Abstract
Recent studies have demonstrated great therapeutic potential of educating and unleashing our own immune system for cancer treatment. However, there are still major challenges in cancer immunotherapy, including poor immunogenicity of cancer vaccines, off-target side effects of immunotherapeutics, as well as suboptimal outcomes of adoptive T cell transfer-based therapies. Nanomaterials with defined physico-biochemical properties are versatile drug delivery platforms that may address these key technical challenges facing cancer vaccines and immunotherapy. Nanoparticle systems have been shown to improve targeted delivery of tumor antigens and therapeutics against immune checkpoint molecules, amplify immune activation via the use of new stimuli-responsive or immunostimulatory materials, and augment the efficacy of adoptive cell therapies. Here, we review the current state-of-the-art in nanoparticle-based strategies designed to potentiate cancer immunotherapies, including cancer vaccines with subunit antigens (e.g., oncoproteins, mutated neo-antigens, DNA and mRNA antigens) and whole-cell tumor antigens, dendritic cell-based vaccines, artificial antigen-presenting cells, and immunotherapeutics based on immunogenic cell death, immune checkpoint blockade, and adoptive T-cell therapy.
1. Introduction
Immunotherapy has been explored for more than a century as a potential therapeutic approach to combat against cancer. Dating back to 1891 when neither chemotherapy nor radiotherapy were developed, a surgeon named William B. Coley successfully treated his cancer patients with bacterial products, which are now recognized to have induced non-specific anti-tumor inflammation []. However, immunotherapeutic strategies for cancer treatment have been doubted for a long time due to disappointing failures in various clinical trials. It was not until very recent years that dendritic cell-based vaccines and immune checkpoint inhibitors have each ushered new line of cancer therapy and raised the hope for unleashing patients’ own immune system to eradicate tumors [,]. Cancer immunity consists of several key steps, including release of antigens from tumor beds, presentation of tumor antigens by antigen-presenting cells (APCs), priming and activation of T cells by activated APCs, migration and infiltration of effector T cells back to the tumor, and finally the recognition and killing of tumor cells by effector T cells []. In theory, each of these steps can be targeted with various therapeutic approaches. The current advancement in cancer immunotherapy is mainly driven by striking results obtained with inhibitors of negative immune checkpoint molecules. However, this approach is mostly aimed at augmenting the potency of pre-existing tumor-specific T cells and benefits only a portion of patients as seen in recent clinical trials [,]. In contrast, cancer vaccines targeting early steps of antigen processing can potentially improve both therapeutic and prophylactic efficacies against not only primary tumor but also inoperable metastasis or relapse, and benefit more patients, especially those that lack sufficient levels of pre-existing anti-tumor T cells and/or immune checkpoint molecules.
However, despite tremendous potential of cancer vaccines, successful treatment and eradication of tumors with cancer vaccines has been elusive due to insufficient induction of immune responses with conventional vaccination approaches []. This highlights the need for new vaccination strategies that can efficiently deliver tumor antigens and adjuvants to APCs and stimulate immune responses strong enough to kill tumor cells. In this regard, nanoparticles have been intensively investigated for over the past three decades as delivery vehicles of traditional chemotherapeutics targeted to solid tumors [,,,,,]. Repurposing these nanomaterials to target the immune system may offer new opportunities to tune immunity and elicit strong anti-tumoral immune responses [,,]. Indeed, multi-functional nanomaterials have several key advantages over conventional therapeutics for cancer immunotherapy (Figure 1): (1) nanoparticles carrying both tumor antigens and adjuvants can stably co-deliver vaccine components to APCs [,]; (2) nanocarriers with finely tuned size and a defined surface chemistry can achieve selective delivery to lymphoid tissues [,,], while nanoparticles composed of biomaterials with immune-stimulating properties may also serve a dual role as a vaccine carrier and an adjuvant, thus simplifying the vaccine design [,]; (3) surfaces of nanomaterials can also be engineered to display antigens and co-stimulatory ligands to serve as artificial APC (aAPC) and potentiate T cell immune responses [,]; (4) delivery systems designed to initiate immunogenic cell death or target immune checkpoint molecules can drive anti-tumoral immune responses and reverse immune suppression; and (5) therapeutics-loaded particles can be utilized to improve anti-tumoral efficacy of adoptive T cell therapy []. This review article covers exciting new developments in each of these key areas of research, highlighting the potential of nanoparticle-based immunotherapy against cancer.
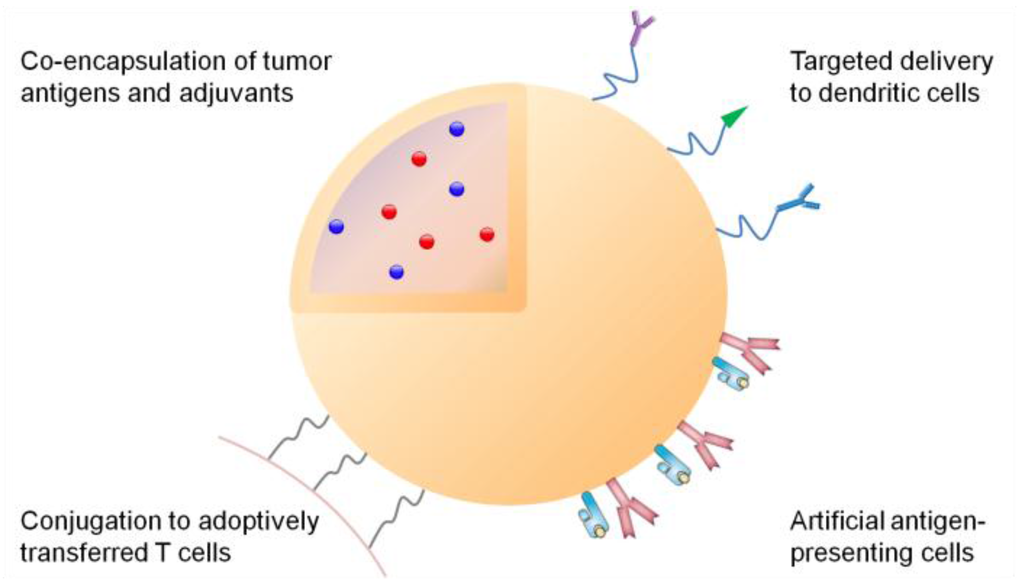
Figure 1.
Multi-functional nanoparticles for cancer immunotherapy. Tumor antigens and adjuvants can be co-loaded into the particle core, while the particle surface can be modified with antibodies or ligands specific to dendritic cells; or major histocompatibility complex/antigen complexes and co-stimulatory ligands as artificial antigen-presenting cells. Additionally, nanoparticles loaded with immune potentiators can be conjugated on T cells to improve adoptive T cell therapy.
2. Principles of Adaptive Immunity against Cancer
Vertebrates are protected by the immune system from pathogens such as viruses, bacteria, fungi and parasites. Immune responses to foreign pathogens can be classified into two categories, namely innate and adaptive immunity. Innate immunity provides rapid defense against pathogens while adaptive immunity requires processing of pathogens by APCs, presentation of immunogenic antigens to T cells and B cells, and elicitation of cellular and humoral immune responses. APCs play a pivotal role at the interface of innate and adaptive immune responses. Professional APCs include B cells, macrophages, and dendritic cells (DCs), among which DCs have been considered as the most efficient APC population []. DCs can process endogenous or exogenous antigens in the context of major histocompatibility complex (MHC) class I or II, and present the MHC/antigen peptide complex as the activation “signal 1” to CD8+ and CD4+ T cells, respectively. Activation of T cells requires an additional “signal 2” induced by ligation of co-stimulatory markers CD80/86 on DCs with CD28 on T cells, as well as a T-cell polarizing “signal 3” provided by cytokines secreted by DCs. Although MHC-I is constitutively expressed by the majority of mammalian cells, non-professional APCs cannot provide “signal 2 and 3” to alert the immune system when infected with pathogens. Therefore, antigen processing and presentation by APCs are a crucial first step in initiation of adaptive immune responses. In particular, DCs have a unique ability to process exogenous pathogens and activate CD8+ T cells via a process known as cross-presentation. Although the exact mechanisms of cross-presentation are still under investigation [,], vacuolar and cytosolic pathways have been identified []. The main difference between the two lies in the intracellular location for processing and loading of antigens to MHC-I: the vacuolar pathway utilizes endosomes while the cytosolic pathway utilizes endoplasmic reticulum for formation of MHC-I/antigen peptide complexes. Notably, increase in endosomal pH is thought to prevent excessive protease-mediated degradation of antigens in endosomes, thus promoting cross-presentation []. In addition, certain DC subsets such as tissue-resident CD8+ and migratory CD103+ DCs are known to be more efficient at antigen cross-presentation than other DC subtypes in mice [], and human CD141+/BDCA-3+ DCs have been recently proposed as the functional equivalent of the murine CD8+ DCs [].
