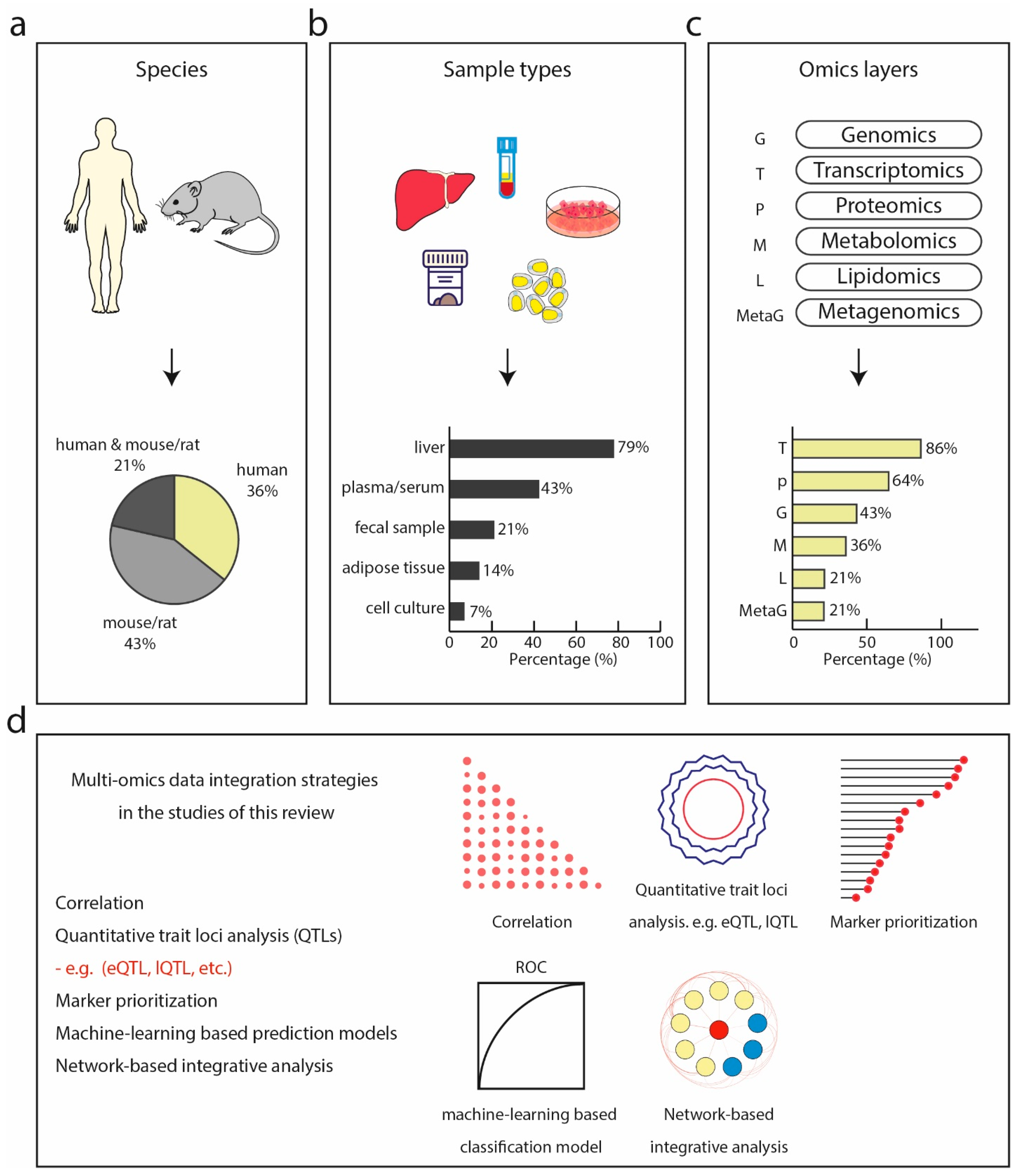
4.1. Characteristics of Studies
4.2. Overview of Data Integration Strategies
4.3. Multi-Omics Classifiers and Discriminative Disease Signatures
5. Conclusions and Prospects
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Targher, G. Increased risk of cardiovascular disease and chronic kidney disease in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 372–381. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Gentile, C.L.; Pagliassotti, M.J. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2008, 19, 567–576. [Google Scholar] [CrossRef]
- Long, M.T.; Gandhi, S.; Loomba, R. Advances in non-invasive biomarkers for the diagnosis and monitoring of non-alcoholic fatty liver disease. Metab. Clin. Exp. 2020, 111, 154259. [Google Scholar] [CrossRef]
- Lambrecht, J.; Tacke, F. Controversies and Opportunities in the Use of Inflammatory Markers for Diagnosis or Risk Prediction in Fatty Liver Disease. Front. Immunol. 2020, 11, 634409. [Google Scholar] [CrossRef]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Belardinilli, F.; Bailetti, D.; Sponziello, M.; D’Erasmo, L.; Polimeni, L.; Baratta, F.; Pastori, D.; Ceci, F.; Montali, A.; et al. Evaluation of Polygenic Determinants of Non-Alcoholic Fatty Liver Disease (NAFLD) By a Candidate Genes Resequencing Strategy. Sci. Rep. 2018, 8, 3702. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Aprile, M.; Esposito, R.; Ciccodicola, A. RNA-Seq and human complex diseases: Recent accomplishments and future perspectives. Eur. J. Hum. Genet. 2013, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef]
- Frantzi, M.; Latosinska, A.; Mischak, H. Proteomics in Drug Development: The Dawn of a New Era? Proteom. Clin. Appl 2019, 13, e1800087. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Kartsoli, S.; Kostara, C.E.; Tsimihodimos, V.; Bairaktari, E.T.; Christodoulou, D.K. Lipidomics in non-alcoholic fatty liver disease. World J. Hepatol. 2020, 12, 436–450. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Pierantonelli, I.; Torquato, P.; Marinelli, R.; Ferreri, C.; Chatgilialoglu, C.; Bartolini, D.; Galli, F. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019, 144, 293–309. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd; Speicher, S.; Griffin, P.R.; Hunkapiller, T. Peptide mass maps: A highly informative approach to protein identification. Anal. Biochem. 1993, 214, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar] [CrossRef]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Kelleher, N.L.; Lin, H.Y.; Valaskovic, G.A.; Aaserud, D.J.; Fridriksson, E.K.; McLafferty, F.W. Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry. J. Am. Chem. Soc. 1999, 121, 806–812. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sorensen, K.D.; Hoyer, S.; Orntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599.e584. [Google Scholar] [CrossRef]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Ahmad Khan, A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014, 42, D959–D965. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. MCP 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Muthusamy, B.; Hanumanthu, G.; Suresh, S.; Rekha, B.; Srinivas, D.; Karthick, L.; Vrushabendra, B.M.; Sharma, S.; Mishra, G.; Chatterjee, P. Plasma Proteome Database as a resource for proteomics research. Proteomics 2005, 5, 3531–3536. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Burgess, M.W.; Gillette, M.A.; Mertins, P.; Clauser, K.R.; Mani, D.R.; Kuhn, E.W.; Farrell, L.A.; Gerszten, R.E.; Carr, S.A. Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol. Cell. Proteom. 2015, 14, 2375–2393. [Google Scholar] [CrossRef]
- Bruderer, R.; Muntel, J.; Müller, S.; Bernhardt, O.M.; Gandhi, T.; Cominetti, O.; Macron, C.; Carayol, J.; Rinner, O.; Astrup, A.; et al. Analysis of 1508 Plasma Samples by Capillary-Flow Data-Independent Acquisition Profiles Proteomics of Weight Loss and Maintenance. Mol. Cell. Proteom. MCP 2019, 18, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Thiele, M.; Geyer, P.E.; Rasmussen, D.N.; Webel, H.E.; Santos, A.; Gupta, R.; Meier, F.; Strauss, M.; Kjaergaard, M.; et al. A paired liver biopsy and plasma proteomics study reveals circulating biomarkers for alcohol-related liver disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef] [PubMed]
- Hensley, P. SOMAmers and SOMAscan—A Protein Biomarker Discovery Platform for Rapid Analysis of Sample Collections From Bench Top to the Clinic. J. Biomol. Tech. JBT 2013, 24, S5. [Google Scholar]