Once activated in lymphoid tissues, cytotoxic CD8+ T lymphocytes (CTLs) enter the systemic circulation and patrol peripheral tissues in search of target cells. When CTLs identify target cells displaying a specific antigen epitope in the context of MHC-I, they secrete perforin and granzymes to lyse the target cells, and within minutes they move on to kill the next target []. CD4+ T cells mainly play an indirect/helper role. Following activation by MHC-II/antigen peptide complex presented by DCs, naïve CD4+ T cells differentiate into distinctive subtypes of helper T (TH) cells depending on the polarizing cytokines []. The TH1 population induced by IL-12 secretes IL-2 and IFN-γ and drives CD8+ T cell responses while TH2 and regulatory T cells (Tregs) induced by IL-4 and TGF-β are involved in humoral immune responses and immune suppression, respectively. In addition, CD4+ helper T cells express CD40L, which feeds back to DCs to further amplify immune activation and aid in establishment of memory CD8+ T cell responses [,].
In theory, immune system can inhibit oncogenesis by actively identifying and eliminating cancerous cells, a process referred to as immunosurveillance []. However, tumor cells have devised mechanisms to evade immune responses, including down-regulation of tumor antigens and promotion of immunosuppression [,]. Established tumor microenvironment is generally immunosuppressive due to up-regulation of inhibitory molecules against T cells. Activated T cells up-regulate cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) that binds to co-stimulatory molecules on DCs with higher affinity than CD28. Although CTLA-4 naturally serves as a peripheral inhibitory signal to prevent over-reactivity of T cells, it also dampens anti-tumor immune responses. Besides, subsets of tumor cells highly express programmed death-ligand 1 (PD-L1) that binds to programmed death-1 (PD-1) on T cells and inhibit their effector functions []. Tumor cells can also secrete cytokines such as IL-10 and TGF-β, which both directly inhibit the proliferation of CTLs and drive the differentiation of Tregs that provide an additional source of these immunosuppressive cytokines. In addition to Tregs, tumor cells can recruit other inhibitory immune cells such as macrophages and myeloid-derived suppressor cells (MDSCs) to further dampen cytotoxic functions of CTLs []. Thus, tumor cells can promote immunosuppressive tumor microenvironment and shield themselves from CTLs by hijacking normal negative feedback loops designed to guard against excessive activation of T cell responses.
3. Synthetic Systems for Delivery of Tumor Antigens
Tumor antigens can be categorized broadly into subunit antigens and whole-cell antigens. Subunit antigens include altered cell-surface polysaccharides, peptides, oncoproteins, and DNA and mRNA that encode those proteins, while tumor-cell lysate and immunogenically dying tumor cells can serve as the source of whole-cell antigens. Table 1 presents major advantages and challenges for each class of tumor antigens utilized for cancer vaccination. Some viruses (e.g., Epstein-Barr virus (EBV), human papilloma virus (HPV) and hepatitis B and C viruses) contribute to cancer development, and virally encoded gene products can also serve as the potential targets of immunotherapy []. Among different types of tumor antigens, oncoproteins, which are either mutated or over-expressed normal or embryonic proteins from fetal development, are intensively investigated for cancer vaccines due to their potential to elicit broad-epitope CD8+ and CD4+ T-cell responses. In comparison to full-length protein antigens that require cellular uptake and processing, peptide epitopes can directly bind to MHC molecules, and their stability is less affected during the preparation and storage of vaccine products. In line with these advantages, there are many ongoing clinical trials on peptide-based cancer vaccines []. However, the major challenge facing cancer vaccination based on subunit antigens is their poor immunogenicity and limited therapeutic efficacy. For example, in the case of melanoma-associated antigens, β-catenin, survivin, tyrosinase, gp100, MAGE, melan-A (MART1), and NY-ESO-1, have been identified and tested in clinical trials []. In a Phase III trial of gp100 peptide in combination with IL-2 and MontanideTM ISA 51 as adjuvants, the response rate and overall survival improved from 6% to 16% and 11.1 months to 17.8 month, respectively, in comparison to the IL-2 alone treatment group []. However, another Phase III trial with MAGE-A3 peptide has failed to prolong disease-free survival []. Overall, therapeutic efficacy of cancer vaccines remain suboptimal, partially due to the fact that many tumor antigens evaluated in clinical trials are self-antigens against which auto-reactive T-cells are eliminated or tolerized. In addition, conventional vaccine/adjuvant delivery systems have limited capability to target delivery of tumor antigens and adjuvants to proper APCs and intracellular compartments. In this regard, nanotechnology-based vaccine systems are poised to address these challenges as described below.

Table 1.
Major advantages and remaining challenges for tumor antigens.
| Tumor antigens | Advantages | Challenges | |
|---|---|---|---|
| Subunit antigens | Polysaccharides | Defined chemical synthesis | Elicitation of humoral rather than cellular immune responses |
| Peptides | Ease of production | Poor delivery efficiency | |
| Stable vaccine formulations | Monovalent immune response | ||
| May not require antigen-processing by APCs | Subject to HLA-specificity | ||
| Proteins | Broad-epitope immune responses Wide HLA-specificity | Poor delivery efficiency | |
| Suboptimal for CD8+ T cell responses | |||
| Weak immunogenicity of self-antigens | |||
| DNA and mRNA | Ease of production | Poor delivery efficiency | |
| In situ expression of full-length antigens | Poor in vivo stability | ||
| Flexible to encode immune stimulators | Limited transfection efficiency | ||
| Whole-cell antigens | Tumor-cell lysate | Broad-epitope immune responses Potential for “personalized” therapy | Requires tissue biopsy |
| Manufacturing challenges | |||
| Loss of antigenicity during production | |||
| Presence of self-antigens | |||
| Immunogenically dying tumor cells | Broad-epitope immune responses | Requires additional therapeutic interventions Presence of self-antigens and immunosuppressive molecules, e.g., PD-L1 | |
| Full preservation of tumor antigens | |||
| Potential for “personalized” therapy | |||
3.1. Efficient Draining to Lymphoid Tissues
Nanocarriers can improve the efficacy of subunit cancer vaccines by facilitating antigen presentation and T-cell activation. This is achieved by exploiting efficient draining of nanocarriers to lymphoid tissues and their prolonged tissue residence as well as controlled release of antigens and adjuvants. Particle size is one of the primary factors determining the efficiency of lymphatic draining. Large particles (>500 nm in diameter) can be physically trapped at the injection site by interaction with extracellular matrix proteins, whereas ultra-small nanoparticles (<10 nm in diameter) or soluble antigen molecules can rapidly diffuse into and out of lymph nodes, thus minimizing the chance of APCs phagocytizing sufficient amount of vaccine particles []. On the other hand, particles of an intermediate size (10–100 nm in diameter) can both efficiently drain to regional draining lymph nodes and become retained there, thereby increasing the chance of antigen uptake and presentation by APCs [,]. Indeed, one study has compared the immunogenicity of protein or peptide antigen-conjugated nanobeads with sizes ranging from 0.02 to 2 μm. Following intradermal injection of these nanobeads into mice, a 40 nm nanovaccine formulation drained most efficiently to lymph nodes and elicited stronger antigen-specific T-cell immune responses than other formulations, including vaccines with conventional adjuvants such as Alum, monophosphoryl lipid A (MPLA), or Quil-A []. In another study, following subcutaneous injection into mice, nanogels with a mean diameter of 60 nm self-assembled by polysaccharide cholesteryl pullulan delivered synthetic long tumor-antigen peptides to medullary macrophages, which primed anti-tumoral CD8+ T cell responses in prophylactic and therapeutic settings (Figure 2) []. Furthermore, enhanced cytotoxic CD8+ T-cell responses and inhibition of tumor growth have been achieved by targeting nanovaccines to tumor-draining lymph nodes rather than non-tumor draining lymph nodes, suggesting that antigen-primed but immune-suppressed lymphoid tissues can serve as ideal sites of immune activation []. These studies rely on various imaging techniques with nanoparticles labeled with fluorescent dyes or contrast agents to track and quantify lymphatic draining. For example, a polyester nanoparticle system loaded with ovalbumin (OVA) and labeled with a near-infrared probe has been utilized to demonstrate co-transport of the antigen and nanoparticles to draining lymph nodes []. In an alternative approach, poly(lactic-co-glycolic acid) (PLGA) nanoparticles designed to carry iron oxide particles conjugated with fluorophore-labeled peptide antigen permitted bimodal tracking of the nanocarriers with MRI and fluorescent imaging []. Notably, it remains to be seen whether these delivery systems can be successfully translated into clinics since most studies were performed on murine models. Indeed, upon subdermal injection into the breast region of breast cancer patients, large (>300 nm) radio-labeled colloids were drained slowly through the lymph vessels and retained longer in sentinel lymph nodes, compared with their small (<50 nm) counterparts that rapidly visualized lymphatic vessels, sentinel lymph nodes, and second- and third-tier lymph nodes []. In addition, lymphatic draining of particulate vaccines also depends on the material composition, morphology, and surface chemistry of particles [].
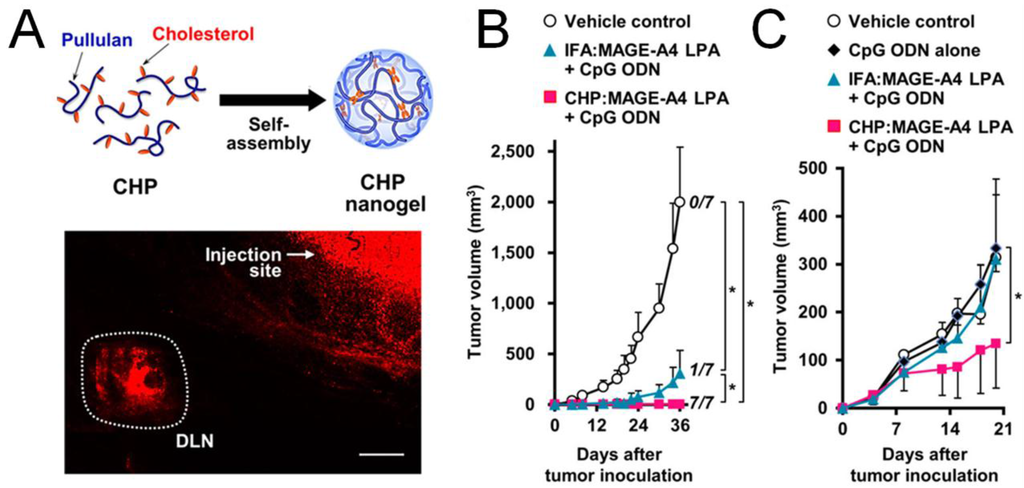
Figure 2.
Anti-tumor efficacy improved by efficient lymphoid draining and retention of nanoparticle-based cancer vaccines. (A) Lymphoid draining of a fluorophore-labeled pullulan nanogel 6 h post subcutaneous injection to mice. Scale bar, 1 mm. The nanogel loaded with a long peptide antigen (LPA) MAGE-A4 achieved better prophylactic (B) and therapeutic (C) efficacy compared to the soluble antigen; (B) Mice were immunized on day -7, followed by inoculation of tumor on day 0; (C) mice were inoculated with tumor on day 0, followed by immunization on day 4 and 11. Reproduced with permission [].
3.2. Targeted Delivery to Dendritic Cells
Once in contact with immune cells, tumor antigens have to be engulfed and processed by APCs, preferably DCs, to activate adaptive immune responses. Therefore, vaccine delivery targeted to DCs may be beneficial. Indeed, a protein vaccine composed of the full-length NY-ESO-1 fused to human mAb specific to DEC-205 (CD205), which is a C-type lectin receptor expressed on DCs, has been shown to elicit robust antigen-specific humoral and cellular immune responses with a good safety profile in a recent Phase I clinical trial []. Nanoparticles encapsulating tumor antigens can also be modified with targeting moieties on their surfaces to achieve DC-specific delivery. Such delivery systems specifically targeted to DEC-205, DC-SIGN, mannose receptor, Fc receptor, CD40, or CD11c have been reported [,,]. Although these systems have been shown to induce stronger DC activation, compared with their non-targeted counterparts, it remains to be determined whether a particular targeting ligand is optimal for the DC-targeting approach. A recent study has addressed this issue by comparing PLGA nanovaccines modified with antibodies against CD40, DEC-205, or CD11c, and showed that the CD40 Ab-modified nanoparticles achieved the greatest binding to and uptake by DCs []. Interestingly, despite differential levels of DC-targeting, these various nanovaccine formulations induced similar levels of CD8+ T cell responses []. It should be noted that the extent of DC-targeting, particle uptake, and subsequent immune activation depends on specific physico-chemical properties of the nanocarrier itself as well as adjuvants employed in the vaccine system. Thus, efficiencies of DC-targeting and induction of adaptive immune responses by nanovaccines may have to be optimized in the context of each vaccine carrier and adjuvant. In particular, different DC subsets have distinctive sites of tissue residence, receptor expression profiles, and functions [], and nanovaccines designed to target DC subsets with high efficiency of antigen cross-presentation, such as murine lymphoid tissue-resident CD8+ DCs and human CD141+/BDCA-3+ DCs and Langerhans cells [,], should be explored further.
3.3. Promotion of Cross-Presentation
Extracellular antigens are usually processed and presented via MHC-II by APCs to CD4+ T cells; however, tumor antigens engulfed by APCs need to be presented via MHC-I to activate CTLs, which are the main effector cells against tumor cells. Thus, traditional vaccine approaches relying on soluble protein or peptide tumor antigens may skew immune responses to CD4+ T cell responses, while failing to induce sufficient CTL responses. In contrast, tumor antigens delivered by functional nanomaterials designed to promote endosomal escape (i.e., translocation of antigens from endosomes/phagosomes to cytosol) may induce cross-presentation and favorably elicit CD8+ T cell responses []. To this end, extensive research efforts have been focused on pH-sensitive delivery systems that can retain their cargo under the physiological pH condition while triggering release of antigens and disruption of endocytic vacuoles at the acidic (~pH 6) endosomal microenvironment []. For example, a liposomal antigen delivery system modified with a pH-sensitive dextran derivative has been shown to promote cytosolic delivery of antigens []. In addition, a micellar system composed of an amphiphilic polymer with a pH-sensitive building block forming the particle core has been devised to induce fusion of the nanomaterials to endosomal vesicles, thus transporting protein antigen surface-displayed on micelles from endosome to cytosol and promoting antigen cross-presentation and CD8+ T cell responses []. An alternative approach includes an oxidation-sensitive polymersome that can respond to the oxidative environment of endosomes and deliver antigens and adjuvants to cytosol []. Additionally, liposomes modified with a cell-penetrating peptide octaarginine or gold nanoparticles displaying tumor antigens were also shown to promote cross-presentation [,].
3.4. Co-Delivery of Adjuvants
Another major advantage of nanoparticle delivery systems lies in their ability to co-deliver antigens together with adjuvants, thereby enhancing cross-presentation and/or skewing immune responses to desired CD4+ T helper phenotypes. Agonists for Toll-like receptors (TLRs) have been widely investigated as adjuvants for cancer vaccines []. Although TLRs are mainly involved in innate immunity by sensing pathogenic danger signals, they are crucial for induction of adaptive immune responses as they can promote cross-presentation in APCs to activate CD8+ T cells or prime APCs to release cytokines that can polarize CD4+ TH cells to specific phenotypes []. Since the TH1 responses elicited by activation of TLR3, TLR7, or TLR9 contribute to CD8+ T cell responses [,], agonists of these TLRs have been widely examined for cancer nanovaccines. CpG, which is an unmethylated oligonucleotide containing CpG motif, is a potent TLR9 agonist. CpG has been complexed with cationic polymers via the electrostatic interaction or conjugated with nanocarriers, which improved immune activation compared with administration of free soluble adjuvant (Figure 3A) [,]. The charge-mediated entrapment was also exploited to co-load an anionic TLR3 agonist poly I:C and cationic antigen peptides onto gold nanoparticles via the “layer-by-layer” strategy, leading to elicitation of robust antigen-specific CD8+ T cells when tested with a model antigen in vivo (Figure 3B) []. In addition to efficient loading of adjuvants, co-entrapment of an antigen and adjuvant within the same particles can also enhance the efficiency of cross-presentation and induction of CD8+ T cells, compared with soluble vaccine components admixed together [,,]. Moreover, nanoparticles designed for multifaceted drug loading can support a combinational use of adjuvants, thus permitting exploitation of synergy among certain TLR agonists []. For example, CpG and poly I:C have been co-loaded into polyester nanoparticles [], while the TLR4 agonist glucopyranosyl lipid A and TLR7 agonist imiquimod have been co-encapsulated into liposomes []. In both cases, the TH1 response was significantly improved by the dual TLR agonists-loaded particles, compared with that elicited by a single adjuvant. Alternatively, TLR agonists can be combined with siRNAs inhibiting the immunosuppressive pathways. Co-delivery of CpG and siRNA targeting IL-10, the inducer of TH2 and Treg cells, skewed immune responses to the TH1 type []. The combination of peptide epitope of tyrosine-related protein 2 (Trp2) and CpG-based nanovaccine with siRNA against TGF-β, which is one of the major cytokines responsible for induction and maintenance of immunosuppressive tumor microenvironment, has significantly improved the therapeutic efficacy of nanoparticle-based cancer vaccine in a late-stage murine melanoma model [].