- Billing, A.M.; Ben Hamidane, H.; Bhagwat, A.M.; Cotton, R.J.; Dib, S.S.; Kumar, P.; Hayat, S.; Goswami, N.; Suhre, K.; Rafii, A.; et al. Complementarity of SOMAscan to LC-MS/MS and RNA-seq for quantitative profiling of human embryonic and mesenchymal stem cells. J. Proteom. 2017, 150, 86–97. [Google Scholar] [CrossRef]
- Berggrund, M.; Ekman, D.; Gustavsson, I.; Sundfeldt, K.; Olovsson, M.; Enroth, S.; Gyllensten, U. Protein Detection Using the Multiplexed Proximity Extension Assay (PEA) from Plasma and Vaginal Fluid Applied to the Indicating FTA Elute Micro Card™. J. Circ. Biomark. 2016, 5, 9. [Google Scholar] [CrossRef]
- Petrera, A.; von Toerne, C.; Behler, J.; Huth, C.; Thorand, B.; Hilgendorff, A.; Hauck, S.M. Multiplatform Approach for Plasma Proteomics: Complementarity of Olink Proximity Extension Assay Technology to Mass Spectrometry-Based Protein Profiling. J. Proteome Res. 2021, 20, 751–762. [Google Scholar] [CrossRef]
- Finkernagel, F.; Reinartz, S.; Schuldner, M.; Malz, A.; Jansen, J.M.; Wagner, U.; Worzfeld, T.; Graumann, J.; von Strandmann, E.P.; Müller, R. Dual-platform affinity proteomics identifies links between the recurrence of ovarian carcinoma and proteins released into the tumor microenvironment. Theranostics 2019, 9, 6601–6617. [Google Scholar]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Züllig, T.; Trötzmüller, M.; Köfeler, H.C. Lipidomics from sample preparation to data analysis: A primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef]
- Wenk, M.R. Lipidomics: New tools and applications. Cell 2010, 143, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Han, X. Lipidomics for studying metabolism. Nat. Rev. Endocrinol. 2016, 12, 668–679. [Google Scholar] [CrossRef]
- Pradas, I.; Huynh, K.; Cabré, R.; Ayala, V.; Meikle, P.J.; Jové, M.; Pamplona, R. Lipidomics Reveals a Tissue-Specific Fingerprint. Front. Physiol. 2018, 9, 1165. [Google Scholar] [CrossRef]
- Molinaro, A.; Wahlström, A.; Marschall, H.U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. TEM 2018, 29, 31–41. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients with Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.e211. [Google Scholar] [CrossRef]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients with Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Rampler, E.; Abiead, Y.E.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent Topics in Mass Spectrometry-Based Metabolomics and Lipidomics—Standardization, Coverage, and Throughput. Anal. Chem. 2021, 93, 519–545. [Google Scholar] [CrossRef]
- Dunn, W.B.; Bailey, N.J.C.; Johnson, H.E. Measuring the metabolome: Current analytical technologies. Analyst 2005, 130, 606–625. [Google Scholar] [CrossRef]
- Domenick, T.M.; Gill, E.L.; Vedam-Mai, V.; Yost, R.A. Mass Spectrometry-Based Cellular Metabolomics: Current Approaches, Applications, and Future Directions. Anal. Chem. 2021, 93, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current analytical platforms and methodologies. TrAC Trends Anal. Chem. 2005, 24, 285–294. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- García-Cañaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. A comprehensive untargeted metabonomic analysis of human steatotic liver tissue by RP and HILIC chromatography coupled to mass spectrometry reveals important metabolic alterations. J. Proteome Res. 2011, 10, 4825–4834. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab Dis 2018, 41, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Huan, T.; Forsberg, E.M.; Rinehart, D.; Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Fang, M.; Aisporna, A.; Hilmers, B.; Poole, F.L.; et al. Systems biology guided by XCMS Online metabolomics. Nat. Methods 2017, 14, 461–462. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Vasilopoulou, C.G.; Sulek, K.; Brunner, A.-D.; Meitei, N.S.; Schweiger-Hufnagel, U.; Meyer, S.W.; Barsch, A.; Mann, M.; Meier, F. Trapped ion mobility spectrometry and PASEF enable in-depth lipidomics from minimal sample amounts. Nat. Commun. 2020, 11, 331. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Nothias, L.-F.; Fleischauer, M.; Reher, R.; Ludwig, M.; Hoffmann, M.A.; Petras, D.; Gerwick, W.H.; Rousu, J.; Dorrestein, P.C.; et al. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nat. Biotechnol. 2021, 39, 462–471. [Google Scholar] [CrossRef]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Löfgren, L.; Forsberg, G.B.; Ståhlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Sci. Rep. 2016, 6, 27688. [Google Scholar] [CrossRef]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef]