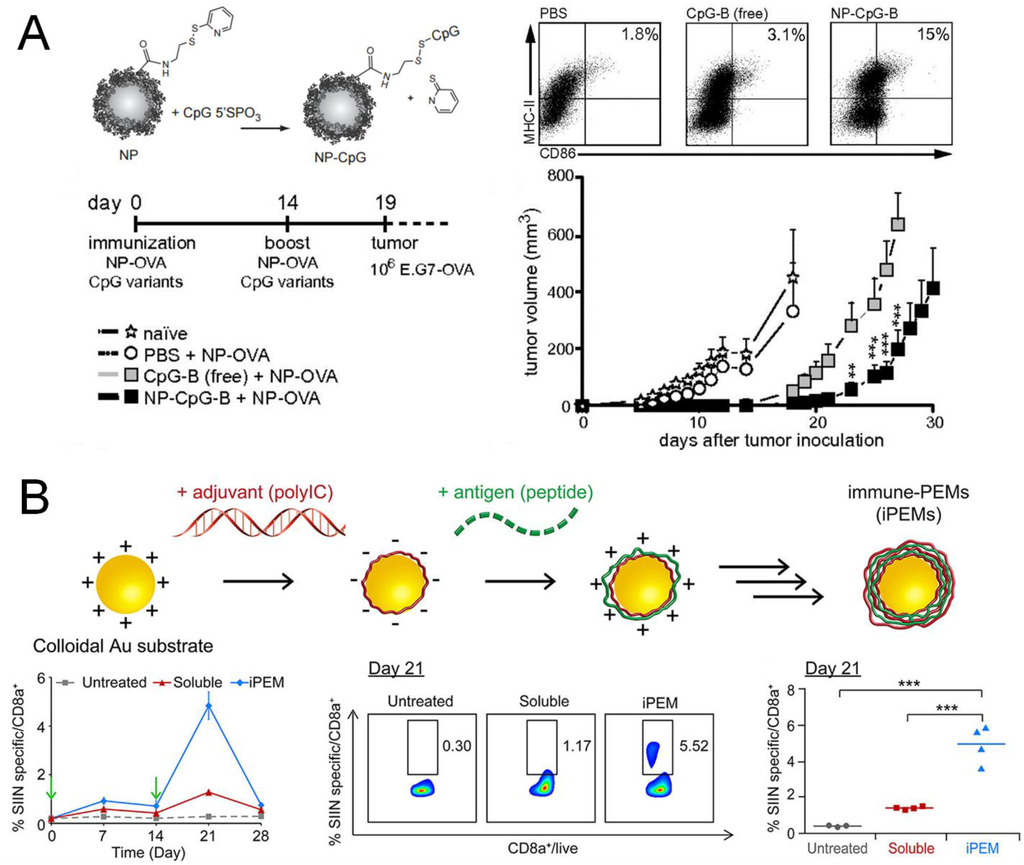
Figure 3.
Co-delivery of antigens and adjuvants by nanoparticles. (A) TLR9 agonist CpG was conjugated on the surface of polymeric nanoparticles via disulfide exchange. The particulate adjuvant improved DC activation in vitro as well as prophylactic efficacy against tumor in vivo, compared with the soluble CpG; (B) TLR3 agonist poly I:C and an antigen peptide were complexed onto gold nanoparticles via electrostatic interactions, and elicited more antigen-specific CD8+ T cells compared with the soluble vaccine. Panel (A) reproduced with permission []; Panel (B) reproduced with permission [].
3.5. Delivery of DNA and mRNA Tumor Antigens
DNA and mRNA encoding oncogenic proteins or peptides are appealing tumor antigens due to the ease of manufacturing scale-up and their potential for further modification with nucleic acid sequences that encode for proteins with immunostimulatory functions (e.g., flagellin). However, previous clinical trials on DNA cancer vaccines, majority of which were administered as naked DNA via the intramuscular route, showed generally poor response rates []. Although viral vectors and electroporation have been employed to improve the transfection of DNA vaccines, they are subject to safety and compliance issues []. Alternatively, synthetic delivery systems can be used to deliver DNA and mRNA therapeutics due to several advantages: (1) synthetic materials such as cationic lipids and polymers are safer alternatives to viral vectors; (2) gene therapeutics can be stabilized and protected from nuclease-mediated degradation by particulate carriers, and DNA and RNA have also been designed to self-assemble into distinctive nanostructures with improved colloidal stability [,]; (3) injection-free gene delivery routes can be exploited with DNA- and RNA-loaded nanocarriers, such as microneedles, pH-sensitive polymeric nanoparticles, or lipoplexes for non-parenteral routes of delivery [,,]; and (4) nanocarriers can be modified with targeting moieties, e.g., mannose, to achieve DC-targeted delivery and transfection [].
3.6. Delivery of Whole-Cell Cancer Vaccines
Compared with a single peptide or protein antigen, whole-cell cancer vaccines may elicit multivalent immune responses by broadening epitope recognition and help to realize personalized immunotherapy. Whole-cell antigen can be obtained from tumor cell lysates with necrotic features or inactivated whole tumor cells with apoptotic features. Similar to subunit vaccine nanocarriers, tumor cell lysates and TLR agonists have been co-encapsulated into particulate delivery systems, including liposomes or PLGA micro/nanoparticles [,]. Whole-cell cancer vaccine has also been delivered by a biodegradable, “infection-mimicking” PLGA matrix containing tumor lysate as the source of tumor antigens, granulocyte macrophage colony-stimulating factor (GM-CSF) for recruitment of DCs in situ, and CpG for activation of recruited DCs []. This PLGA matrix-based whole-cell cancer vaccine successfully elicited antigen-specific CD8+ T cells and improved both prophylactic and therapeutic anti-tumor efficacy, compared with a conventional whole-cell vaccine GVAX, composed of irradiated, GM-CSF-secreting tumor cells. In an alternative approach, plasma membrane of tumor cells has been extracted and coated onto polymeric nanoparticle cores along with the TLR4 agonist MPLA as a tumor cell-mimicking cancer vaccine [].
4. Synthetic Delivery Systems for DC-Based Cancer Vaccines
Direct administration of autologous DCs activated by tumor antigens ex vivo may be an efficient approach for vaccination against cancers, as exemplified by the first and only approved therapeutic DC-based cancer vaccine, Sipuleucel-T (Provenge), which has improved the median survival of patients with metastatic castrate-resistant prostate cancer by 4.1 months, compared with the placebo group []. As introduced earlier, delivery of tumor antigens by particulate systems has been shown to enhance antigen processing and presentation by DCs; therefore, particle-based vaccine delivery applied to DC-based vaccines may improve their anti-tumoral efficacy. One study has employed antigen-loaded poly(γ-glutamic acid) nanoparticles to show that DCs activated by these particles released TH1 cytokines, elicited robust T-cell activation in vitro, and enhanced protection against tumor challenge in mice []. In another approach, antigen delivery by porous silica particles induced secretion of type I IFN cytokine from DCs, leading to reduced tumor growth in both therapeutic and prophylactic conditions []. The benefits of multi-drug loading within nanoparticles were also demonstrated in a TriMix delivery system []: the mixture of antigen and adjuvant mRNAs was encapsulated in cationic liposomes which were then conjugated to microbubbles to allow ultrasound-triggered transfection of DCs. DCs activated by this strategy exhibited enhanced therapeutic efficacy against established tumors, when compared with DCs primed with antigen mRNA with or without lipopolysaccharide (LPS).
There has been increasing interest in artificial APCs (aAPCs) surface-decorated with covalently conjugated tumor antigen/MHC complex and anti-CD28 antibody. The rationale is that direct activation of antigen-specific T cells by aAPCs will obviate the need for antigen delivery to APCs and antigen processing and presentation, while also avoiding potential activation of immune checkpoint molecules, such as CTLA-4, expressed on T cells via the use of agonist antibodies directed toward co-stimulatory pathways. Indeed, various particle platforms have been explored for aAPCs, including PLGA microparticles [], liposomes [], iron/dextran nanoparticles [], and carbon nanotubes []. In particular, aAPCs composed of an iron nanoparticle core and stimulatory molecules on the dextran shell were shown to induce T-cell receptor clustering when incubated with T cells under magnetic field, thus allowing external stimulus-induced proliferation of antigen-specific T cells in vitro and in vivo (Figure 4) []. Moreover, carbon nanotubes loaded with activation signals have been also developed as aAPCs to expand antigen-specific CD8+ T cells, which were then successfully used for adoptive T cell therapy []. Interestingly, recent studies have revealed that ellipsoidal PLGA nano/microparticles were more efficient aAPCs than their spherical counterparts [,], demonstrating that biophysical parameters of aAPCs may play a crucial role in induction of T cell responses.
5. Synthetic Delivery Approaches to Induce Immunogenic Cell Death (ICD)
Recent studies have demonstrated immunogenicity of dying cancer cells under certain chemotherapies or radiotherapy []. Although systemic administration of chemotherapeutics is generally immunosuppressive, in situ treatments with certain chemodrugs especially anthracyclines, such as doxorubicin and mitoxantrone, have been shown to induce ICD [,]. In addition, the abscopal effect observed during radiotherapy, i.e., regression of distant, non-irradiated tumors, is also believed to be caused by systemic immune responses elicited by dying primary tumor cells []. Since the initial discovery of ICD, anthracycline chemodrugs have been investigated widely for immune-mediated anti-tumor efficacy in addition to their direct tumor-killing effects, especially in combination with other cancer immunotherapies, such as vaccines, adoptive cell transfer, and checkpoint inhibitors []. Indeed, ICD suggests an alternative approach for whole-cell vaccination based on ex vivo generated immunogenically dying tumor cells or induction of ICD in situ. In addition, co-delivery of adjuvants with ICD inducers may be helpful to potentiate anti-tumor immune responses, thus motivating the use of adjuvant-carrying particulate delivery systems for further enhancing ICD. For example, PLGA microparticles have been employed to encapsulate doxorubicin and CpG and intratumorally injected for induction of ICD []. In addition, doxorubicin-based in situ vaccination strategy combined with anti-CTLA-4 and anti-OX40, an agonistic antibody against the stimulatory checkpoint molecule OX40, has been shown to improve T cell infiltration into distant tumors, leading to tumor eradication and increased survival [].
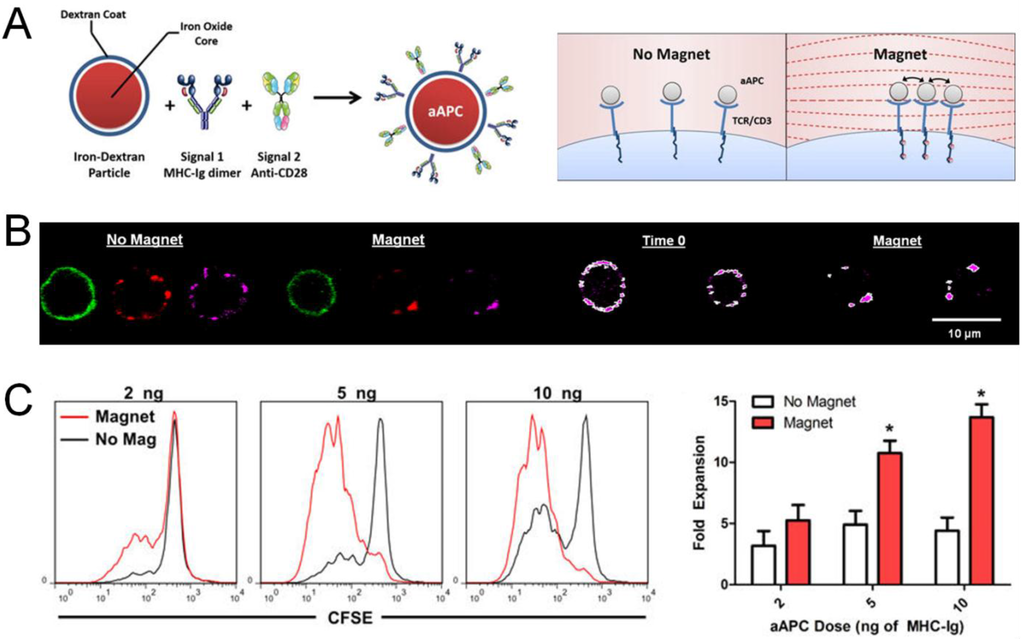
Figure 4.
Artificial antigen-presenting cells (aAPCs) for activation of T cells. (A) aAPCs composed of an iron nanoparticle core and dextran shell conjugated with stimulatory molecules induced clustering of T-cell receptors (TCRs) under magnetic field; (B) TCR clustering was visualized by fluorescence imaging. Green, lymphocyte marker on T cells; red: aAPCs; magenta: CD3ε on T cells; (C) Proliferation of T cells was enhanced by aAPC-induced TCR clustering in vitro. Reproduced with permission [].
6. Synthetic Delivery Systems Targeted to Immune Checkpoints
Cancer immunotherapy aiming to reverse immunosuppression has achieved striking success in recent years. The CTLA-4 inhibitory antibody Ipilimumab has improved the survival of patients with advanced, untreatable melanoma by 3.7 months, and gained FDA-approval as a new category of cancer immunotherapy []. However, treatment with Ipilimumab was also accompanied with adverse events and moderate response rates []. The PD-1 inhibitory antibodies Nivolumab and Pembrolizumab were also approved for the treatment of malignant melanoma in late 2014. PD-1 is now considered a better target than CTLA-4 because antibody-mediated blockade of PD-1 among tumor-infiltrating T cells within the tumor microenvironment leads to mitigated side effects and higher response rates [], especially among patients with PD-L1 positive tumors [,]. In addition, dual inhibition of CTLA-4 and PD-1 recently has been shown to be more efficacious than a single therapy, mainly due to their distinctive mechanisms of action: the main sites of action for antibodies against CTLA-4 and PD-1 are thought to be within lymphoid tissues and tumor regions, respectively. However, the current systemic administration route for these therapeutic antibodies may still cause off-target toxicity. A previous study has addressed this issue by intratumoral administration of anti-CTLA-4 antibody with mesoporous silica []. The micron-size carrier with nanopores achieved high loading efficiency and enhanced anti-tumor efficacy, compared with soluble antibody injected intraperitoneally in a murine melanoma model, possibly due to controlled release of antibody from an in situ depot. Alternatively, targeted delivery of siRNA against PD-L1 has also been investigated with cationic lipoid and polymeric nanoparticles [,]. PD-L1 expressed on cancer cells were efficiently silenced by siRNA complexed with folic acid-modified polyethylenimine, resulting in enhanced in vitro T-cell activation [].
In addition to immune checkpoint inhibitors covered above, there are also stimulatory checkpoint targets, such as OX40 (CD134) and 4-1BB (CD137), which can be activated to improve anti-tumor immunity. Both molecules belong to the receptor family of tumor necrosis factor (TNF) and directly induce T-cell activation. Ligation of OX40 on T cells with its ligand on APCs results in activation of both CD4+ and CD8+ T cells, leading to inhibition of tumor growth []. Notably, expansion of Tregs following activation of OX40 remains a controversial topic [,]. Since all CD4+ T cell subtypes can be activated by the OX40 pathway, it is likely that induction of Tregs depends on the particular polarizing cytokine milieus that the cells are exposed to. In contrast to OX40, 4-1BB preferentially activates CD8+ rather than CD4+ T cells []. 4-1BB is up-regulated as a surrogate for CD28 which cannot compete against CTLA-4 in binding to co-stimulatory molecules during the late or secondary immune response []. Nanoparticle delivery systems have been developed for these antibody therapeutics, aiming to mimic the natural immune activation by antibodies surface-displayed on particles or to reduce systemic toxicity by localized administration. Anti-OX40 antibody was conjugated to the surfaces of PLGA nanoparticles via EDC/NHS chemistry and promoted antigen-specific killing by CD8+ T cells in vitro []. In another combinational therapeutic approach, anti-4-1BB antibody and IL-2 were separately displayed on liposomal surfaces for localized tumor therapy, inducing robust anti-tumor CTL responses, while minimizing off-target side effects and preventing cytokine storm typically observed after systemic administration of immunotherapeutics [].
7. Synthetic Delivery Systems for Adoptively Transferred T Cells
Adoptive cell therapy (ACT), based on autologous T cells expanded with tumor antigens and IL-2 ex vivo, is envisioned to induce tumor regression as a “live drug”. However, this approach is limited by moderate responses, due to insufficient expansion of transferred T cells and inefficient trafficking to tumor regions [], as well as potential severe side effects characterized by “cytokine storm” with TNF, IFN-γ and IL-6 when IL-2 is systemically co-administered with T cells []. To address these limitations, nanoparticle-based cellular engineering approaches have been examined to improve the therapeutic efficacy of ACT. Maleimide-modified synthetic nanoparticles were conjugated to the surfaces of CD8+ T cells via sulfhydryl groups exposed by cell-surface proteins []. T-cell stimulating cytokine complexes, IL-15/IL-15Ra and IL-21, were encapsulated into nanoparticles to provide signals for T cell expansion in situ. This strategy resulted in potent proliferation of transferred T cells and eradicated metastatic melanoma tumors in mice, whereas co-administration of T cells mixed with those cytokines failed to eliminate tumors. Tumor-specific T cells “equipped” with small-molecule inhibitors against T-cell exhaustion using the same method also showed therapeutic benefits []. In an alternative strategy, ex vivo expanded antigen-specific CD8+ T cells and T-cell activating particles were co-delivered via an implantable biodegradable hydrogel system to treat residual tumors as well as metastases in murine breast cancer models []. Within the local implant, lipid-coated mesoporous silica particles encapsulating IL-15 and IL-15Ra while displaying stimulatory antibodies against CD3, CD28, and CD137 efficiently activated T cells, which were gradually released from the depot and more efficacious than systemic or local administration of T cells without stimulatory signals. In addition to CTL-mediated tumor-specific killing, T cells also have been utilized to shuttle therapeutics to diseased tissues. As shown by a recent study on a metastatic murine lymphoma model, autologous polyclonal T cells with the potential to home to tumor-bearing lymphoid tissues were primed ex vivo and conjugated with chemodrug-loaded nanoparticles on the cell surfaces (Figure 5A) []. This “Trojan horse” approach exploiting innate tropism of T cells to lymphoid tissues allowed selective delivery of the chemotherapeutic to disseminated lymphoma tumors, with 90-fold greater concentration of the drug accumulated in lymph nodes than free drug systemically injected at 10-fold higher doses (Figure 5B). This T cell-mediated delivery significantly reduced tumor burden and improved survival, compared with administration of free drug or drug-loaded nanoparticles alone.