- Holčapek, M.; Liebisch, G.; Ekroos, K. Lipidomic Analysis. Anal. Chem. 2018, 90, 4249–4257. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Smith, A.J.; Astarita, G. Ion mobility mass spectrometry in the omics era: Challenges and opportunities for metabolomics and lipidomics. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Arita, M. The importance of bioinformatics for connecting data-driven lipidomics and biological insights. Biochim. Et Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 762–765. [Google Scholar] [CrossRef]
- Van Meer, G.; De Kroon, A.I.P.M. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef]
- Doll, S.; Dreßen, M.; Geyer, P.E.; Itzhak, D.N.; Braun, C.; Doppler, S.A.; Meier, F.; Deutsch, M.-A.; Lahm, H.; Lange, R.; et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017, 8, 1469. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Telu, K.H.; Yan, X.; Wallace, W.E.; Stein, S.E.; Simón-Manso, Y. Analysis of human plasma metabolites across different liquid chromatography/mass spectrometry platforms: Cross-platform transferable chemical signatures. Rapid Commun. Mass Spectrom. RCM 2016, 30, 581–593. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, S.H.; Kim, J.H.; Hwang, S.; Yoo, H.J. Understanding Metabolomics in Biomedical Research. Endocrinol. Metab. 2016, 31, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef]
- Thompson, J.W.; Adams, K.J.; Adamski, J.; Asad, Y.; Borts, D.; Bowden, J.A.; Byram, G.; Dang, V.; Dunn, W.B.; Fernandez, F.; et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal. Chem. 2019, 91, 14407–14416. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Geyer, P.E.; Mann, M. Proteomics in the Study of Liver Diseases. In The Human Gut-Liver-Axis in Health and Disease; Krag, A., Hansen, T., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 165–193. [Google Scholar] [CrossRef]
- Ladaru, A.; Balanescu, P.; Stan, M.; Codreanu, I.; Anca, I.A. Candidate proteomic biomarkers for non-alcoholic fatty liver disease (steatosis and non-alcoholic steatohepatitis) discovered with mass-spectrometry: A systematic review. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2016, 21, 102–114. [Google Scholar] [CrossRef]
- Atabaki-Pasdar, N.; Ohlsson, M.; Viñuela, A.; Frau, F.; Pomares-Millan, H.; Haid, M.; Jones, A.G.; Thomas, E.L.; Koivula, R.W.; Kurbasic, A.; et al. Predicting and elucidating the etiology of fatty liver disease: A machine learning modeling and validation study in the IMI DIRECT cohorts. PLoS Med. 2020, 17, e1003149. [Google Scholar] [CrossRef]
- Wood, G.C.; Chu, X.; Argyropoulos, G.; Benotti, P.; Rolston, D.; Mirshahi, T.; Petrick, A.; Gabrielson, J.; Carey, D.J.; DiStefano, J.K.; et al. A multi-component classifier for nonalcoholic fatty liver disease (NAFLD) based on genomic, proteomic, and phenomic data domains. Sci. Rep. 2017, 7, 43238. [Google Scholar] [CrossRef]
- Luo, Y.; Wadhawan, S.; Greenfield, A.; Decato, B.E.; Oseini, A.M.; Collen, R.; Shevell, D.E.; Thompson, J.; Jarai, G.; Charles, E.D.; et al. SOMAscan Proteomics Identifies Serum Biomarkers Associated with Liver Fibrosis in Patients With NASH. Hepatol. Commun. 2021, 5, 760–773. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Janech, M.G.; Sobolesky, P.M.; Bland, A.M.; Samsuddin, S.; Alazawi, W.; Syn, W.K. Proteomic screening of plasma identifies potential noninvasive biomarkers associated with significant/advanced fibrosis in patients with nonalcoholic fatty liver disease. Biosci. Rep. 2020, 40, BSR20190395. [Google Scholar] [CrossRef] [PubMed]
- Veyel, D.; Wenger, K.; Broermann, A.; Bretschneider, T.; Luippold, A.H.; Krawczyk, B.; Rist, W.; Simon, E. Biomarker discovery for chronic liver diseases by multi-omics—A preclinical case study. Sci. Rep. 2020, 10, 1314. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24.e14. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Systems biology elucidates common pathogenic mechanisms between nonalcoholic and alcoholic-fatty liver disease. PLoS ONE 2013, 8, e58895. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Nidhina Haridas, P.A.; Dwivedi, O.P.; Hyötyläinen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136. [Google Scholar] [CrossRef]