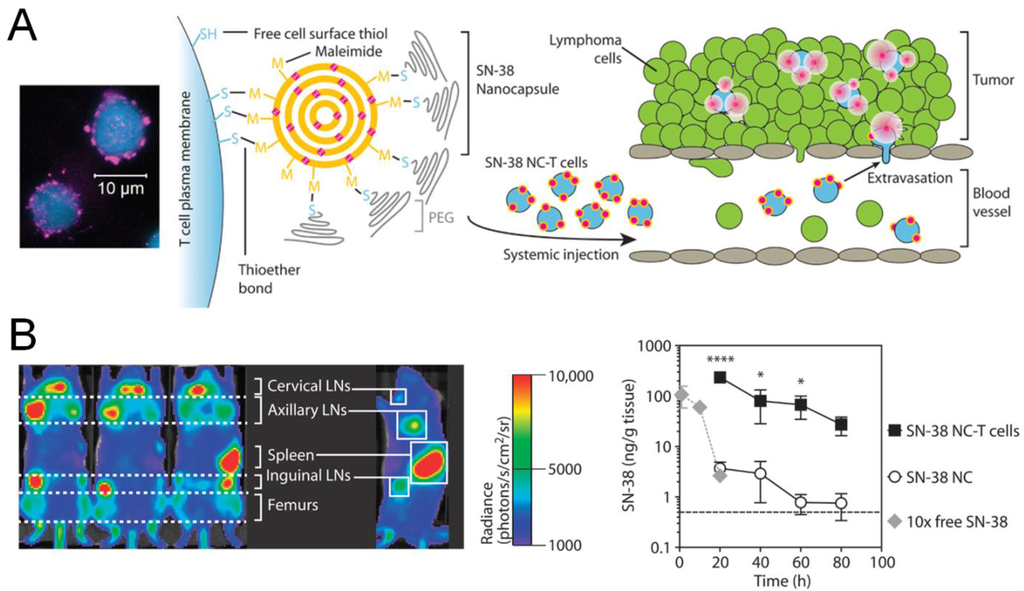
Figure 5.
Engineered T cells for cancer therapy. (A) T cells (blue) conjugated with nanoparticles (magenta) loaded with a chemodrug (SN-38) were used for drug delivery to lymphoma; (B) T cells with surface-bound chemodrug preferentially accumulated in tumor-bearing lymphoid tissues following systemic administration, and significantly improved the drug distribution in lymph nodes, compared with equivalent or 10-fold higher dose of the nanoparticulate or soluble drug, respectively. Reproduced with permission [].
8. Conclusions
This review article has covered various applications of nanotechnology in cancer vaccination and immunotherapy. Therapeutic strategies based on nanomaterials can enhance the efficacy of cancer vaccines by improving lymphatic delivery of tumor antigens or by incorporating targeting approaches and/or stimuli-responsive materials to modulate immune activation. Moreover, adjuvants loaded into nanocarriers via hydrophobic or electrostatic interactions can further increase immunogenicity of tumor antigens. In addition, nanoparticulate drug delivery systems have been applied to augment the therapeutic potential of autologous DCs and synthetic artificial APCs, immunogenic cell death, immune checkpoint inhibitors, and adoptive T-cell therapy.
New horizons in cancer immunotherapy include identification of tumor neo-antigens by tumor exomes sequencing and utilization of neo-antigens for “precision medicine” by tailoring the cancer vaccine for each patient []. As mutated neo-antigens should be more immunogenic than self-antigens over-expressed on tumor cells, this approach may dramatically improve the therapeutic efficacy of cancer vaccines, as demonstrated by tantalizing results in recent studies [,]. However, these vaccination strategies based on neo-antigens are still subjected to delivery issues facing conventional subunit vaccination; therefore, vaccine nanoparticles loaded with neo-antigens may address these challenges and offer personalized nanomedicines designed to elicit anti-tumoral immune responses. In addition, combination therapies utilizing cancer vaccines and immune checkpoint inhibitors merit further exploration as they could simultaneously drive potent anti-tumor immune responses and reverse immunosuppression in tumor microenvironment. Indeed, recent remarkable clinical results observed after dual blockade of PD-1 and CTLA-4 have highlighted advantages of combinational immunotherapies with distinct targets or mechanisms. Furthermore, numerous nanocarriers designed for delivery of conventional chemotherapeutics can be potentially applied to initiate and enhance immunogenic cell death and may further provoke the immune system against tumor cells when co-administered with cancer immunotherapeutics. In conclusion, nanoparticle delivery systems are versatile platforms that can be rationally designed to address key technical challenges in cancer vaccination and immunotherapy and may offer cutting-edge material-based strategies to advance this exciting field of cancer therapy.
Acknowledgments
This work is supported in part by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000433, the John S. and Suzanne C. Munn Cancer Fund of the University of Michigan Comprehensive Cancer Center, and the Melanoma Research Alliance Young Investigator Award.
Author Contributions
Yuchen Fan and James Moon both wrote this review article.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| aAPC | Artificial antigen-presenting cell |
| ACT | Adoptive cell therapy |
| APCs | Antigen-presenting cells |
| CpG | Unmethylated oligonucleotide containing CpG motif |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CTL | Cytotoxic T lymphocyte |
| DCs | Dendritic cells |
| EBV | Epstein-Barr virus |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| HLA | Human leukocyte antigen |
| HPV | Human papilloma virus |
| ICD | Immunogenic cell death |
| LPS | Lipopolysaccharide |
| MDSCs | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| MPLA | Monophosphoryl lipid A |
| OVA | Ovalbumin |
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death ligand-1 |
| PLGA | Poly(lactic-co-glycolic acid) |
| siRNA | Small-interfering RNA |
| TCRs | T-cell receptors |
| TNF | Tumor necrosis factor |
| TH cells | Helper T cells |
| TLRs | Toll-like receptors |
| TNF | Tumor necrosis factor |
| Tregs | Regulatory T cells |
| Trp-2 | Tyrosine-related protein 2 |
References
- Starnes, C.O. Coley’s toxins in perspective. Nature 1992, 357, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Downey, S.G.; Klapper, J.A.; Smith, F.O.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Kammula, U.S.; Hughes, M.S.; Allen, T.E.; Levy, C.L.; et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clin. Cancer Res. 2007, 13, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.S.; Mohammad, F. Magnetic nanomaterials for hyperthermia-based therapy and controlled drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Du, W.; He, B.; Fu, F.; Yuan, L.; Wu, H.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; et al. The reduction of tumor interstitial fluid pressure by liposomal imatinib and its effect on combination therapy with liposomal doxorubicin. Biomaterials 2013, 34, 2277–2288. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Miura, S.; Na, K.; Bae, Y.H. pH-responsive and charge shielded cationic micelle of poly(L-histidine)-block-short branched PEI for acidic cancer treatment. J. Control. Release 2013, 172, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Van der Meel, R.; Vehmeijer, L.J.; Kok, R.J.; Storm, G.; van Gaal, E.V. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: Current status. Adv. Drug Deliv. Rev. 2013, 65, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, S.; Shi, Y.; Chuan, X.; Li, J.; Zhong, T.; Zhang, H.; Dai, W.; He, B.; Zhang, Q. The development of site-specific drug delivery nanocarriers based on receptor mediation. J. Control. Release 2014, 193, 139–153. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Schulz, A.; Wan, X.; Seitz, J.; Bludau, H.; Alakhova, D.Y.; Darr, D.B.; Perou, C.M.; Jordan, R.; Ojima, I.; et al. Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: Preparation, in vitro and in vivo evaluation. J. Control. Release 2015, 208, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Huang, B.; Irvine, D.J. Engineering nano- and microparticles to tune immunity. Adv. Mater. 2012, 24, 3724–3746. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Swartz, M.A.; Szeto, G.L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013, 12, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Immunoengineering: How nanotechnology can enhance cancer immunotherapy. Cell 2015, 161, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Videira, M.; Gaspar, R.; Preat, V.; Florindo, H.F. Immune system targeting by biodegradable nanoparticles for cancer vaccines. J. Control. Release 2013, 168, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Dewitte, H.; Verbeke, R.; Breckpot, K.; de Smedt, S.C.; Lentacker, I. Nanoparticle design to induce tumor immunity and challenge the suppressive tumor microenvironment. Nano Today 2014, 9, 743–758. [Google Scholar] [CrossRef]
- Nune, S.K.; Gunda, P.; Majeti, B.K.; Thallapally, P.K.; Forrest, M.L. Advances in lymphatic imaging and drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Hirosue, S.; Hubbell, J.A. Engineering approaches to immunotherapy. Sci. Trans. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Hanson, M.C.; Rakhra, K.; Tokatlian, T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Leleux, J.; Roy, K. Micro and nanoparticle-based delivery systems for vaccine immunotherapy: An immunological and materials perspective. Adv. Healthc. Mater. 2013, 2, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Sahdev, P.; Ochyl, L.J.; Moon, J.J. Biomaterials for nanoparticle vaccine delivery systems. Pharm. Res. 2014, 31, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Green, J.J. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine 2013, 8, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Singha, S.; Clemente-Casares, X.; Tsai, S.; Yang, Y.; Santamaria, P. Nanoparticle-based immunotherapy for cancer. ACS Nano 2015, 9, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Irvine, D.J. Enhancing cell therapies from the outside in: Cell surface engineering using synthetic nanomaterials. Nano Today 2011, 6, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.; de los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef]