- Zhong, G.; Kirkwood, J.; Won, K.J.; Tjota, N.; Jeong, H.; Isoherranen, N. Characterization of Vitamin A Metabolome in Human Livers with and Without Nonalcoholic Fatty Liver Disease. J. Pharmacol. Exp. Ther. 2019, 370, 92–103. [Google Scholar] [CrossRef]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Lavín Trueba, J.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461.e1447. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Ajmera, V.H.; Puri, P.; Hsu, C.L.; Bassirian, S.; Mgdsyan, M.; Singh, S.; Faulkner, C.; Valasek, M.A.; Rizo, E.; et al. Serum metabolites detect the presence of advanced fibrosis in derivation and validation cohorts of patients with non-alcoholic fatty liver disease. Gut 2019, 68, 1884–1892. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Liu, L.; Guo, F.; Li, S.; Huang, L.; Sun, C.; Feng, R. Serum metabonomics of NAFLD plus T2DM based on liquid chromatography–mass spectrometry. Clin. Biochem. 2016, 49, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Mayo, R.; Crespo, J.; Martínez-Arranz, I.; Banales, J.M.; Arias, M.; Mincholé, I.; Aller de la Fuente, R.; Jimenez-Agüero, R.; Alonso, C.; de Luis, D.A.; et al. Metabolomic-based noninvasive serum test to diagnose nonalcoholic steatohepatitis: Results from discovery and validation cohorts. Hepatol. Commun. 2018, 2, 807–820. [Google Scholar] [CrossRef]
- Sansone, S.-A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; Morrison, N.; et al. The Metabolomics Standards Initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [CrossRef]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Perakakis, N.; Polyzos, S.A.; Yazdani, A.; Sala-Vila, A.; Kountouras, J.; Anastasilakis, A.D.; Mantzoros, C.S. Non-invasive diagnosis of non-alcoholic steatohepatitis and fibrosis with the use of omics and supervised learning: A proof of concept study. Metab. Clin. Exp. 2019, 101, 154005. [Google Scholar] [CrossRef]
- Khusial, R.D.; Cioffi, C.E.; Caltharp, S.A.; Krasinskas, A.M.; Alazraki, A.; Knight-Scott, J.; Cleeton, R.; Castillo-Leon, E.; Jones, D.P.; Pierpont, B.; et al. Development of a Plasma Screening Panel for Pediatric Nonalcoholic Fatty Liver Disease Using Metabolomics. Hepatol. Commun. 2019, 3, 1311–1321. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Scian, R.; Fernández Gianotti, T.; Dopazo, H.; Rohr, C.; Gaj, G.; San Martino, J.; Sevic, I.; Flichman, D.; et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. Am. J. Clin. Nutr. 2016, 103, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Banton, S.; Tran, V.T.; Konomi, J.V.; Li, S.; Jones, D.P.; Vos, M.B. Amino Acid Metabolism is Altered in Adolescents with Nonalcoholic Fatty Liver Disease-An Untargeted, High Resolution Metabolomics Study. J. Pediatrics 2016, 172, 14–19.e15. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Liu, X.; Zhou, K.; He, X.; Lu, C.; He, B.; Niu, X.; Xiao, C.; Xu, G.; Bian, Z.; et al. The Potential Biomarkers to Identify the Development of Steatosis in Hyperuricemia. PLoS ONE 2016, 11, e0149043. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.; Eder, S.K.; Felder, T.K.; Kedenko, L.; Paulweber, B.; Stadlmayr, A.; Huber-Schönauer, U.; Niederseer, D.; Stickel, F.; Auer, S.; et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects with Non-alcoholic Fatty Liver. Am. J. Gastroenterol 2017, 112, 102–110. [Google Scholar] [CrossRef]
- Zhou, Y.; Orešič, M.; Leivonen, M.; Gopalacharyulu, P.; Hyysalo, J.; Arola, J.; Verrijken, A.; Francque, S.; Van Gaal, L.; Hyötyläinen, T.; et al. Noninvasive Detection of Nonalcoholic Steatohepatitis Using Clinical Markers and Circulating Levels of Lipids and Metabolites. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2016, 14, 1463–1472.e1466. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallée, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zhan, Z.Y.; Cao, H.Y.; Wu, C.; Bian, Y.Q.; Li, J.Y.; Cheng, G.H.; Liu, P.; Sun, M.Y. Urinary metabolomics analysis identifies key biomarkers of different stages of nonalcoholic fatty liver disease. World J. Gastroenterol. WJG 2017, 23, 2771–2784. [Google Scholar] [CrossRef] [PubMed]