- Cebrian, I.; Visentin, G.; Blanchard, N.; Jouve, M.; Bobard, A.; Moita, C.; Enninga, J.; Moita, L.F.; Amigorena, S.; Savina, A. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 2011, 147, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Samie, M.; Cresswell, P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat. Immunol. 2015, 16, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L. The ins and outs of cross-presentation. Nat. Immunol. 2003, 4, 941–943. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Perez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CDL interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Huster, K.M.; Busch, V.; Schiemann, M.; Linkemann, K.; Kerksiek, K.M.; Wagner, H.; Busch, D.H. Selective expression of IL-7 receptor on memory T cells identifies early CDL-dependent generation of distinct CD8+ memory T cell subsets. Proc. Natl. Acad. Sci. USA. 2004, 101, 5610–5615. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Maeurer, M.J.; Ferrone, S. Antigen-processing machinery breakdown and tumor growth. Immunol. Today 2000, 21, 455–464. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Bloy, N.; Buque, A.; Eggermont, A.; Cremer, I.; Sautes-Fridman, C.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; et al. Trial Watch: Peptide-based anticancer vaccines. Oncoimmunology 2015, 4, e974411. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, D.; Harada, N.; Hayashi, T.; Tahara, Y.; Momose, F.; Sawada, S.; Mukai, S.A.; Akiyoshi, K.; Shiku, H. Nanogel-based immunologically stealth vaccine targets macrophages in the medulla of lymph node and induces potent antitumor immunity. ACS Nano 2014, 8, 9209–9218. [Google Scholar] [CrossRef] [PubMed]
- Jeanbart, L.; Ballester, M.; de Titta, A.; Corthesy, P.; Romero, P.; Hubbell, J.A.; Swartz, M.A. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res. 2014, 2, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, S.; Kleinovink, J.W.; Fransen, M.F.; Mezzanotte, L.; Gold, H.; Wisse, P.; Overkleeft, H.; Amidi, M.; Jiskoot, W.; Lowik, C.W.; et al. Near-infrared labeled, ovalbumin loaded polymeric nanoparticles based on a hydrophilic polyester as model vaccine: In vivo tracking and evaluation of antigen-specific CD8(+) T cell immune response. Biomaterials 2015, 37, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Zeelenberg, I.S.; Srinivas, M.; Bonetto, F.; Weigelin, B.; Eich, C.; de Vries, I.J.; Figdor, C.G. Tracking targeted bimodal nanovaccines: Immune responses and routing in cells, tissue, and whole organism. Mol. Pharm. 2014, 11, 4299–4313. [Google Scholar] [CrossRef] [PubMed]
- Mariani, G.; Moresco, L.; Viale, G.; Villa, G.; Bagnasco, M.; Canavese, G.; Buscombe, J.; Strauss, H.W.; Paganelli, G. Radioguided sentinel lymph node biopsy in breast cancer surgery. J. Nucl. Med. 2001, 42, 1198–1215. [Google Scholar] [PubMed]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Trans. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Unger, W.J.; Storm, G.; van Kooyk, Y.; Mastrobattista, E. Targeting tumor antigens to dendritic cells using particulate carriers. J. Control. Release 2012, 161, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.L.; Lustgarten, J. Targeting the tumor microenvironment with anti-neu/anti-CD40 conjugated nanoparticles for the induction of antitumor immune responses. Vaccine 2010, 28, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Rosalia, R.A.; Cruz, L.J.; van Duikeren, S.; Tromp, A.T.; Silva, A.L.; Jiskoot, W.; de Gruijl, T.; Lowik, C.; Oostendorp, J.; van der Burg, S.H.; et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials 2015, 40, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Rosalia, R.A.; Kleinovink, J.W.; Rueda, F.; Lowik, C.W.; Ossendorp, F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: A comparative study. J. Control. Release 2014, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, K.; Dhodapkar, K.M.; Dhodapkar, M.V. Targeting human dendritic cells in situ to improve vaccines. Immunol. Lett. 2014, 162, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; von Zastrow, M. Signal transduction and endocytosis: Close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 2002, 3, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E.; Tajima, N.; Yoshizaki, Y.; Harada, A.; Hayashi, H.; Kono, K. Dextran derivative-based pH-sensitive liposomes for cancer immunotherapy. Biomaterials 2014, 35, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Wilson, J.T.; Patilea, G.I.; Kern, H.B.; Convertine, A.J.; Stayton, P.S. Neutral polymer micelle carriers with pH-responsive, endosome-releasing activity modulate antigen trafficking to enhance CD8(+) T cell responses. J. Control. Release 2014, 191, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.A.; Stano, A.; Gillard, M.; Maio-Liu, A.C.; Swartz, M.A.; Hubbell, J.A. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials 2012, 33, 6211–6219. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ono, K.; Suzuki, Y.; Moriguchi, R.; Kogure, K.; Harashima, H. Octaarginine-modified liposomes enhance cross-presentation by promoting the C-terminal trimming of antigen peptide. Mol. Pharm. 2014, 11, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lee, I.H.; Kang, S.; Kim, D.; Choi, M.; Saw, P.E.; Shin, E.C.; Jon, S. Gold nanoparticles displaying tumor-associated self-antigens as a potential vaccine for cancer immunotherapy. Adv. Healthc. Mater. 2014, 3, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Mandraju, R.; Murray, S.; Forman, J.; Pasare, C. Differential ability of surface and endosomal TLRs to induce CD8 T cell responses in vivo. J. Immunol. 2014, 192, 4303–4315. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Z.; Kurche, J.S.; Burchill, M.A.; Kedl, R.M. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 2011, 118, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- De Titta, A.; Ballester, M.; Julier, Z.; Nembrini, C.; Jeanbart, L.; van der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc. Natl. Acad Sci. USA 2013, 110, 19902–19907. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.T.; Keller, S.; Manganiello, M.J.; Cheng, C.; Lee, C.C.; Opara, C.; Convertine, A.; Stayton, P.S. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 2013, 7, 3912–3925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chiu, Y.C.; Tostanoski, L.H.; Jewell, C.M. Polyelectrolyte multilayers assembled entirely from immune signals on gold nanoparticle templates promote antigen-specific T cell response. ACS Nano 2015, 9, 6465–6477. [Google Scholar] [CrossRef] [PubMed]
- Molino, N.M.; Anderson, A.K.; Nelson, E.L.; Wang, S.W. Biomimetic protein nanoparticles facilitate enhanced dendritic cell activation and cross-presentation. ACS Nano 2013, 7, 9743–9752. [Google Scholar] [CrossRef] [PubMed]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Zupancic, E.; Vandermeulen, G.; Oliveira, V.G.; Salgado, A.; Videira, M.; Gaspar, M.; Graca, L.; Preat, V.; Florindo, H.F. In vivo delivery of peptides and Toll-like receptor ligands by mannose-functionalized polymeric nanoparticles induces prophylactic and therapeutic anti-tumor immune responses in a melanoma model. J. Control Release 2015, 198, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Sivananthan, S.J.; Duthie, M.S.; Vergara, J.; Guderian, J.A.; Moon, E.; Coblentz, D.; Reed, S.G.; Carter, D. A nanoliposome delivery system to synergistically trigger TLR4 AND TLR7. J. Nanobiotechnol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, P.; Qin, H.; Leleux, J.A.; Gwak, D.; Sakamaki, I.; Kwak, L.W.; Roy, K. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials 2014, 35, 5491–5504. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Ottensmeier, C.H.; Stevenson, F.K. DNA vaccines: Precision tools for activating effective immunity against cancer. Nat. Rev. Cancer 2008, 8, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Senovilla, L.; Vacchelli, E.; Garcia, P.; Eggermont, A.; Fridman, W.H.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: DNA vaccines for cancer therapy. Oncoimmunology 2013, 2, e23803. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Hogberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Shu, Y.; Haque, F.; Abdelmawla, S.; Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011, 6, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xu, B.H.; Xu, J.J.; Shou, D.; Liu, E.G.; Gao, J.Q.; Liang, W.Q.; Huang, Y.Z. Microneedle-assisted dendritic cell-targeted nanoparticles for transcutaneous DNA immunization. Polym. Chem. 2015, 6, 373–379. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, D.; Liu, S.; Gong, J.; Wang, D.; Xiong, M.; Chen, X.; Xiang, R.; Tan, X. Alginic acid-coated chitosan nanoparticles loaded with legumain DNA vaccine: Effect against breast cancer in mice. PLoS ONE 2013, 8, e60190. [Google Scholar] [CrossRef] [PubMed]