- Troisi, J.; Pierri, L.; Landolfi, A.; Marciano, F.; Bisogno, A.; Belmonte, F.; Palladino, C.; Guercio Nuzio, S.; Campiglia, P.; Vajro, P. Urinary Metabolomics in Pediatric Obesity and NAFLD Identifies Metabolic Pathways/Metabolites Related to Dietary Habits and Gut-Liver Axis Perturbations. Nutrients 2017, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Notarnicola, M.; Caruso, M.G.; Tutino, V.; Bonfiglio, C.; Cozzolongo, R.; Giannuzzi, V.; De Nunzio, V.; De Leonardis, G.; Abbrescia, D.I.; Franco, I.; et al. Significant decrease of saturation index in erythrocytes membrane from subjects with non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. 2017, 16, 160. [Google Scholar] [CrossRef]
- Qi, S.; Xu, D.; Li, Q.; Xie, N.; Xia, J.; Huo, Q.; Li, P.; Chen, Q.; Huang, S. Metabonomics screening of serum identifies pyroglutamate as a diagnostic biomarker for nonalcoholic steatohepatitis. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 473, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.; Bullò, M.; Tinahones, F.J.; Martínez-González, M.; Corella, D.; Fragkiadakis, G.A.; López-Miranda, J.; Estruch, R.; Fitó, M.; Salas-Salvadó, J. Serum metabolites in non-alcoholic fatty-liver disease development or reversion; a targeted metabolomic approach within the PREDIMED trial. Nutr. Metab. 2017, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.-X.; Hu, C.-X.; Sun, W.-L.; Pan, Q.; Shen, F.; Yang, Z.; Su, Q.; Xu, G.-W.; Fan, J.-G. Serum Monounsaturated Triacylglycerol Predicts Steatohepatitis in Patients with Non-alcoholic Fatty Liver Disease and Chronic Hepatitis B. Sci. Rep. 2017, 7, 10517. [Google Scholar] [CrossRef]
- Hu, X.-Y.; Li, Y.; Li, L.-Q.; Zheng, Y.; Lv, J.-H.; Huang, S.-C.; Zhang, W.; Liu, L.; Zhao, L.; Liu, Z.; et al. Risk factors and biomarkers of non-alcoholic fatty liver disease: An observational cross-sectional population survey. BMJ Open 2018, 8, e019974. [Google Scholar] [CrossRef]
- Tiwari-Heckler, S.; Gan-Schreier, H.; Stremmel, W.; Chamulitrat, W.; Pathil, A. Circulating Phospholipid Patterns in NAFLD Patients Associated with a Combination of Metabolic Risk Factors. Nutrients 2018, 10, 649. [Google Scholar] [CrossRef]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- de Mello, V.D.; Sehgal, R.; Männistö, V.; Klåvus, A.; Nilsson, E.; Perfilyev, A.; Kaminska, D.; Miao, Z.; Pajukanta, P.; Ling, C.; et al. Serum aromatic and branched-chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int. 2020, 41, 754–763. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Räsänen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietiläinen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e555. [Google Scholar] [CrossRef] [PubMed]
- Ægidius, H.M.; Veidal, S.S.; Feigh, M.; Hallenborg, P.; Puglia, M.; Pers, T.H.; Vrang, N.; Jelsing, J.; Kornum, B.R.; Blagoev, B.; et al. Multi-omics characterization of a diet-induced obese model of non-alcoholic steatohepatitis. Sci. Rep. 2020, 10, 1148. [Google Scholar] [CrossRef] [PubMed]
- Wruck, W.; Kashofer, K.; Rehman, S.; Daskalaki, A.; Berg, D.; Gralka, E.; Jozefczuk, J.; Drews, K.; Pandey, V.; Regenbrecht, C.; et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci. Data 2015, 2, 150068. [Google Scholar] [CrossRef]
- Mesnage, R.; Biserni, M.; Balu, S.; Frainay, C.; Poupin, N.; Jourdan, F.; Wozniak, E.; Xenakis, T.; Mein, C.A.; Antoniou, M.N. Integrated transcriptomics and metabolomics reveal signatures of lipid metabolism dysregulation in HepaRG liver cells exposed to PCB 126. Arch. Toxicol 2018, 92, 2533–2547. [Google Scholar] [CrossRef]
- Mesnage, R.; Renney, G.; Séralini, G.E.; Ward, M.; Antoniou, M.N. Multiomics reveal non-alcoholic fatty liver disease in rats following chronic exposure to an ultra-low dose of Roundup herbicide. Sci. Rep. 2017, 7, 39328. [Google Scholar] [CrossRef]
- Qian, M.; Hu, H.; Yao, Y.; Zhao, D.; Wang, S.; Pan, C.; Duan, X.; Gao, Y.; Liu, J.; Zhang, Y.; et al. Coordinated changes of gut microbiome and lipidome differentiates nonalcoholic steatohepatitis (NASH) from isolated steatosis. Liver Int. 2020, 40, 622–637. [Google Scholar] [CrossRef]
- Jha, P.; McDevitt, M.T.; Gupta, R.; Quiros, P.M.; Williams, E.G.; Gariani, K.; Sleiman, M.B.; Diserens, L.; Jochem, A.; Ulbrich, A.; et al. Systems Analyses Reveal Physiological Roles and Genetic Regulators of Liver Lipid Species. Cell Syst. 2018, 6, 722–733.e726. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Liu, Z.; Klevstig, M.; Mukhopadhyay, B.; Bergentall, M.; Cinar, R.; Ståhlman, M.; Sikanic, N.; Park, J.K.; et al. Network analyses identify liver-specific targets for treating liver diseases. Mol. Syst. Biol. 2017, 13, 938. [Google Scholar] [CrossRef]
- Chella Krishnan, K.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S.; et al. Integration of Multi-omics Data from Mouse Diversity Panel Highlights Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 103–115.e107. [Google Scholar] [CrossRef]
- Kurt, Z.; Barrere-Cain, R.; LaGuardia, J.; Mehrabian, M.; Pan, C.; Hui, S.T.; Norheim, F.; Zhou, Z.; Hasin, Y.; Lusis, A.J.; et al. Tissue-specific pathways and networks underlying sexual dimorphism in non-alcoholic fatty liver disease. Biol. Sex Differ. 2018, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.-Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e645. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Graw, S.; Chappell, K.; Washam, C.L.; Gies, A.; Bird, J.; Robeson, M.S., 2nd; Byrum, S.D. Multi-omics data integration considerations and study design for biological systems and disease. Mol. Omics 2021, 17, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Artrith, N.; Butler, K.T.; Coudert, F.-X.; Han, S.; Isayev, O.; Jain, A.; Walsh, A. Best practices in machine learning for chemistry. Nat. Chem. 2021, 13, 505–508. [Google Scholar] [CrossRef]
- Alexandrov, T. Spatial Metabolomics and Imaging Mass Spectrometry in the Age of Artificial Intelligence. Annu. Rev. Biomed. Data Sci. 2020, 3, 61–87. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Aim of the Analysis | Proteomics | Proteomics | Proteomics | Metabolomics | Lipidomics |
|---|---|---|---|---|---|
| Platform | Mass spectrometry | Proximity Extension Assay (Olink) | Aptamer-based platform (SomaScan) | Mass spectrometry | Mass spectrometry |
| Analytes | Proteins | Proteins | Proteins | Polar small molecules (below 1500 Da), including carbohydrates, ketones, aminoacids andbiogenic amides | Apolar molecules, including glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, polyketides, sterol and prenol lipids [47] |
| Analyte separation | Liquid chromatography | No | No | Liquid chromatography | Liquid chromatography (no for direct infusion) |
| Analysis of body fluids (e.g., plasma, serum, urine, saliva) | Yes | Yes | Yes | Yes | Yes |
| Analysis of tissue/cell culture | Yes | Not optimal | Not optimal | Yes | Yes |
| Analysis of proteoforms (PTMs, isoforms, coding-variants) | Yes | No | No | Not applicable | Not applicable |
| Analyte multi-plexing | Varies depending on the workflow and specimen, up to >10,000 proteins [29,81] (300–5000 in plasma) [33,35,36] | 1472 proteins (targeted panels of 48–384 proteins) a | 7000 proteins b | Varies depending on the workflow and specimen [82] (expected up to 10% of molecular features identification) [83] | Varies depending on the workflow and specimen [82], currently up to 1108 lipids in plasma [64] |
| Dynamic range | High-medium abundance in case of plasma | (pg/mL to µg/mL) c | (femtomolar to micromolar) d | (picomolar to milimolar) [42,84] | (picomolar to nanomolar) [85] |
| Reproducibility | Medium | Medium to High (Intra-assay CV 10–12% for the 384-protein panels) e | High (Median intra-assay CV 4.6% for the 1300-protein platform) f | Medium to high [83,86] | Medium to high [86] |
| Throughput (per day per system) | Low to medium (10 s–100 s) | High (100 s) | High (100 s) | Medium to high (10 s–100 s) | Medium to high (10 s–100 s) |
| Quantification | Relative or absolute for targeted assays | Relative or absolute for targeted assays | Relative | Relative or absolute for targeted assays | Relative or absolute for targeted assays |
| Study Number | Study Aim | Sample Type | Data Release | Omics Type | Analytical Method | Metabolic Alterations | Reference |
|---|---|---|---|---|---|---|---|
| 1 | D | liver | NA | UL | LC-MS | TG, FA, CER | (Luukkonen et al., 2016) [107] |
| 2 | D | serum | NA | TM | LC-MS | AA, TCA | (Sookoian et al., 2016) [108] |
| 3 | D | plasma | NA | UM | LC-MS | AA | (Jin et al., 2016) [109] |
| 4 | D | serum | NA | UM | LC-MS | PL, PN, PTA, CE, I | (Tan et al., 2016) [110] |
| 5 | DV | serum | NA | UM | LC-MS | AA, OA, B, CR, PC, lyso-PC | (Chen et al., 2016) [101] |
| 6 | D | serum | NA | TM, TL | LC-MS | Lyso-PC, PC, AA, SM | (Feldman et al., 2017) [111] |
| 7 | DV | plasma | NA | UM, UL | LC-MS, GC-MS | AA, lyso-PC, PE | (Zhou et al.,, 2016) [112] |
| 8 | D | serum, liver | NA | UL | LC-MS, GC-MS | FA, TG, PC | (Luukkonen et al., 2017) [97] |
| 9 | D | serum | NA | UM, UL | LC-MS | TG, DG, FA, CER | (Alonso et al., 2017) [99] |
| 10 | D | liver | NA | UL | LC-MS | PS, TG, CER, PE, PC, PI, SM, CE, DG, FA | (Chiappini et al., 2017) [113] |