- Phua, K.K.; Staats, H.F.; Leong, K.W.; Nair, S.K. Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffres, P.A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccination against BF10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 2011, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhuang, Y.; Zhang, Y.; Luo, Z.; Gao, N.; Li, P.; Pan, H.; Cai, L.; Ma, Y. Toll-like receptor 3 agonist complexed with cationic liposome augments vaccine-elicited antitumor immunity by enhancing TLR3-IRF3 signaling and type I interferons in dendritic cells. Vaccine 2012, 30, 4790–4799. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.P.; Wongrakpanich, A.; Francis, M.B.; Salem, A.K.; Norian, L.A. A therapeutic microparticle-based tumor lysate vaccine reduces spontaneous metastases in murine breast cancer. AAPS J. 2014, 16, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.A.; Huebsch, N.; Cao, L.; Dranoff, G.; Mooney, D.J. Infection-mimicking materials to program dendritic cells in situ. Nat. Mater. 2009, 8, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Hu, C.M.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Ishii, Y.; Matsuo, K.; Yoshinaga, T.; Akashi, M.; Mukai, Y.; Yoshioka, Y.; Okada, N.; Nakagawa, S. The utility of poly(gamma-glutamic acid) nanoparticles as antigen delivery carriers in dendritic cell-based cancer immunotherapy. Biol. Pharm. Bull. 2010, 33, 2003–2007. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Mai, J.; Xu, R.; Perez, J.E.; Guevara, M.L.; Shen, Q.; Mu, C.; Tung, H.Y.; Corry, D.B.; Evans, S.E.; et al. Porous silicon microparticle potentiates anti-tumor immunity by enhancing cross-presentation and inducing type I interferon response. Cell Rep. 2015, 11, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Dewitte, H.; Van Lint, S.; Heirman, C.; Thielemans, K.; de Smedt, S.C.; Breckpot, K.; Lentacker, I. The potential of antigen and TriMix sonoporation using mRNA-loaded microbubbles for ultrasound-triggered cancer immunotherapy. J. Control. Release 2014, 194, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Steenblock, E.R.; Fahmy, T.M. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol. Ther. 2008, 16, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Prakken, B.; Wauben, M.; Genini, D.; Samodal, R.; Barnett, J.; Mendivil, A.; Leoni, L.; Albani, S. Artificial antigen-presenting cells as a tool to exploit the immune “synapse”. Nat. Med. 2000, 6, 1406–1410. [Google Scholar] [PubMed]
- Perica, K.; Tu, A.; Richter, A.; Bieler, J.G.; Edidin, M.; Schneck, J.P. Magnetic field-induced T cell receptor clustering by nanoparticles enhances T cell activation and stimulates antitumor activity. ACS Nano 2014, 8, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Fadel, T.R.; Sharp, F.A.; Vudattu, N.; Ragheb, R.; Garyu, J.; Kim, D.; Hong, E.; Li, N.; Haller, G.L.; Pfefferle, L.D.; et al. A carbon nanotube-polymer composite for T-cell therapy. Nat. Nanotechnol. 2014, 9, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A.; Sunshine, J.C.; Perica, K.; Kosmides, A.K.; Aje, K.; Schneck, J.P.; Green, J.J. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small 2015, 11, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Perica, K.; Schneck, J.P.; Green, J.J. Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials 2014, 35, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spisek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Vitale, I.; Tartour, E.; Eggermont, A.; Sautes-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Anticancer radioimmunotherapy. Oncoimmunology 2013, e25595. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Galluzzi, L.; Fridman, W.H.; Galon, J.; Sautes-Fridman, C.; Tartour, E.; Kroemer, G. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2012, 1, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Joshi, V.B.; Wongrakpanich, A.; Lemke, C.D.; Gross, B.P.; Salem, A.K.; Weiner, G.J. Biodegradable microparticles loaded with doxorubicin and CpG ODN for in situ immunization against cancer. AAPS J. 2015, 17, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Joshi, V.B.; Lemke, C.D.; Wongrakpanich, A.; Olivier, A.K.; Blackwell, S.E.; Salem, A.K.; Weiner, G.J. Three steps to breaking immune tolerance to lymphoma: A microparticle approach. Cancer Immunol. Res. 2015, 3, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Liu, P.; Chen, B.; Mao, Y.; Engelmann, H.; Shin, Y.; Jaffar, J.; Hellstrom, I.; Liu, J.; Hellstrom, K.E. Local release of highly loaded antibodies from functionalized nanoporous support for cancer immunotherapy. J. Am. Chem. Soc. 2010, 132, 6906–6907. [Google Scholar] [CrossRef] [PubMed]
- Roeven, M.W.; Hobo, W.; van der Voort, R.; Fredrix, H.; Norde, W.J.; Teijgeler, K.; Ruiters, M.H.; Schaap, N.; Dolstra, H. Efficient nontoxic delivery of PD-L1 and PD-L2 siRNA into dendritic cell vaccines using the cationic lipid SAINT-18. J. Immunother. 2015, 38, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Teo, P.Y.; Yang, C.; Whilding, L.M.; Parente-Pereira, A.C.; Maher, J.; George, A.J.; Hedrick, J.L.; Yang, Y.Y.; Ghaem-Maghami, S. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: Strategies to enhance T cell killing. Adv. Healthc. Mater. 2015, 4, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.E.; Kovacsovics-Bankowski, M.; Weinberg, A.D. The TNFRs OX40, 4–1BB, and CD40 as targets for cancer immunotherapy. Curr. Opin. Immunol. 2013, 25, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Gong, W.; Demirci, G.; Liu, W.; Spoerl, S.; Chu, X.; Bishop, D.K.; Turka, L.A.; Li, X.C. New insights on OX40 in the control of T cell immunity and immune tolerance in vivo. J. Immunol. 2012, 188, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Park, Y.; Song, A.; Cheroutre, H.; Kwon, B.S.; Croft, M. Functional dichotomy between OX40 and 4–1BB in modulating effector CD8 T cell responses. J. Immunol. 2006, 177, 4464–4472. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, A.T.; Mufti, G.J.; Guinn, B.A. Role of 4-1BB:4-1BB ligand in cancer immunotherapy. Cancer Gene Ther. 2004, 11, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ouyang, H.; Zhou, S.; Li, J.; Ye, Y. PLGA-nanoparticle mediated delivery of anti-OX40 monoclonal antibody enhances anti-tumor cytotoxic T cell responses. Cellular Immunol. 2014, 287, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Kwong, B.; Gai, S.A.; Elkhader, J.; Wittrup, K.D.; Irvine, D.J. Localized immunotherapy via liposome-anchored Anti-CD137+ IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 2013, 73, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Res. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Moon, J.J.; Um, S.H.; Bershteyn, A.; Irvine, D.J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 2010, 16, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Stephan, S.B.; Bak, P.; Chen, J.; Irvine, D.J. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials 2012, 33, 5776–5787. [Google Scholar] [CrossRef] [PubMed]
- Stephan, S.B.; Taber, A.M.; Jileaeva, I.; Pegues, E.P.; Sentman, C.L.; Stephan, M.T. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 2015, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Abraham, W.D.; Zheng, Y.; Bustamante Lopez, S.C.; Luo, S.S.; Irvine, D.J. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Trans. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Lower, M.; Diekmann, J.; Boegel, S.; Schrors, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).