| 11 | D | urine | NA | UM | LC-MS | NAC, AA | (Dong et al., 2017) [114] |
| 12 | D | urine | NA | UM | GC-MS | G; AAD; X | (Troisi et al., 2017) [115] |
| 13 | D | RBC | UR | TL | GC-MS | FA | (Notarnicola et al., 2017) [116] |
| 14 | D | serum | NA | UM | LC-MS | AAD | (Qi et al., 2017) [117] |
| 15 | D | serum | NA | TM, TL | LC-MS | AA, CE, SM, CER, GPC | (Papandreou et al., 2017) [118] |
| 16 | D | serum | NA | UL | LC-MS | TG | (Yang et al., 2017) [119] |
| 17 | D | serum | NA | TL | LC-MS | NS | (Hu et al., 2018) [120] |
| 18 | DV | serum | NA | UM, UL | LC-MS | TG | (Mayo et al., 2018) [102] |
| 19 | D | serum | NA | UL | LC-MS | PC, SM | (Tiwari-Heckler et al., 2018) [121] |
| 20 | D | plasma, liver | NA | UL | LC-MS | PC, CL, CoQ, ACR | (Peng et al., 2018) [122] |
| 21 | DV | serum | NA | UM | LC-MS, GC-MS | AA, PT, FA, BA, ST | (Caussy et al., 2018) [100] |
| 22 | D | serum | NA | UM | LC-MS | AA, PC, UR | (de Mello et al., 2020) [123] |
| 23 | D | plasma | NA | UM | LC-MS | AA, lyso-PC | (Khusial et al., 2019) [106] |
| 24 | D | liver | NA | TM | LC-MS | RPD | (Zhong et al., 2019) [98] |
| 25 | D | serum | NA | UL, TM | LC-MS, GC-MS | DG, PC, PG, SM, PE, FA, GL | (Perakakis et al., 2019) [105] |
| Platform | Sample Type | Number of Analytes Quantified in Total | Sample Size (Discovery Cohort) | Sample Size (Validation Cohort) | Classifier | Prediction Target | Markers | AUROC | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Omics | Serum | 1129 proteins (SomaScan), 1 genotype, >200 clinical variables | n = 443 | n = 133 | Logistic regression | Hepatic steatosis in obesity | 8 proteins + 1 genotype + 12 clinical variables: ACY1, SHBG, CTSZ, MET, GSN, LGALS3BP, CHL1, SERPINC1, PNPLA3 variant. | 0.935 (0.914 in validation cohort) | (Wood et al., 2017) [90] |
| Omics | Serum | 860 proteins, 288 metabolites, 108 SNPs, 16,209 protein-coding genes, 58 clinical variables | n = 1049 | No for the omics model | Random forest | Fatty liver | 185 clinical and omics features | 0.84 | (Atabaki-Pasdar et al., 2020) [89] |
| SOMAscan proteomics | serum | 1305 proteins | n = 113 | n = 71, n = 32 | Elastic-Net | Fibrosis F0–1 against F2–4 | serum amyloid P, fibrinogen, olfactomedin, and SHBG | 0.74 (0.52–0.78 in validation cohorts) | (Luo et al., 2021) [91] |
| SOMAscan proteomics | serum | 1305 proteins | n = 113 | n = 71, n = 32 | Elastic-Net | Fibrosis (F3–4 against F0–2) | latent transforming growth factor beta binding protein 4, IGF-1, vascular cell adhesion molecule 1, interleukin-1 soluble receptor type-1, IL18BP, thrombospondin-2, collectin kidney 1, SHBG, interleukin-27 receptor subunit alpha, leukemia inhibitory factor receptor, soluble, fibulin-3, and plexin-B2 | 0.83 (0.74–0.78 in validation cohorts) | (Luo et al., 2021) [91] |
| MS-based proteomics | Plasma | 235–277 proteins | n = 19 | NA | Unclear | Fibrosis F2–4 against F0–1 | Complement component C7, α-2-macroglobulin, Fibulin-1, Complement component C8 γ chain; α-1-antichymotrypsin | 0.79–1 for each individual protein | (Hou et al., 2020) [93] |
| Metabolomics | serum | 365 lipids, 61 glycans and 23 fatty acids | n = 31 | NA | support vector machine | Fibrosis F2–4 against F0–1 | 10 lipids: DG(36:3), LPC(18:0), PC(36:2), PC(37:2), PC(40:5), TG(38:0), TG(50:0), TG(51:1), TG(57:1), TG(60:2) | 1 | (Perakakis et al., 2019) [105] |
| Metabolomics | Serum | 365 lipids, 61 glycans and 23 fatty acids | n = 80 | NA | Support vector machine | NASH vs. NAFL vs. Healthy | 29 lipids: AcCa(10:0), Cer(d34:2), DG(34:1), DG(36:4), LPC(20:0e), LPC(22:5), LPE(16:0), PC(32:0), PC(32:1e), PC(34:0), PC(34:2e), PC(35:3), PC(36:4), PC(36:5e), PC(37:2), PC(40:6e), PC(40:7), PC(40:8), PC(42:6), PE(38:1), PE(38:6), PI(36:1), SM(d32:0), SM(d32:2), SM(d40:1), TG(38:0), TG(38:2), TG(43:1), TG(53:5) | 0.94–0.99 (one vs. rest) | (Perakakis et al., 2019) [105]. |
| Metabolomics | Plasma | 13,008 metabolic features | n = 559 | NA | Random forest | NAFLD vs. non-NAFLD | 11 metabolite features + 3 clinical variables: serine, leucine/isoleucine, tryptophan, three putatively annotated compounds, two unknowns, lysoPE(20:0), lysoPC(18:1), WC, WBISI, and triglycerides | 0.94 | (Khusial et al., 2019) [106] |
| Metabolomics | Serum | 652 metabolites | n = 156 | n = 142 | Logistic regression | Fibrosis F3–4 vs. F0–2 in NAFLD | 8 lipids + 1 amino acid + 1 carbohydrate: 5alpha-androstan-3beta monosulfate, pregnanediol-3-glucuronide, androsterone sulfate, epiandrosterone sulfate, palmitoleate, dehydroisoandrosterone sulfate, 5alpha-androstan-3beta disulfate, glycocholate, taurine, fucose | 0.94 (0.84–0.94 in validation cohort) | (Caussy et al., 2019) [100] |
| Metabolomics | Serum | 540 lipids and amino acids | n = 467 | n = 192 | Logistic regression | NAFLD vs. Healthy | 11 triglycerides | 0.9 (0.88 in validation cohort) | (Mayo et al., 2018) [102] |
| Metabolomics | Serum | 540 lipids and amino acids | n = 467 | n = 192 | Logistic regression | NASH against NAFL | 20 triglycerides | 0.95 (0.79 in validation cohort) | (Mayo et al., 2018) [102] |
| Metabolomics | Serum | Sphingolipids and branched fatty acid esters of hydroxy fatty acids | n = 1479 | NA | Logistic regression | oleic acid-hydroxy oleic acid | 0.61 | (Hu et al., 2018) [120] | |
| Metabolomics | Serum | 1761 metabolic features | n = 59 | NA | Unclear | NASH against NAFL | pyroglutamate | 0.846 | (Qi et al., 2017) [117] |
| Metabolomics | Urine | Unclear | n = 78 | NA | Unclear | NASH against NAFL | Pyroglutamic acid | 0.65 | (Dong et al., 2017) [114] |
| Metabolomics | Serum | Unclear | n = 223 | n = 95 | Logistic regression | NASH against non-NASH | glutamate, isoleucine, glycine, lysophosphatidylcholine 16:0, phosphoethanolamine 40:6, AST, and fasting insulin | 0.882 (0.856 in validation cohort) | (Zhou et al., 2016) [112] |
| Lipidomics | Serum | 239 lipids | n = 42 | n = 22 | Logistic regression | NASH in NAFLD | Monounsaturated triglycerol | 0.83 in both discovery and validation cohorts | (Yang et al., 2017) [119] |
| Study Aim | Sample Type | Species | New Data | Data Release | Sample Size | Omics Integration | G | T | P | M | L | MetaG | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | Liver, serum, fecal samples | Human | Y | Y | n = 10, 7 | Data | Y | Y | Y | Y | Y | (Mardinoglu et al., 2018) [124] | |
| C | liver | Mice | Y | n = 9 for TP, n = 6 for scRNAseq | Data and results | Y | Y | (Ægidius et al., 2020) [125] | |||||
| C | Liver, serum | Human | Y | Y | n = 18 for plasma, n = 9 for liver T | Y | Y | Y | (Wruck et al., 2015) [126] | ||||
| C | Cell culture | Human | Y | n = 20 | Results | Y | Y | (Mesnage et al., 2018) [127] | |||||
| C | Liver | Rat | Y | n = 10, 10 | Results | Y | Y | (Mesnage et al., 2017) [128] | |||||
| C | Liver, plasma, feces | Mice, human | Y | Y | n = 10 in mice, n = 14 in patients | Data | Y | Y | Y | (Qian et al., 2020) [129] | |||
| B | Serum | Human | Y | n = 795 for T2D, n = 2234 for high risk of T2D | Data | Y | Y | Y | Y | Y | (Atabaki-Pasdar et al., 2020) [89] | ||
| B | Liver, plasma | Human | Y | n = 576 | Data | Y | Y | (Wood et al., 2017) [90] | |||||
| B | Liver | Mice | Y | Y | n = 48 for liver, n = 16 for plasma | Data and results | Y | Y | (Veyel et al., 2020) [94] | ||||
| C | Liver | Mice | Y | Y | n = 385 | Data | Y | Y | Y | (Jha et al., 2018) [130] | |||
| C | Liver | Human, mice | Data | Y | Y | (Lee et al., 2017) [131] | |||||||
| C | Liver, adipose tissue | Mice | Y | Y | n = 228 from 113 mouse strains | Data | Y | Y | (Krishnan et al., 2018) [132] | ||||
| C | Liver, adipose tissue | Mice | Data | Y | Y | (Kurt et al., 2018) [133] | |||||||
| C | Liver | Human, mice | Y | Y | n = 144 in human, n = 6 in mice | Data and results | Y | Y | (Xiong et al., 2019) [134] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, L.; Sulek, K.; Vasilopoulou, C.G.; Santos, A.; Wewer Albrechtsen, N.J.; Rasmussen, S.; Meier, F.; Mann, M. Defining NASH from a Multi-Omics Systems Biology Perspective. J. Clin. Med. 2021, 10, 4673. https://doi.org/10.3390/jcm10204673
Niu L, Sulek K, Vasilopoulou CG, Santos A, Wewer Albrechtsen NJ, Rasmussen S, Meier F, Mann M. Defining NASH from a Multi-Omics Systems Biology Perspective. Journal of Clinical Medicine. 2021; 10(20):4673. https://doi.org/10.3390/jcm10204673
Chicago/Turabian StyleNiu, Lili, Karolina Sulek, Catherine G. Vasilopoulou, Alberto Santos, Nicolai J. Wewer Albrechtsen, Simon Rasmussen, Florian Meier, and Matthias Mann. 2021. "Defining NASH from a Multi-Omics Systems Biology Perspective" Journal of Clinical Medicine 10, no. 20: 4673. https://doi.org/10.3390/jcm10204673
APA StyleNiu, L., Sulek, K., Vasilopoulou, C. G., Santos, A., Wewer Albrechtsen, N. J., Rasmussen, S., Meier, F., & Mann, M. (2021). Defining NASH from a Multi-Omics Systems Biology Perspective. Journal of Clinical Medicine, 10(20), 4673. https://doi.org/10.3390/jcm10204673